

Abstract
Anaplastic thyroid carcinoma (ATC) is one of the most virulent of all human malignancies, with a mean survival time among patients of less than 1 year after diagnosis. To date, however, cytogenetic information on this disease has been very limited. During the course of a program to screen a panel of ATC cell lines for genomic copy‐number aberrations using array‐based comparative genomic hybridization, we identified a high‐level amplification of the ITCH gene, which is mapped to 20q11.22 and belongs to the homologous to the E6‐associated protein carboxylterminus ubiquitin ligase family. The expression of ITCH was increased in 4 of 14 ATC cell lines (28.6%), including 8305C in which there was a copy‐number amplification of this gene, and six of seven primary cases (85.7%). Among the primary thyroid tumors, a considerable number of ITCH high expressers was found in ATC (40/45, 88.9%), papillary thyroid carcinoma (25/25, 100%), and papillary microcarcinoma (25/25, 100%). Furthermore, knockdown of ITCH by specific small interfering RNA significantly inhibited the growth of ITCH‐overexpressing cells, whereas ectopic overexpression of ITCH promoted growth of ATC cell lines with relatively weak expression. These observations indicate ITCH to be the most likely target for 20q11.22 amplification and to play a crucial role in the progression of thyroid carcinoma. (Cancer Sci 2008; 99: 1940–1949)
Anaplastic thyroid carcinoma (ATC), which accounts for 2–5% of all thyroid cancers, ranks among the most lethal solid tumors, with a mean survival time of only 4–12 months,( 1 , 2 , 3 , 4 ) whereas most patients with well‐differentiated thyroid carcinomas (WDTC), including papillary thyroid carcinoma (PTC) and follicular thyroid carcinoma, have an excellent prognosis. ATC shares several genetic alterations with WDTC, such as point mutations in RAS and BRAF,( 2 , 3 , 4 , 5 ) and point mutations or gene amplification of PIK3CA.( 6 , 7 ) In contrast, TP53 is rarely mutated in WDTC, although more than 70% of ATC are associated with a TP53 mutation.( 3 , 5 ) Together with the circumstantial evidence that ATC is sometimes topographically associated with WDTC,( 8 ) the current consensus holds that most ATC arise from a pre‐existing WDTC, even though some ATC may arise de novo.( 5 , 6 ) However, the molecular basis for the highly malignant phenotype of ATC remains largely unknown. The recent success of targeted therapies for a limited subset of cancer genotypes has solidified the view that a more detailed knowledge of the spectrum of genetic lesions will lead to meaningful progress in ATC therapy.
Significant genomic copy‐number alterations, such as amplifications and homozygous losses, are useful landmarks for identifying oncogenes and tumor‐suppressor genes (TSG), respectively.( 9 , 10 , 11 ) We have carried out bacterial artificial chromosome (BAC) array‐based comparative genomic hybridization (array‐CGH), which allows high resolution, high throughput, and quantitative copy‐number analysis throughout the genome in various cancers, and identified many novel cancer‐related genes.( 11 , 12 , 13 , 14 , 15 ) In ATC, we have reported DUSP26 as a novel growth‐promoting gene from an amplified region detected in ATC cell lines by array‐CGH using an in‐house low‐density cancer‐targeting array (MCG Cancer Array‐800).( 11 , 12 ) It is likely that more genes related to the pathogenesis of ATC remain to be identified, prompting us to screen cryptic copy‐number aberrations in a panel of ATC cell lines with higher resolution.
The itchy homolog ITCH, also known as atrophin 1‐interacting protein 4, belongs to the Nedd4‐like family of E3 ubiquitin ligases.( 16 ) ITCH is characterized by a modular organization that includes: a protein kinase C‐related C2 domain at the N‐terminus, four WW domains, and a HECT (homologous to the E6‐associated protein carboxyl‐terminus) ubiquitin protein ligase domain at the C‐terminus. Itch‐deficient mice develop a fatal disease characterized by constant itching of the skin and the development of severe inflammatory and immune disorders.( 17 ) Nedd4‐like E3 ligases play a crucial role in carcinogenesis, for example, overexpression of Smurf2 in esophageal squamous cell carcinoma,( 18 ) WWP1 in prostate and breast cancer,( 19 , 20 ) Nedd4 in prostate and bladder cancer,( 21 ) and Smurf1 in pancreatic cancer.( 22 , 23 ) However, the relationship between ITCH and cancer remains unknown. Recently, depletion of ITCH by small interfering RNA (siRNA) augmented apoptosis upon treatment with chemotherapeutic drugs.( 24 )
In the course of a program to screen a panel of ATC cell lines by array‐CGH using in‐house high‐density BAC array (MCG Whole Genome Array‐4500) covering the entire human genome with a total of 4523 BAC,( 11 ) we identified novel amplifications at 20q11.22 containing ITCH as a possible amplification target. Compared with the normal thyroid epithelia, overexpression of ITCH protein in primary thyroid tumors, including ATC, was observed. Knockdown of ITCH by siRNA suppressed the growth of ATC cells highly expressing ITCH, whereas ectopic overexpression of ITCH promoted the growth of ATC cells with relatively weak expression. Together, these observations suggest that ITCH contributes to the pathogenesis of thyroid tumors, especially ATC, through overexpression.
Materials and Methods
Cell lines and primary tumor samples. Fourteen ATC cell lines were used in the present study.( 12 ) Each cell line was maintained in appropriate medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 µg/mL streptomycin.
Primary thyroid tumor samples were obtained during surgery from 112 patients being treated at the Ito Hospital (Tokyo, Japan), with written consent from each patient after approval by the local ethics committee. Samples from seven of these patients with ATC were frozen immediately in liquid nitrogen and stored at –80°C until required. Samples from 105 of the patients were embedded in paraffin for immunohistochemistry: 45 ATC, 25 PTC, 25 papillary microcarcinomas (PMC), and 10 adenomatous goiters. None of the patients had received preoperative radiation or chemotherapy.
Array‐CGH analysis. MCG Whole Genome Array‐4500( 11 ) was used for the array‐CGH. Hybridizations were carried out as described elsewhere.( 15 ) Acquired images of hybridized slides were analyzed with GenePix Pro 6.0 imaging software (Axon Instruments, Foster City, CA, USA). Average ratios that deviated significantly (>2 SD) from 0 (log2 ratio, <–0.4 and >0.4) were considered abnormal.
Fluorescence in situ hybridization. Metaphase chromosomes were prepared from normal male lymphocytes and from ATC cell lines. Fluorescence in situ hybridization (FISH) analyses were carried out as described previously,( 12 ) using BAC clones located in the region of interest as probes.
Reverse transcription–polymerase chain reaction. Single‐stranded cDNA was amplified with primers specific for each gene. Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) was amplified to allow estimation of the efficiency of cDNA synthesis. In conventional reverse transcription–polymerase chain reaction (RT‐PCR), products were separated on 3% agarose gels, and band quantification was done using LAS‐3000 and Multi Gauge software (Fuji Film, Tokyo, Japan). Quantitative real‐time RT‐PCR was done with an ABI Prism 7500 Fast Real‐time PCR System and Taqman Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions. Each assay was conducted in duplicate. The expression level of ITCH, normalized to GAPDH, in each sample was divided by that in normal thyroid and recorded as a ‘fold increase’. All of the relevant primer sequences are listed in Suppl. Table S1.
Western blotting. Cells were lysed in protein extraction buffer and analyzed as described elsewhere.( 12 ) Anti‐ITCH and anti‐p21 antibodies were purchased from SantaCruz Biotechnology (Santa Cruz, CA, USA), and anti‐Myc‐tag, anti‐GFP‐tag, anti‐p27, and anti‐β‐actin antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA), MBL (Nagoya, Japan), BD Biosciences Pharmingen (San Jose, CA, USA), and Sigma (St Louis, MO, USA), respectively.
