

Abstract
Telomerase is a ribonucleoprotein enzyme complex that adds telomeric repeats to the ends of chromosomes. The core telomerase components are the telomerase reverse transcriptase (TERT) catalytic subunit, and the telomerase RNA (TR) template subunit. In most cancers, telomerase is expressed at levels that are substantially higher than in normal cells. A known consequence of telomerase up‐regulation which is considered to play a critical role in oncogenesis is maintenance of telomere length, and thus evasion by cancer cells of the normal limits on proliferation that are associated with the steady decrease in telomere length that accompanies proliferation of normal cells. It has also been suggested that telomerase up‐regulation confers other advantages on cancer cells independent of its enzymatic activity. The mechanisms responsible for up‐regulation of telomerase in cancer are incompletely understood. Here we review evidence suggesting that this frequently results from increased copy number of the genes encoding telomerase components. The TERT gene is located at human chromosome band 5p15.33, and the telomerase RNA component (TERC) gene that encodes TR is at 3q26.3. Chromosomal gains and gene amplifications involving chromosome arms 5p and 3q are among the most frequent in human tumors. Increased TERT and TERC gene dosage has been detected frequently in a variety of human cancers, and clonal evolution of cells with increased TERT or TERC copy number has been observed, suggesting a growth advantage in cells with increased TERT or TERC gene dosage. (Cancer Sci 2008; 99: 1092–1099)
Telomerase is a ribonucleoprotein enzyme complex that adds telomeric repeats to the ends of chromosomes.( 1 ) The active human telomerase enzyme is composed of human telomerase reverse transcriptase (hTERT), human telomerase RNA (hTR) and dyskerin.( 2 ) hTERT (encoded by the TERT gene) is the catalytic reverse transcriptase component( 3 ), hTR (encoded by the TERC gene) serves as the RNA template for the addition of telomeric repeats( 4 ) and dyskerin (encoded by the DKC1 gene) is an RNA binding protein.( 5 ) Mutations in any of these components may result in dyskeratosis congenita, a human disease syndrome associated with short telomeres (reviewed in Kirwan et al. ( 6 )).
Telomerase activity has been detected in more than 85% of human tumors( 7 ) whereas in normal human somatic cells it is either undetectable or present at low levels. In normal cells, telomeres shorten with every cell division, and this eventually results in senescence, a state characterized by permanent withdrawal from the cell division cycle. The increased telomerase activity found in cancers prevents telomere shortening, and allows cancer cells to escape the normal limits on cellular proliferation (reviewed in Colgin et al.( 8 )). When exogenous hTERT is expressed in normal cells, telomere shortening is prevented, and immortalization may occur.( 9 , 10 ) Furthermore, inhibition of telomerase activity leads to senescence or apoptosis of tumor cells( 11 , 12 , 13 ) indicating that telomerase activity is required for their long‐term viability. Up‐regulation of telomerase activity resulting in telomere length maintenance is therefore thought to be critical for oncogenesis. There is evidence, however, that TERT can also promote cell proliferation independently of the telomere‐lengthening enzymatic activity of telomerase. For example, mouse TERT (mTERT) overexpression in mouse skin stimulates the proliferation of hair‐follicle stem cells and facilitates robust hair growth; this effect is independent of telomerase activity because mTERT overexpression in mice that lack the RNA component results in the same effect.( 14 )
In view of the observations that hTR is ubiquitously expressed,( 4 , 15 ) whereas hTERT is expressed only in telomerase‐positive cells( 3 ) and that expression of exogenous hTERT alone can immortalize normal human cells,( 9 , 10 ) abundance of hTERT was previously thought to be the sole limiting factor for telomerase activity. However, evidence is increasing in support of the notion that hTR levels can also be limiting for telomerase activity. For example, overexpression of both hTERT and hTR substantially increased telomerase activity, whereas overexpression of either hTR or hTERT alone induced telomerase activity to a lesser extent.( 16 ) The consequences for telomerase activity of overexpression of the other known telomerase component, dyskerin, are currently unknown, and there is no information available to indicate whether availability of this subunit is also limiting.
Changes in the copy number (gains and losses) of whole chromosomes or chromosome arms have been observed in a large number of human tumors (reviewed in Rooney et el. ( 17 )). The chromosome arms that are most frequently gained include 8q (27.7% of tumors), 1q (25.1%), 7q (23.1%), 7p (21.5%), 17q (18.5%), 3q (16.4%), 20q (15.5%) and 5p (13.2%).( 17 ) TERT has been mapped to chromosome 5 at 5p15.33( 18 ) and TERC has been mapped to chromosome 3 at 3q26.3;( 19 ) that is, both the TERT and TERC genes are located in regions frequently involved in chromosomal gains. The gene encoding dyskerin, DKC1, is located on Xq28, a region that is also known to be involved in amplification or chromosome gains in cancer cells.( 17 , 20 ) However, there is no information available about whether the DKC1 gene itself is amplified in human cancer.
Gene amplification refers to the situation where there is an array of copies of a restricted region of a chromosome( 21 ) and the region of the chromosome that becomes amplified is referred to as the ‘amplicon’. Gene amplification is common in human cancers and is considered to be one of the mechanisms of oncogene activation. Both the TERT and TERC gene loci are located on chromosome regions that are frequently amplified in human cancers. For example, chromosome 5p is often amplified in neuroblastoma, lung cancer, squamous cell carcinoma of the head and neck (SCC‐HN), carcinoma of the cervix, medulloblastoma and osteosarcoma.( 20 , 22 , 23 ) Amplifications involving 3q have been consistently detected in ovarian carcinoma,( 24 ) carcinoma of the cervix, lung cancer and SCC‐HN (reviewed in Knuutila et al.( 20 )).
This present study reviews the evidence that the gene dosage of TERT and TERC are often increased in human tumors. It should be noted that most studies reviewed here do not distinguish between chromosomal gain and gene amplification. Nevertheless, the outcome for the cell could be similar: both lead to an increase in gene dosage. We propose that increased TERT and TERC gene dosage may promote oncogenesis both through telomerase enzyme activity and possibly in activity‐independent ways. The possible clinical implications are also discussed.
Increased TERT copy number in cancer
Fluorescence in situ hybridization (FISH) and Southern blot analysis using probes containing TERT sequences or polymerase chain reaction (PCR) using primers specific for TERT have demonstrated copy number increases of the TERT gene in multiple tumors or immortalized cell lines (Table 1). For example, FISH analysis using a probe that covered the genomic region encoding TERT together with a specific sequence at 5q31 as a marker probe detected two TERT gene copies located on band 5p15.33 and a 1:1 ratio of TERT/5q31 signal in normal cells. However, only 5 of 26 human tumor cell lines from different origins and 28 of 58 human primary tumors carried two TERT and two 5q31 marker copies. The remainder of the cell lines and primary tumors had more than two copies of TERT and a TERT/5q31 ratio ≥1.( 25 ) Some of these (50% of cell lines and 22% of primary tumors) displayed 3–4 TERT copies/cell( 25 ) while 31% of cell lines and 30% of human primary tumors had ≥5 copies of TERT per cell.( 25 )
Table 1.