Plasmids. Plasmids expressing Myc‐ or green fluorescent protein (GFP)‐tagged wild‐type ITCH (pCMV‐Tag3‐ITCH WT or pEGFP‐ITCH WT) and mutant‐type ITCH (pCMV‐Tag3‐ITCH MUT or pEGFP‐ITCH MUT) were obtained by cloning the full coding sequences for wild‐type ITCH and a ubiquitin ligase‐inactive mutant form of ITCH, in which Cys‐830 was replaced by Ala,( 25 ) in‐frame into the pCMV‐Tag3 (Stratagene, La Jolla, CA, USA) or pEGFP (Takara, Tokyo, Japan) vector along with the Myc or GFP epitope.
Transfection with synthetic siRNA and cell growth assay. siRNA for ITCH was synthesized by Sigma. The target sequences were GGUGACAAAGAGCCAACAAGAG (siRNA‐ITCH #1), ACAUUAAAGUCAAACAAUAUG (siRNA‐ITCH #2), and GGAGCAACAUCUGGAUUAAUA (siRNA‐ITCH #3).( 24 , 26 ) A control siRNA for the luciferase gene (CGUACGCGGAAUACUUCGA, siRNA‐Luc) was also synthesized by Sigma. Each ATC cell line was transfected with 16.7 nmol/L synthetic siRNA using Lipofectamine RNAiMAX Reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer's protocol. The numbers of viable cells after treatment were assessed by a colorimetric water‐soluble tetrazolium salt assay (cell counting kit‐8; Dojindo, Kumamoto, Japan).( 12 )
Flow cytometry. For the fluorescence‐activated cell sorting (FACS) analysis, cells were trypsinized, fixed in 70% ethanol overnight, and treated sequentially with RNase A (40 U/mL, 30 min) and propidium iodide (20 g/mL, 30 min) in phosphate‐buffered saline. Cells were analyzed for DNA content using a FACSCaliber cytometer and Cell Quest software (BD Biosciences). Experiments were repeated twice.
Colony‐formation assays. pCMV‐Tag3‐ or pEGFP‐ITCH WT, pCMV‐Tag3‐ or pEGFP‐ITCH MUT, or the empty vector (pCMV‐Tag3‐mock or pEGFP‐mock) as a control was transfected into ATC cells as described previously.( 12 ) Expression of ITCH protein in transfected cells was confirmed by western blotting and/or fluorescence microscopy. After 3 weeks of incubation with appropriate concentrations of G418, cells were fixed with 70% ethanol and stained with crystal violet.
Immunohistochemistry and scoring method. Indirect immunohistochemistry was done with formalin‐fixed, paraffin‐embedded tissue sections as described elsewhere.( 27 ) Briefly, antigens were retrieved by microwave pretreatment in citrate buffer (pH 6.0) for 15 min. After blocking in 2% normal swine serum, the slides were incubated with an anti‐ITCH antibody (1:200; SantaCruz) overnight at 4°C, and then reacted with a Histofine simple stain (MAX PO(G); Nichirei, Tokyo, Japan). Antigen–antibody reactions were visualized with 0.2% diaminobenzidine tetrahydrochloride and hydrogen peroxide. The slides were counterstained with Mayer's hematoxylin. The results of immunostaining were evaluated by two reviewers (T.I. and H.T.) independently. The grade of staining intensity was determined by comparing the results for epithelial lesions and a built‐in positive control of smooth muscle cells or vascular endothelial cells in the same section. Grades for epithelial lesions were defined as follows: negative staining (grade 0), stained with an intensity weaker than the built‐in positive control (grade 1), stained with a similar intensity to the built‐in positive control (grade 2), and stained with a stronger intensity than the built‐in positive control (grade 3). Proportions were defined as follows: negative staining (proportion 0); 10–30% of tumor cells stained (proportion I); 31–60% of tumor cells stained (proportion II); and 61–90% of tumor cells stained (proportion III).
Results
High density in‐house BAC array‐based CGH analysis of ATC cell lines. In the genome‐wide copy‐number analysis of 14 ATC cell lines by array‐CGH using MCG Whole Genome Array‐4500,( 11 ) copy‐number gains and losses were seen to some degree in all lines examined as expected from our previous study.( 12 ) As the most common genetic aberrations had already been identified in ATC cell lines,( 12 ) we paid attention to newly detected remarkable patterns of chromosomal abnormalities, such as high‐level amplifications and homozygous deletions, which are likely to be landmarks of novel oncogenes and TSG, respectively. 1, 2 summarize the clones showing high‐level amplifications (log2 ratio > 2.0) and homozygous deletions (log2 ratio < –2.0), respectively. Homozygous deletions were detected in 6 of the 14 ATC cell lines (ARO, KTA‐4, TCO‐1, TTA‐1, TTA‐2, and TTA‐3), and three loci were represented (Table 2). Among them, a homozygous deletion of the region containing CDKN2A/p16 at 9p21.3 is the most frequently (four lines) observed by means of array‐CGH. The region 16q23 harboring WWOX as a possible TSG was detected only in one cell line (KTA‐4), whereas 16q13.2 was identified as a novel region without known TSG in one cell line (ARO). High‐level amplifications were detected in four of the 14 ATC cell lines (ARO, KTA‐3, TTA‐1, and 8305C), and 10 loci were represented. Possible cancer‐related genes, such as MYC (8q24), BIRC2, and YAP1 (11q22), are located at these loci.( 27 , 28 , 29 ) The 8p12 amplicon had also been detected by our low‐density BAC array‐CGH in the same panel of ATC cell lines, and DUSP26 and RNF122 had already been identified as its possible target genes.( 12 ) Among those loci, 20q11.22 has never been reported as a region with high‐level amplification and not only 8305C cells but another ATC cell line, KTA‐4, also showed a moderate level of amplification (log2 ratio = 1.6 in BAC RP11–318 N1) around the same region (Fig. 1a,b). Therefore, we focused on this region to identify novel amplification targets.
Table 1.
Loci of high‐level amplification (log2 ratio > 2.0) detected in anaplastic thyroid carcinoma cell lines by array‐based comparative genomic hybridization analysis using MCG Whole Genome Array‐4500
| Locus no. | Bacterial artificial chromosome | Locus † | Cell line name | Possible candidate gene ‡ | Number of known genes within each locus | |
|---|---|---|---|---|---|---|
| Chromosome band | Position | |||||
| 1 | RP11‐124A11 | 1q41 | 213,712,056–213,886,235 | TTA‐1 | 3 | |
| RP11‐5F19 | 1q41 | 213,843,688–214,005,804 | TTA‐1 | |||
| RP11‐79H5 | 1q41 | 214,152,848–214,333,834 | TTA‐1 | |||
| RP11‐45L21 | 1q41 | 215,048,055–215,223,099 | TTA‐1TTA‐1TTA‐1TTA‐1 | |||
| RP11‐66M7 | 1q41 | 215,209,941–215,373,866 | ||||
| 2 | RP11‐54L9 | 3q28 | 191,443,377–191,627,661 | 8 | ||
| RP11‐455C22 | 3q28 | 191,341,029–191,525,722 | ||||
| RP11‐88H6 | 3q28 | 192,493,851–192,676,936 | TTA‐1TTA‐18305C8305C8305CKTA‐3TTA‐1TTA‐1KTA‐3KTA‐3KTA‐3KTA‐3ARO8305C | |||
| 3 | RP11‐51M22 | 7q31.2 | 115,628,304–115,790,622 | 1 | ||
| 4 | RP11‐451O18 | 8p12 | 33,148,264–33,325,914 | DUSP26, RNF122 | 5 | |
| RP11‐258M15 | 8p12 | 33,641,086–33,802,158 | ||||
| RP11‐91P13 | 8p12 | 33,915,031–34,077,419 | ||||
| 5 | RP11‐142F22 | 8q22.2 | 100,624,093–100,777,928 | 1 | ||
| 6 | RP11‐89K10 | 8q24.21 | 127,636,647–127,799,456 | MYC | 2 | |
| RP11‐89L16 | 8q24.21 | 129,633,607–129,784,954 | ||||
| 7 | RP11‐91M10 | 11q14.1 | 84,821,843–84,975,001 | 0 | ||
| 8 | RP11‐134G19 | 11q22.2 | 101,600,032–101,600,600 | YAP1, BIRC2, BIRC3 | 8 | |
| RP11‐28I24 | 11q22.2 | 101,722,105–101,886,737 | ||||
| RP11‐817J15 | 11q22.2 | 101,922,842–102,095,829 | ||||
| 9 | RP11‐19G24 | 17q12 | 32,528,965–32,674,877 | 1 | ||
| 10 | RP11‐318N1 | 20q11.22 | 32,349,241–32,542,223 | 2 | ||
Based on UCSC Genome Browser, March 2006 Assembly (http://genome.ucsc.edu/cgi‐bin/hgGateway).
Representative candidate oncogene located around the bacterial artificial chromosome.
Table 2.
Loci of homozygous deletion (log2 ratio < –2.0) detected in anaplastic thyroid carcinoma cell lines by array‐based comparative genomic hybridization analysis using MCG Whole Genome Array‐4500
| Locus no. | Bacterial artificial chromosome | Locus † | Cell line | Possible candidate gene ‡ | Number of known genes within each locus | ||
|---|---|---|---|---|---|---|---|
| Chromosome band | Position | n | Name | ||||
| 1 | RP11–113D19 | 9p21.3 | 20,996,401‐21,158,464 | 1 | TTA‐1 | MTAP, CDKN2A, CDK2B | 23 |
| RP11–344A7 | 9p21.3 | 21,506,374‐21,676,227 | 2 | TTA‐1, TTA‐3 | |||
| RP11–408N14 | 9p21.3 | 22,155,847‐22,309,629 | 2 | TTA‐2, TCO‐1 | |||
| RP11–441I5 | 9p21.3 | 22,309,530‐22,479,595 | 2 | TTA‐1, TTA‐2 | |||
| RP11–11J1 | 9p21.3 | 22,479,496‐22,579,721 | 2 | TTA‐1, TTA‐2 | |||
| RP11–782K2 | 9p21.3 | 22,584,981‐22,585,358 | 2 | TTA‐1, TTA‐2 | |||
| RP11–346N23 | 9p21.3 | 22,604,594‐22,795,769 | 2 | TTA‐1, TTA‐2 | |||
| RP11–33O15 | 9p21.3 | 22,823,087‐22,823,490 | 1 | TTA‐1 | |||
| RP11–482I10 | 9p21.3 | 24,547,905‐24,738,555 | 1 | TTA‐1 | |||
| 2 | RP11–185J20 | 16q13.2 | 6,588,011‐6756,741 | 1 | ARO | 1 | |
| 3 | RP11–61L1 | 16q23.1 | 77,344,719‐77,345,302 | 1 | KTA‐4 | WWOX | 1 |
Based on UCSC Genome Browser, March 2006 Assembly (http://genome.ucsc.edu/cgi‐bin/hgGateway).
Representative candidate tumor suppressor located around the bacterial artificial chromosome.
Figure 1.
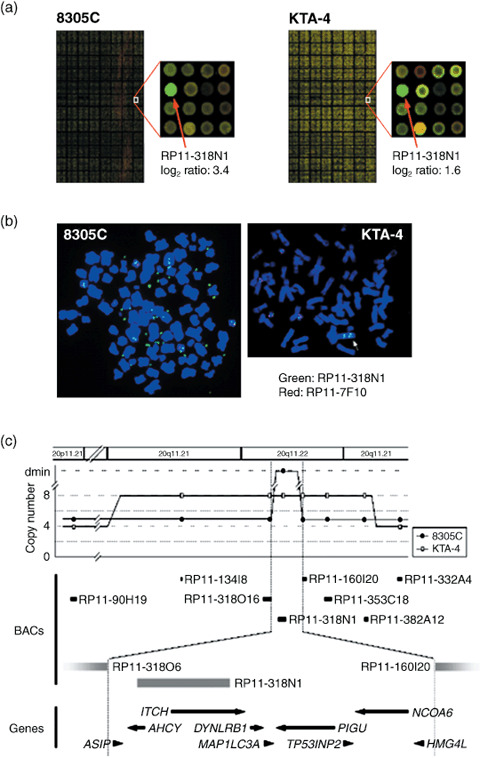
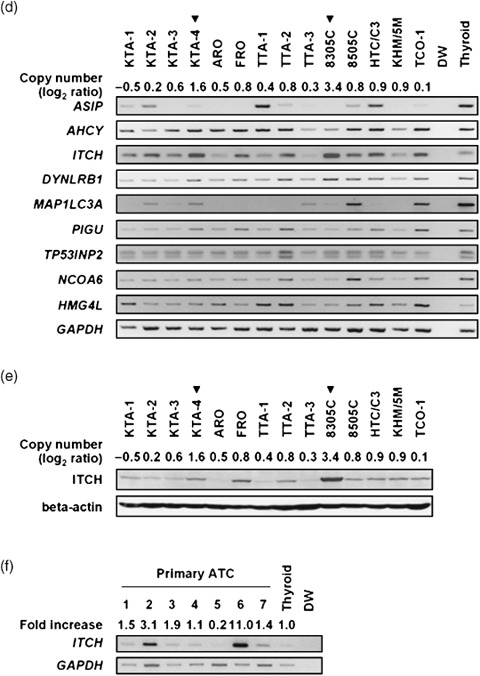
Amplification and overexpression of ITCH in anaplastic thyroid carcinoma (ATC) cell lines. (a) Representative image of array‐based comparative genomic hybridization (array‐CGH) using MCG Whole Genome Array‐4500. A remarkable increase in the copy‐number ratio (log2 ratio) at 20q11.22 was detected as a clear green signal in two ATC cell lines: 8305C and KTA‐4 (red arrows). (b) Representative images of fluorescence in situ hybridization (FISH) analysis on metaphase chromosomes prepared from 8305C and KTA‐4 cells. Bacterial artificial chromosome (BAC) clone RP11‐318 N1 (green signals) was used as a probe for the amplified region together with a chromosome 20 control probe (RP11‐7F10, 20p11.22; red signals). The 8305C cells showed remarkable amplification with a double‐minute chromosome (dmin) pattern, whereas the KTA‐4 cells showed amplification with a tandem‐repeat pattern (arrowhead). (c) Map of 20q11.22 covering the region amplified in ATC cell lines. Eight BAC used for FISH are indicated as black bars. Nine transcripts present in the amplicon are indicated. All markers and transcripts located within the smallest region of overlap (SRO) are positioned according to the human genome databases (http://www.ncbi.nlm.nih.gov/ and http://genome.ucsc.edu/). (d) Expression of genes located on the 20q11.22 amplicon in ATC cell lines and normal thyroid as a control were determined by reverse transcription–polymerase chain reaction (RT‐PCR). DW, distilled water as a negative control. Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used for estimation of the efficiency of cDNA synthesis. Arrowheads indicate the cell lines with 20q11.22 amplification detected by array‐CGH. The expression pattern of ITCH was correlated with its copy‐number status in ATC cell lines. (e) Western blot analysis of the ITCH protein level in ATC cell lines. Expression levels of ITCH protein were also correlated with the copy‐number status of this gene. (f) RT‐PCR analysis of ITCH expression in seven primary ATC and normal thyroid tissue. The band quantification was carried out using LAS‐3000 and Multi Gauge software. The expression level of ITCH, normalized with GAPDH, in each sample was divided by that in normal thyroid, and indicated as a fold increase.