Increased hTERT copy number in cancer
| Human cancer type | Number and types of samples | Method of hTERT copy number analysis | Threshold for increased hTERT copy number | Frequency of increased hTERT copy number | Correlation with expression | Correlation with activity | References |
|---|---|---|---|---|---|---|---|
| Cervical, lung, bladder and epidermal carcinomas | 15 cancer cell lines | FISH using a BAC clone 518C13 which is specific for hTERT | Not defined | 10/15 cell lines had a modal hTERT copy number of 3 or more per cell | – | – | ( 18 ) |
| Neuroblastomas, lung, cervical and breast cancer | 26 cell lines 58 primary tumors | FISH using specific hTERT gene probe isolated from PAC clone and a reference probe located at 5q31 | ≥5 copies of hTERT per cell in ≥40% of the cells (for cell lines) | 8/26 (31%) tumor cell lines | Yes (for neuroblastoma cell lines) | Yes (for neuroblastoma cell lines) | ( 25 ) |
| ≥20% of the cells (for primary tumors) | 17/58 (30%) primary tumors | ||||||
| (1/8 neuroblastomas 8/21 lung tumors, 3/10 cervical tumors 5/19 breast tumors) | |||||||
| Cervical cancer | 4 cervical cancer cell lines | Same as above | Same as above | 1 of 4 (25%) cell lines | Yes | – | ( 27 ) |
| 88 cervical carcinomas | 21 of 88 (24%) primary tumors | ||||||
| Lung cancer | 20 cell lines | FISH and Southern blotting | FISH: not defined | 6/20 (30%) | Yes | Yes | ( 28 ) |
| (14 SCLC, 6 NSCLC) | Southern: intensity of hTERT gene relative to control gene >150% | (5/14 SCLC, 1/6 NSCLC) | |||||
| Melanomas | 48 primary cell cultures from 46 CMM patients | FISH: using a BAC clone containing the hTERT locus | Not defined | Frequencies not given, but increase in copy number of chromosome region detected by CGH in 13/50 cell lines | No | – | ( 32 ) |
| One culture from a dysplastic naevus | Southern: using primers specific for hTERT | ||||||
| Melanoma celll line SK‐Mel‐28 | |||||||
| CNS embryonal tumors | 36 CNS embryonal tumors | Differential PCR: using primers specific for hTERT and primers specific for 5qSTS as the control gene | hTERT/5qSTS ratio ≥2.17 | 15/36 (42%) | Yes | – | ( 26 ) |
| (2.17 = twice the mean of normal +3 standard deviations) | |||||||
| Hepatocellular carcinomas | 46 HCC | qPCR: using primers for hTERT, CSF1R (5q33.35) and GAPDH (12p13) | Ratio of hTERT/GAPDH >2 | 10/46 (22%) | No | ( 31 ) | |
| Colorectal cancer | 64 colorectal carcinomas | FISH: using a hTERT locus specific probe isolated from a PAC clone; using a 5q31 marker probe as the reference nonamplified gene | ≥3 hTERT copies per nucleus in ≥20% of the cells | 31/64 (48%) | No | No | ( 30 ) |
| NSCLC | qPCR on 81 tumors | qPCR: using primers specific for hTERT and primers specific for PIK3R1 (5q13.1) as the reference nonamplified gene | qPCR: hTERT copy number > mean of normal +2 standard deviations | qPCR: 46/81(57%) (30/40 ADC; 13/37 SQCC) | Yes | – | ( 36 ) |
| FISH on a subset of 59 tumors that had been studied by qPCR | FISH: using hTERT/5q dual‐color probe | FISH: presence of tight hTERT gene clusters and a ratio of hTERT to chromosome 5q ≥2 or ≥15 copies of hTERT gene per cell in ≥10% of the cells | FISH: 37/59 (63%) | ||||
| Leukemia | 29 ALL | FISH using probes for hTERT and 5q31 | Not defined | 2–60 copies of hTERT detected | – | – | ( 29 ) |
| 16 ANLL | |||||||
| Myeloblastic cell line K562 |
– , not studied; ADC, adenocarcinoma; ALL, acute lympholastic leukemia; ANLL, non‐lymphoblastic leukemia; BAC, bacterial artificial chromosome; CGH, comparative genomic hybridization; CMM, cutaneous malignant melanoma; CNS, central nervous system; CSF1R, colony‐stimulating factor 1 receptor; FISH, fluorescence in situ hybridization; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; HCC, hepatocellular carcinomas; hTERT, human telomerase reverse transcriptase; NSCLC, non‐small cell lung cancer; PAC, P1‐derived artificial chromosome; qPCR, quantitative polymerase chain reaction; SCLC, small cell lung cancer; SQCC, squamous cell carcinoma.