Amplicon mapping and expression analysis at 20q11.22 in ATC cell lines. To define a map of the 20q11.22 amplicon, we carried out a series of FISH experiments in the 8305C and KTA‐4 cell lines using seven BAC mapped around RP11‐318N1, which showed remarkably high intensity in the array‐CGH analysis in both cell lines (Fig. 1a), and one control BAC at 20p11 (Fig. 1b). In 8305C cells, RP11‐318N1 produced the highest number of signals with a pattern of double‐minute chromosomes (dmins; Fig. 1b). In KTA‐4, on the other hand, six BAC (RP11‐134I8, 318O16, 318 N1, 160I20, 353C18, and 382A12) produced eight signals with a tandem repeat pattern (Fig. 1b). Therefore, the smallest region of overlap (SRO) is approximately 0.6 Mb in extent, and nine genes (ASIP, AHCY, ITCH, DYNLRB1, MAP1LC3A, TP53INP2, PIGU, NCOA6, and HMG4L) locate within this SRO (Fig. 1c), according to the information in the human genome databases (http://www.ncbi.nlm.nih.gov/ and http://genome.ucsc.edu/). To identify the most likely target genes, we determined the correlation between gene amplification and the expression status of each gene. Among nine genes within this SRO, only the ITCH gene was remarkably overexpressed in the 8305C and KTA‐4 cell lines in an amplification‐dependent manner, and also expressed in most of the ATC cell lines compared with normal thyroid by as assessed by RT‐PCR (Fig. 1d). This result was also confirmed by quantitative real‐time RT‐PCR analysis (Suppl. Fig. S1). As targets for amplification seem to show a positive correlation between their copy number and expression status,( 12 ) ITCH might be the most likely target gene for this amplicon. Western blotting confirmed overexpression of ITCH in 8305C and KTA‐4 cells at the protein level as well, although a discrepancy between the mRNA and protein level was observed in some ATC cell lines (Fig. 1d,e). Overexpression of ITCH mRNA was also observed frequently in primary ATC tumors, although only seven tumors were available for conventional RT‐PCR analysis (Fig. 1f).
Overexpression of ITCH in primary thyroid tumor. To assess the expression status of ITCH in primary thyroid tumors including ATC, we carried out immunohistochemical analysis of the ITCH protein in 105 primary tumors: 45 ATC, 25 PTC, 25 PMC, and 10 adenomatous goiters (Suppl. Table S2). The specificity of the anti‐ITCH antibody was confirmed using a cell line overexpressing ITCH (8305C) and another almost lacking ITCH (ARO) as positive and negative controls, respectively (Fig. 2a,b). Representative immunostaining patterns of ITCH are shown in Figure 2 and summarized in Table 3. In ATC (Fig. 2d,e), PMC (Fig. 2f), and PTC (Fig. 2g), positive immunostaining was observed, which was defined as grade 1–3 by comparing the results for epithelial lesions and a built‐in positive control of smooth muscle cells and/or vascular endothelial cells in the same sections, although 5 of 45 ATC tumors (11.1%; Table 3) lacked ITCH immunoreactivity (grade 0; Fig. 2h). In adenomatous goiter, weak (grade 1, Fig. 2i) or no (grade 0) staining for ITCH was observed. Whereas the proportion of immunopositive tumor cells was similar among the three malignant tumors, less than 60% of tumor cells showed immunoreactivity in most cases of adenomatous goiter.
Figure 2.
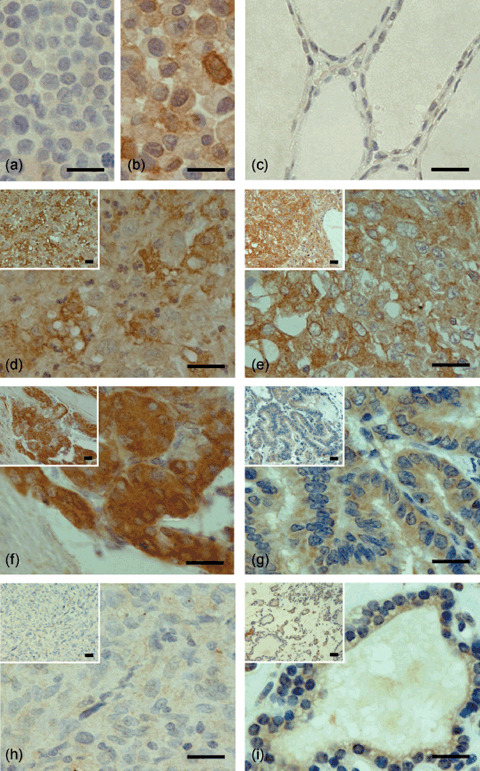
Examples of ITCH immunostaining in primary thyroid carcinomas and benign thyroid tumor (also see Table 3). (a) ARO cell line; (b) 8305C cell line; (c) normal thyroid; (d) anaplastic thyroid carcinoma (ATC) (grade 3 intensity); (e) ATC (grade 2 intensity); (f) papillary microcarcinoma (grade 3 intensity); (g) papillary thyroid carcinoma (grade 2 intensity); (h) ATC (grade 0 intensity); and (i) adenomatous goiter (grade 1 intensity). To evaluate the expression level of ITCH protein, we classified the intensity of its immunoreactivity into four levels (grade 0–3) and the proportion of immunopositive cells into four levels (proportion 0–III). Scale bars = 20 µm.
Table 3.
Expression pattern of ITCH in primary thyroid tumor
| Tumor type | n | ITCH immunoreactivity | |||||
|---|---|---|---|---|---|---|---|
| Intensity (grade) † | n | % | Proportion ‡ | n | % | ||
| Malignant tumor | |||||||
| Anaplastic thyroid carcinoma | 45 | 3 | 3 | 6.7 | III | 21 | 46.7 |
| 2 | 15 | 33.3 | II | 12 | 26.7 | ||
| 1 | 22 | 48.9 | I | 7 | 15.6 | ||
| 0 | 5 | 11.1 | 0 | 5 | 11.1 | ||
| Papillary thyroid carcinoma | 25 | 3 | 0 | 0.0 | III | 16 | 64.0 |
| 2 | 14 | 56.0 | II | 1 | 4.0 | ||
| 1 | 11 | 44.0 | I | 8 | 32.0 | ||
| 0 | 0 | 0.0 | 0 | 0 | 0.0 | ||
| Papillary microcarcinoma | 25 | 3 | 1 | 4.0 | III | 20 | 80.0 |
| 2 | 10 | 40.0 | II | 3 | 12.0 | ||
| 1 | 14 | 56.0 | I | 2 | 8.0 | ||
| 0 | 0 | 0.0 | 0 | 0 | 0.0 | ||
| Benign tumor | |||||||
| Adenomatous goiter | 10 | 3 | 0 | 0.0 | III | 1 | 10.0 |
| 2 | 0 | 0.0 | II | 5 | 50.0 | ||
| 1 | 9 | 90.0 | I | 3 | 30.0 | ||
| 0 | 1 | 10.0 | 0 | 1 | 10.0 | ||
| Normal thyroid | 0 | 0 | |||||
Intensity (grade): 0, negative staining; 1, stained with an intensity weaker than the built‐in positive control; 2, stained with a similar intensity to the built‐in positive control; and 3, stained with a stronger intensity than the built‐in positive control.
Proportion: 0, negative staining; I, 10–30% of tumor cells stained; II, 31–60% of tumor cells stained; and III, 61–90% of tumor cells stained.
Note that normal thyroid epithelial cells observed in each tumor tissue sample showed no immunoreactivity.
Oncogenic activity of ITCH in ATC cells. To investigate the effect of ITCH overexpression on the growth of ATC cells, we first carried out a cell‐growth assay after knockdown of ITCH using specific siRNA. In 8305C cells as a preliminary experiment, all three of the ITCH siRNA (siRNA‐ITCH #1–3) induced knockdown of ITCH at the protein level 24–72 h after transfection and inhibited cell growth compared with their non‐specific control siRNA‐ (siRNA‐Luc‐) and mock‐transfected counterparts at least 72 h after transfection (Suppl. Fig. S2A,B). Endogenous ITCH protein expression was decreased 24–72 h after the introduction of siRNA‐ITCH #1, the most effective siRNA, compared with control siRNA‐Luc in both 8305C and KTA‐4 cell lines having ITCH amplification and overexpression (Fig. 3a,b). Cell growth was also inhibited by siRNA‐ITCH #1 compared with its control counterparts (Fig. 3a,b). The growth‐inhibitory effect of siRNA‐ITCH #1 in KTA‐4 cells was less than that in 8305C cells (69 and 77% inhibition compared with siRNA‐Luc at 72 h after transfection respectively; Fig. 3a,b), suggesting that the effect may depend on the ITCH protein expression level (Fig. 1e). To further examine the mode of action of ITCH on the growth of ATC cells, we analyzed the cell cycle in siRNA‐ITCH #1‐introduced 8305C cells and their control counterparts by FACS. Suppression of ITCH expression resulted in G1 arrest, represented by an increase in the proportion of cells in G0–G1 phase and a decrease in the proportion of cells in S phase (Fig. 3c). Consistent with this observation, the protein expression levels of two cyclin‐dependent kinases, p27 and p21, were also increased in siRNA‐ITCH #1‐transfected 8305C cells compared with their control counterparts (Suppl. Fig. S3). Together with these findings and no morphological changes, such as nuclear condensation and fragmentation (data not shown), suppression of ITCH alone inhibits cell growth through induction of G1 arrest without inducing apoptosis.
Figure 3.
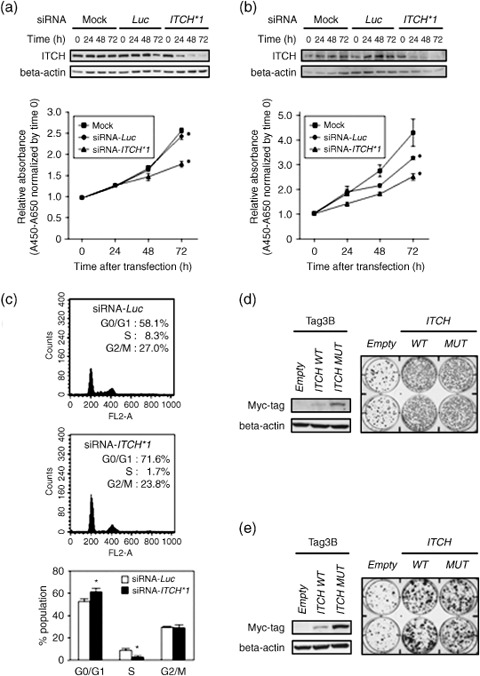
(a–c) Effect of ITCH downregulation on the growth of anaplastic thyroid carcinoma (ATC) cell lines. Representative results of western blotting and growth curves of (a) 8305C and (b) KTA‐4 cells transfected with ITCH‐specific small interfering RNA (siRNA) (siRNA‐ITCH #1) compared with their counterparts transfected with siRNA‐Luc or vehicle alone (mock; Lipofectamine RNAiMAX). The knockdown effects of siRNA were examined 24–72 h after transfection in these cell lines. The number of viable cells 24–72 h after transfection was assessed using a water‐soluble tetrazolium salt assay. Each data point represents the mean of triplicate determinations (bars, SD) in these experiments. *P < 0.05; statistical analysis used the Mann–Whitney U‐test. (c) Representative results of the population in each phase of the cell cycle assessed by fluorescence‐activated cell sorting (FACS) using 8305C cells 72 h after transfection of (top) control siRNA‐Luc or (middle) siRNA‐ITCH #1. siRNA‐ITCH #1‐transfected cells accumulated in G0–G1 phase with a decrease in the S‐phase population compared with their siRNA‐Luc‐transfected counterparts. (Bottom) Note that the transfection of ITCH‐specific siRNA mainly induced the accumulation of G0–G1‐phase cells and a decrease in S‐phase cells. *P < 0.05 versus siRNA‐Luc; statistical analysis used the Mann–Whitney U‐test in three different experiments. Colony‐formation assays using the (d) TTA‐1 and (e) 8505C cell lines. Cells with relatively weak ITCH expression were transiently transfected with Myc‐tagged constructs containing empty vector (pCMV‐Tag3‐empty), wild‐type ITCH (pCMV‐Tag3‐ITCH WT), or a ubiquitin ligase‐inactive mutant of ITCH (pCMV‐Tag3‐ITCH MUT), and selected with appropriate concentrations of G418 for 3 weeks. (Left) Western blotting prepared with 30 µg of protein extract and anti‐Myc tag‐specific antibody, demonstrating that cells transiently transfected with pCMV‐Tag3‐ITCH WT or pCMV‐Tag3‐ITCH MUT expressed Myc‐tagged wild‐type or mutant ITCH. (Right) Three weeks after transfection and subsequent selection of drug‐resistant colonies, the colonies formed by ITCH WT‐ or ITCH MUT‐transfected cells were more numerous than those formed by empty vector‐transfected cells.
To confirm the growth‐promoting effect of ITCH in ATC cells and investigate whether it is dependent on the ubiquitin ligase activity of ITCH, we carried out a colony‐formation assay by transiently transfecting an expression construct of wild‐type or a ubiquitin ligase‐inactive mutant (Cys830Ala) of ITCH into the TTA‐1 and 8505C ATC cell lines, which exhibit relatively lower expression levels of ITCH (Fig. 1d,e, Suppl. Fig. S1). Ectopic expression of wild‐type and mutant ITCH with a Myc or GFP tag in these cell lines was verified by western blotting using epitope tag‐specific antibodies (Fig. 3d,e, Suppl. Fig. S4A,B). Both the wild‐type and ubiquitin ligase‐inactive form of ITCH produced remarkably more colonies than did the empty plasmid, suggesting that ITCH may promote cell proliferation in a ubiquitin ligase‐independent manner in ATC cells (Fig. 3d,e, Suppl. Fig. S4A,B). As Myc‐tagged wild‐type ITCH expressed a lower level of protein compared with mutant ITCH, we analyzed the expression of epitope‐tagged transcripts at the mRNA level using tagged transcript‐specific primer sets (Suppl. Fig. S5). In the Myc‐ and GFP‐tagged constructs, no difference was observed in the level of mRNA between wild‐type and mutant ITCH transfectants, suggesting that the difference observed in the protein levels may be caused by post‐transcriptional or post‐translational mechanisms including autoubiquitination.
Discussion
Despite the increasing efforts of clinicians in their fight against the disease, ATC remains one of the deadliest of malignancies. Therefore, novel treatment strategies are necessary if we are to make any progress in treating ATC. Through the genetic copy‐number screening‐assisted approach, we identified a member of the HECT subclass of the E3 ubiquitin ligase family, ITCH,( 30 ) from a novel amplicon detected in ATC cell lines as a frequently overexpressed amplification target, which may be associated with the progression of thyroid cancer including ATC, and a candidate therapeutic target. Gene expression levels in cancer cells can be abnormally regulated through multiple mechanisms, such as genomic structural alterations, epigenetic modifications, and post‐transcriptional and post‐translational modifications. In some genes involved in tumor progression, including ITCH, overexpression through copy‐number gain or amplification might be rare but can be a useful landmark for their identification.( 9 , 10 , 11 , 12 )
Using conventional CGH, gain of 20q has been detected as a chromosomal abnormality present exclusively in poorly differentiated thyroid cancer and ATC, suggesting it to be a transitional event in the progression of WDTC to poorly differentiated thyroid cancer( 31 ) or ATC.( 32 ) However, possible target genes on 20q associated with the malignant progression of thyroid cancers remain largely unknown, although remarkable overexpression of one putative oncogene located at 20q13.12, UBE2C (also known as UbcH10), was found in ATC through the screening of ATC cell lines using expression array.( 33 ) Genetic and cytogenetic studies have revealed frequent amplification of the chromosomal arm 20q in various human cancers, and of the nine genes located within the 20q11.22 amplicon observed in ATC cell lines in the present study, NCOA6 ( 34 ) and PIGU ( 35 ) have been reported as putative target oncogenes in breast and bladder cancers, respectively. However, our expression analysis demonstrated that these two genes were not overexpressed in ATC cells compared with normal thyroid in an amplification‐dependent manner,( 12 ) suggesting that NCOA6 and PIGU may not work as oncogenes in this disease.
Among the genes involved in ATC, several molecules, such as those for BRAF serine‐threonine kinase, PLK1 tyrosine kinase, and the CXCR4 chemokine receptor, have been exploited as potential therapeutic targets because of experimental evidence showing growth inhibition after their knockdown in ATC cells.( 36 , 37 , 38 ) Here, we demonstrated that downregulation of ITCH expression by gene‐specific siRNA also inhibits the growth of ATC cell lines overexpressing this gene. The growth‐inhibitory effect of ITCH‐specific siRNA depends on the expression levels of this gene in ATC lines, indicating that ITCH promotes cell growth in a dose‐dependent manner. Together with the overexpression of ITCH protein in some primary thyroid cancers, including ATC, our results suggest that at least a subset of thyroid cancers, including ATC, may be ‘addicted’ to ITCH for their progression,( 39 , 40 ) although the growth‐inhibitory effect of ITCH knockdown on normal thyroid follicular cells was not evaluated.
Our immunohistochemical analysis demonstrated that not only ATC but also PTC and PMC show overexpression of ITCH. Although both PTC and ATC develop in the human thyroid, there are some significant differences between them regarding epidemiology, pathology, and prognosis. For example, most cases of PTC occur at a reproductive age (30–49 years old), especially in women,( 41 ) whereas ATC usually occurs beyond the sixth decade of life and without a sex preference.( 42 ) In addition, the prognosis of PTC is much better than that of ATC. Such evidence suggests that ITCH is not specifically involved in the malignant phenotype of ATC. Recently, the CXCR4 chemokine receptor was shown to be overexpressed in primary tumors and cell lines of ATC, and downregulation of CXCR4 expression or activity inhibited ATC cell growth, although CXCR4 overexpression had also been reported in primary PTC.( 43 ) Therefore, at least some ATC may share the same or a similar pathway or lineage of carcinogenesis with PTC, although it remains unknown whether mechanisms of overexpression of ITCH and CXCR4 are different or not between ATC and PTC.( 38 , 43 )
As ITCH is a member of the HECT subclass of E3 ubiquitin ligases( 30 ) and the proteasome inhibitor bortezomib has been shown to exhibit antitumor effects on ATC cell lines,( 44 ) we speculated that the growth‐promoting effect of ITCH in ATC cells is mediated through its ubiquitin ligase activity. However, our colony‐formation assay using ATC cell lines expressing low levels of ITCH demonstrated an a similar increase in the number of colonies after ectopic overexpression of ITCH in both wild‐type cells and a catalytic mutant unable to induce ubiquitination (Cys830Ala).( 25 ) As the same results were obtained in different epitope‐tagged constructs (Fig. 3, Suppl. Fig. S4), it is suggested that the growth‐promoting effect of ITCH is independent of its ubiquitin ligase activity. Indeed, it was shown that ITCH functions to turn off transforming growth factor (TGF)‐β signaling independent of its ubiquitin ligase activity,( 45 ) and some ATC cells lose sensitivity to TGF‐β, which generally inhibits the growth of epithelial cells,( 46 ) suggesting that unknown mechanisms that inhibit TGF‐β signaling by ITCH contribute to the pathogenesis of thyroid cancers, especially ATC. Recently, Mitsiades et al. reported that a growth‐inhibitory effect of the proteasome inhibitor bortezomib was observed in anaplastic and medullary thyroid cancer cell lines, but some anaplastic and papillary thyroid cancer cell lines were resistant to this inhibitor.( 44 ) However, they did not use 8305C and KTA‐4 and did not verify the expression level of ITCH or other specific components in the ubiquitin–proteasome pathway. However, as UbcH10 (belonging to the E2 gene family), which is involved in ubiquitin‐dependent proteolysis and located at 20q13.12, is also known to be overexpressed frequently in ATC,( 33 ) it is still possible that endogenously overexpressed ITCH acts as an oncogene with overexpressed UbcH10 synergistically through its ubiquitin ligase activity in this tumor. It will be of interest to explore the additional functions of the ITCH protein and their contribution to the carcinogenesis of various human tissues, including the thyroid.
In summary, we have shown that ITCH, belonging to the HECT ubiquitin ligase family, is the most likely target for 20q11.22 amplification in ATC. This is the first report to present data on the overexpression of ITCH in ATC cell lines as well as primary thyroid carcinomas, indicating that ITCH plays a crucial role in the progression of thyroid carcinoma. Because ATC cells with amplification and overexpression of ITCH showed ‘addiction’ to this molecule in cell growth, it is suggested that ITCH may be a useful target for developing novel therapeutic agents.
Supporting information
Fig. S1. Relative ITCH mRNA expression levels in anaplastic thyroid carcinoma (ATC) cell lines and normal thyroid as a control determined by real‐time reverse transcription–polymerase chain reaction. The expression level of ITCH, normalized with glyceraldehyde‐3‐phosphate dehydrogenase, in each sample was divided by that in normal thyroid, and is indicated as a fold increase.
Fig. S2. Effects of different small interfering RNA (siRNA) molecules (siRNA‐ITCH #1–3) on (a) ITCH protein expression and (b) cell growth of the 8305C cell line. Each data point represents the mean of triplicate determinations (bars, SD) in these experiments. *P < 0.05 versus siRNA‐Luc at each time after transfection; statistical analysis used the Mann–Whitney U‐test.
Fig. S3. Effect of ITCH downregulation on the expression levels of p27 and p21 in the 8305C cell line. Expression of p21 and p27 was induced 72 h after transfection of siRNA‐ITCH #1.
Fig. S4. Colony‐formation assays using the (a) TTA‐1 and (b) 8505C cell lines. Cells with relatively lower ITCH expression were transfected transiently with a green fluorescent protein (GFP)‐tagged construct containing empty vector (pEGFP‐mock), wild‐type ITCH (pEGFP‐ITCH WT), or a ubiquitin ligase‐inactive mutant of ITCH (pEGFP‐ITCH MUT), and selected with appropriate concentrations of G418 for 3 weeks. (Left) Western blotting prepared with 30 µg of protein extract and anti‐GFP tag‐specific antibody. (Right) At 3 weeks after transfection and subsequent selection of drug‐resistant colonies, the colonies formed by ITCH WT‐ or ITCH MUT‐transfected cells were more numerous than those formed by empty vector‐transfected cells.
Fig. S5. Representative results of reverse transcription–polymerase chain reaction using tagged transcript‐specific primer sets in (a) TTA‐1 and (b) 8505C cell lines transiently transfected with (top) Myc‐tagged constructs containing empty vector (pCMV‐Tag3‐empty), wild‐type ITCH (pCMV‐Tag3‐ITCH WT), or a ubiquitin ligase‐inactive mutant of ITCH (pCMV‐Tag3‐ITCH MUT), or those with (bottom) a GFP‐tagged constructs containing empty vector (pEGFP‐mock), wild‐type ITCH (pEGFP‐ITCH WT), or a ubiquitin ligase‐inactive mutant of ITCH (pEGFP‐ITCH MUT) for colony‐formation assays. DW, distilled water as a negative control. Glyceraldehyde‐3‐phosphate dehydrogenase was used to allow estimation of the efficiency of cDNA synthesis.
Table S1. Primer sequencies used in reverse transcription–polymerase chain reaction analysis
Table S2. Clinicopathological characteristics of primary thyroid tumors
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
We are grateful to Professor Masaki Noda (Tokyo Medical and Dental University) for continuous encouragement throughout this work. We also thank Ayako Takahashi and Rumi Mori for technical assistance. This study was supported in part by Grants‐in‐Aid for Scientific Research and Scientific Research on Priority Areas, and the 21st Century Center of Excellence Program for Molecular Destruction and Reconstitution of Tooth and Bone from the Ministry of Education, Culture, Sports, Science, and Technology, Japan, a grant from Core Research for Evolutional Science and Technology of Japan Science and Technology Corporation, and a grant from the New Energy and Industrial Technology Development Organization.
References
- 1. Sherman SI. Thyroid carcinoma. Lancet 2003; 361: 501–11. [DOI] [PubMed] [Google Scholar]
- 2. Fagin JA. How thyroid tumors start and why it matters: kinase mutants as targets for solid cancer pharmacotherapy. J Endocrinol 2004; 183: 249–56. [DOI] [PubMed] [Google Scholar]
- 3. Kondo T, Ezzat S, Asa SL. Pathogenetic mechanisms in thyroid follicular‐cell neoplasia. Nat Rev Cancer 2006; 6: 292–306. [DOI] [PubMed] [Google Scholar]
- 4. Espinosa AV, Porchia L, Ringel MD. Targeting BRAF in thyroid cancer. Br J Cancer 2007; 96: 16–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nikiforov YE. Genetic alterations involved in the transition from well‐differentiated to poorly differentiated and anaplastic thyroid carcinomas. Endocr Pathol 2004; 15: 319–27. [DOI] [PubMed] [Google Scholar]
- 6. Garcia‐Rostan G, Costa AM, Pereira‐Castro I et al . Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res 2005; 15: 10 199–207. [DOI] [PubMed] [Google Scholar]
- 7. Wu G, Mambo E, Guo Z et al . Uncommon mutation, but common amplifications, of the PIK3CA gene in thyroid tumors. J Clin Endocrinol Metab 2005; 90: 4688–93. [DOI] [PubMed] [Google Scholar]
- 8. Are C, Shaha AR. Anaplastic thyroid carcinoma: biology, pathogenesis, prognostic factors, and treatment approaches. Ann Surg Oncol 2006; 13: 453–64. [DOI] [PubMed] [Google Scholar]
- 9. Snijders AM, Nowak N, Segraves R et al . Assembly of microarrays for genome‐wide measurement of DNA copy number. Nat Genet 2001; 29: 263–4. [DOI] [PubMed] [Google Scholar]
- 10. Albertson DG, Pinkel D. Genomic microarrays in human genetic disease and cancer. Hum Mol Genet 2003; 12: 145–52. [DOI] [PubMed] [Google Scholar]
- 11. Inazawa J, Inoue J, Imoto I. Comparative genomic hybridization (CGH)‐arrays pave the way for identification of novel cancer‐related genes. Cancer Sci 2004; 95: 559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu W, Imoto I, Inoue J, Onda M, Emi M, Inazawa J. A novel amplification target, DUSP26, promotes anaplastic thyroid cancer cell growth by inhibiting p38 MAPK activity. Oncogene 2007; 26: 1178–87. [DOI] [PubMed] [Google Scholar]
- 13. Sonoda I, Imoto I, Inoue J et al . Frequent silencing of low density lipoprotein receptor‐related protein 1B (LRP1B) expression by genetic and epigenetic mechanisms in esophageal squamous cell carcinoma. Cancer Res 2004; 64: 3741–7. [DOI] [PubMed] [Google Scholar]
- 14. Takada H, Imoto I, Tsuda H et al . Genomic loss and epigenetic silencing of very‐low‐density lipoprotein receptor involved in gastric carcinogenesis. Oncogene 2006; 25: 6554–62. [DOI] [PubMed] [Google Scholar]
- 15. Imoto I, Izumi H, Yokoi S et al . Frequent silencing of the candidate tumor suppressor PCDH20 by an epigenetic mechanism in non‐small‐cell lung cancers. Cancer Res 2006; 66: 4617–26. [DOI] [PubMed] [Google Scholar]
- 16. Perry WL, Hustad CM, Swing DA, O'Sullivan TN, Jenkins NA, Copeland NG. The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18H mice. Nat Genet 1998; 18: 143–6. [DOI] [PubMed] [Google Scholar]
- 17. Fang D, Elly C, Gao B et al . Dysregulation of T lymphocyte function in itchy mice: a role for Itch in TH2 differentiation. Nat Immunol 2002; 3: 281–7. [DOI] [PubMed] [Google Scholar]
- 18. Fukuchi M, Fukai Y, Masuda N et al . High‐level expression of the Smad ubiquitin ligase Smurf2 correlates with poor prognosis in patients with esophageal squamous cell carcinoma. Cancer Res 2002; 62: 7162–75. [PubMed] [Google Scholar]
- 19. Chen C, Sun X, Guo P et al . Ubiquitin E3 ligase WWP1 as an oncogenic factor in human prostate cancer. Oncogene 2007; 26: 2386–94. [DOI] [PubMed] [Google Scholar]
- 20. Chen C, Zhou Z, Ross JS, Zhou W, Dong JT. The amplified WWP1 gene is a potential molecular target in breast cancer. Int J Cancer 2007; 121: 2834–41. [DOI] [PubMed] [Google Scholar]
- 21. Wang X, Trotman LC, Koppie T et al . NEDD4‐1 is a proto‐oncogenic ubiquitin ligase for PTEN. Cell 2007; 128: 129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bashyam MD, Bair R, Kim YH et al . Array‐based comparative genomic hybridization identifies localized DNA amplifications and homozygous deletions in pancreatic cancer. Neoplasia 2005; 7: 556–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Loukopoulos P, Shibata T, Katoh H et al . Genome‐wide array‐based comparative genomic hybridization analysis of pancreatic adenocarcinoma: identification of genetic indicators that predict patient outcome. Cancer Sci 2007; 98: 392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hansen TM, Rossi M, Roperch JP et al . Itch inhibition regulates chemosensitivity in vitro . Biochem Biophys Res Commun 2007; 36: 33–6. [DOI] [PubMed] [Google Scholar]
- 25. Winberg G, Matskova L, Chen F et al . Latent membrane protein 2A of Epstein–Barr virus binds WW domain E3 protein–ubiquitin ligases that ubiquitinate B‐cell tyrosine kinases. Mol Cell Biol 2000; 20: 8526–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marchese A, Raiborg C, Santini F, Keen JH, Stenmark H, Benovic JL. The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein‐coupled receptor CXCR4. Dev Cell 2003; 5: 709–72. [DOI] [PubMed] [Google Scholar]
- 27. Imoto I, Tsuda H, Hirasawa A et al . Expression of cIAP1, a target for 11q22 amplification, correlates with resistance of cervical cancers to radiotherapy. Cancer Res 2002; 62: 4860–6. [PubMed] [Google Scholar]
- 28. Dalla‐Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c‐myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci USA 1982; 79: 7824–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zender L, Spector MS, Xue W et al . Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006; 125: 1253–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huibregtse JM, Scheffner M, Beaudenon S, Howley PM. A family of proteins structurally and functionally related to the E6‐AP ubiquitin–protein ligase. Proc Natl Acad Sci USA 1995; 92: 2563–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wreesmann VB, Ghossein RA, Patel SG et al . Genome‐wide appraisal of thyroid cancer progression. Am J Pathol 2002; 161: 1549–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee JJ, Foukakis T, Hashemi J et al . Molecular cytogenetic profiles of novel and established human anaplastic thyroid carcinoma models. Thyroid 2007; 17: 289–301. [DOI] [PubMed] [Google Scholar]
- 33. Pallante P, Berlingieri MT, Troncone G et al . UbcH10 overexpression may represent a marker of anaplastic thyroid carcinomas. Br J Cancer 2005; 93: 464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee SK, Anzick SL, Choi JE et al . A nuclear factor ASC‐2, as a cancer‐amplified transcriptional coactivator essential for ligand‐dependent transactivation by nuclear receptors in vivo . J Biol Chem 1999; 274: 34 283–93. [DOI] [PubMed] [Google Scholar]
- 35. Guo Z, Linn JF, Wu G et al . CDC91L1 (PIG‐U) is a newly discovered oncogene in human bladder cancer. Nat Med 2004; 10: 374–81. [DOI] [PubMed] [Google Scholar]
- 36. Salvatore G, De Falco V, Salerno P et al . BRAF is a therapeutic target in aggressive thyroid carcinoma. Clin Cancer Res 2006; 12: 1623–9. [DOI] [PubMed] [Google Scholar]
- 37. Salvatore G, Nappi TC, Salerno P et al . A cell proliferation and chromosomal instability signature in anaplastic thyroid carcinoma. Cancer Res 2007; 67: 10 148–58. [DOI] [PubMed] [Google Scholar]
- 38. De Falco V, Guarino V, Avilla E et al . Biological role and potential therapeutic targeting of the chemokine receptor CXCR4 in undifferentiated thyroid cancer. Cancer Res 2007; 67: 11 821–9. [DOI] [PubMed] [Google Scholar]
- 39. Weinstein IB. Addiction to oncogenes – the Achilles heal of cancer. Science 2002; 297: 63–4. [DOI] [PubMed] [Google Scholar]
- 40. Weinstein IB, Joe AK. Mechanisms of disease: oncogene addiction – a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol 2006; 3: 448–57. [DOI] [PubMed] [Google Scholar]
- 41. Hundahl SA, Cady B, Cunningham MP et al . Initial results from a prospective cohort study of 5583 cases of thyroid carcinoma treated in the United States during 1996. Cancer 2000; 89: 202–17. [DOI] [PubMed] [Google Scholar]
- 42. Gimm O. Thyroid cancer. Cancer Lett 2001; 163: 143–56. [DOI] [PubMed] [Google Scholar]
- 43. Castellone MD, Guarino V, De Falco V et al . Functional expression of the CXCR4 chemokine receptor is induced by RET/PTC oncogenes and is a common event in human papillary thyroid carcinomas. Oncogene 2004; 23: 5958–67. [DOI] [PubMed] [Google Scholar]
- 44. Mitsiades CS, McMillin D, Kotoula V et al . Antitumor effects of the proteasome inhibitor bortezomib in medullary and anaplastic thyroid carcinoma cells in vitro . J Clin Endocrinol Metab 2006; 91: 4013–21. [DOI] [PubMed] [Google Scholar]
- 45. Lallemand F, Seo SR, Ferrand N et al . AIP4 restricts transforming growth factor‐β signaling through a ubiquitination‐independent mechanism. J Biol Chem 2005; 280: 27 645–53. [DOI] [PubMed] [Google Scholar]
- 46. Heldin NE, Bergström D, Hermansson A et al . Lack of responsiveness to TGF‐β1 in a thyroid carcinoma cell line with functional type I and type II TGF‐β receptors and Smad proteins, suggests a novel mechanism for TGF‐β insensitivity in carcinoma cells. Mol Cell Endocrinol 1999; 153: 79–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Relative ITCH mRNA expression levels in anaplastic thyroid carcinoma (ATC) cell lines and normal thyroid as a control determined by real‐time reverse transcription–polymerase chain reaction. The expression level of ITCH, normalized with glyceraldehyde‐3‐phosphate dehydrogenase, in each sample was divided by that in normal thyroid, and is indicated as a fold increase.
Fig. S2. Effects of different small interfering RNA (siRNA) molecules (siRNA‐ITCH #1–3) on (a) ITCH protein expression and (b) cell growth of the 8305C cell line. Each data point represents the mean of triplicate determinations (bars, SD) in these experiments. *P < 0.05 versus siRNA‐Luc at each time after transfection; statistical analysis used the Mann–Whitney U‐test.
Fig. S3. Effect of ITCH downregulation on the expression levels of p27 and p21 in the 8305C cell line. Expression of p21 and p27 was induced 72 h after transfection of siRNA‐ITCH #1.
Fig. S4. Colony‐formation assays using the (a) TTA‐1 and (b) 8505C cell lines. Cells with relatively lower ITCH expression were transfected transiently with a green fluorescent protein (GFP)‐tagged construct containing empty vector (pEGFP‐mock), wild‐type ITCH (pEGFP‐ITCH WT), or a ubiquitin ligase‐inactive mutant of ITCH (pEGFP‐ITCH MUT), and selected with appropriate concentrations of G418 for 3 weeks. (Left) Western blotting prepared with 30 µg of protein extract and anti‐GFP tag‐specific antibody. (Right) At 3 weeks after transfection and subsequent selection of drug‐resistant colonies, the colonies formed by ITCH WT‐ or ITCH MUT‐transfected cells were more numerous than those formed by empty vector‐transfected cells.
Fig. S5. Representative results of reverse transcription–polymerase chain reaction using tagged transcript‐specific primer sets in (a) TTA‐1 and (b) 8505C cell lines transiently transfected with (top) Myc‐tagged constructs containing empty vector (pCMV‐Tag3‐empty), wild‐type ITCH (pCMV‐Tag3‐ITCH WT), or a ubiquitin ligase‐inactive mutant of ITCH (pCMV‐Tag3‐ITCH MUT), or those with (bottom) a GFP‐tagged constructs containing empty vector (pEGFP‐mock), wild‐type ITCH (pEGFP‐ITCH WT), or a ubiquitin ligase‐inactive mutant of ITCH (pEGFP‐ITCH MUT) for colony‐formation assays. DW, distilled water as a negative control. Glyceraldehyde‐3‐phosphate dehydrogenase was used to allow estimation of the efficiency of cDNA synthesis.
Table S1. Primer sequencies used in reverse transcription–polymerase chain reaction analysis
Table S2. Clinicopathological characteristics of primary thyroid tumors
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item