In addition to FISH analysis, TERT amplification can also be detected by quantitative PCR. For example, PCR analysis using primer sets specific for TERT and control sequences on 5q (5qSTS) detected a TERT/5qSTS ratio that ranged from 0.1 to 17.6 in 36 central nervous system (CNS) embryonal tumors whereas a mean ratio of 1.02 (ranging from 0.99 to 1.12) was detected in eight normal subjects.( 26 ) In this study, 42% of CNS embryonal tumors had a TERT/5qSTS ratio ≥2.17 and were considered to have TERT amplification.( 26 )
In various studies, it was found that 25–31% of the cell lines examined had ≥5 TERT gene copies/cell in ≥40% of the cells. These included cell lines derived from neuroblastomas (Lan2, Lan5 and SHEP1) and carcinomas of breast (578T), cervix (HeLa and CaSki) and lung (H125, U1285, U1752, SHP77/97, H1688, Colo677/97, H446/97, BEN and H209).( 25 , 27 , 28 ) Cell lines derived from bladder and epidermal carcinomas (5637 and A431) were reported to have ≥3 copies of hTERT per cell.( 18 ) In primary tumors, increased TERT copy number has been detected in neuroblastomas (12%), CNS embryonal tumors (42%), hepatocellular carcinomas (22%) and cancers of the lung (30–63%), cervix (24–30%), breast (26%) and colon (48%) (Table 1). In addition, FISH analysis revealed 2–60 copies of TERT in leukemic cells.( 29 ) It is worth noting that the threshold beyond which the TERT gene is considered to have increased copy number or to be amplified varies between studies. For example, using FISH analysis, Palmqvist et al. defined increased TERT copy number as ≥3 TERT copies/cell in ≥20% of the cells and detected 48% of 64 colorectal tumor samples that had increased TERT copy number.( 30 ) In contrast, using the same technique, Zhang et al. defined TERT amplification as ≥5 TERT copies/cell in ≥20% of the cells and scored 24–30% of primary tumors as having TERT amplification.( 25 , 27 )
It is possible that the different thresholds for detection of TERT amplication may explain why some of the above‐mentioned studies showed a correlation between increased TERT copy number and hTERT mRNA expression, while others showed no association.( 30 , 31 , 32 ) For example, in a colorectal cancer study that used a low threshold (≥3 copies of TERT per cell in ≥20% of the cells), hTERT mRNA expression did not correlate with TERT gene dosage.( 30 ) Alternatively, the discrepancy between TERT copy number and expression levels could be due to the different origins of the tumors and/or cell types. Observations from our laboratory are consistent with the latter explanation. We have identified increased TERT copy number in hTERT‐immortalized human mammary epithelial cells (HMECs) and in hTERT‐immortalized human foreskin fibroblasts. Increased TERT copy number correlated with hTERT mRNA and protein expression in hTERT immortalized HMECs( 33 ) but not in hTERT immortalized human foreskin fibroblasts (Cao et al. unpublished, 2007). Another possible explanation for the lack of correlation between amplification and hTERT expression is that the amplicon might contain an incomplete copy of the gene. Finally, hTERT expression has been shown to be regulated at multiple levels involving transcription, alternative splicing, translation and post‐translational events (for reviews, see Horikawa et al. ( 34 ) and Ducrest et al.( 35 )) which provides additional potential explanations for why TERT gene dosage does not always correlate with hTERT expression.
Similarly, no consistent correlation between TERT copy number and telomerase activity has been observed. While Zhang et al.( 25 ) and Saretzki et al.( 28 ) reported a correlation between TERT gene dosage and telomerase activity in different cell lines and primary tumors, Palmqvist's study of colorectal primary tumor samples( 30 ) and our studies of hTERT‐immortalized HMECs( 33 ) and human foreskin fibroblasts (Cao et al. unpublished, 2007) did not observe any correlation between TERT gene copy number and telomerase activity. This lack of correlation may be explained by the observation that telomerase is an enzyme complex composed of multiple components and the evidence (discussed below) that the levels of two of these components (hTERT and hTR) are limiting for telomerase activity. It should be noted that most of the published studies on increased TERT gene copy number have not investigated hTR levels. It is possible that hTR levels may limit telomerase activity in some tumors where increased TERT copy number does not correlate with telomerase activity. Our observation that hTR levels limit telomerase activity in mammary epithelial cells with TERT amplification( 33 ) supports this possibility.
In various types of tumors, increased TERT gene copy number has been found to have clinical and prognostic correlates. For example, Zhu et al. showed that lung cancer patients with TERT amplification had poorer recurrence‐free survival.( 36 ) In hepatocellular carcinomas, hTERT amplification was found to be associated with poorly differentiated histopathology, which would be expected to correlate with poor outcome.( 31 ) In melanomas, increased TERT copy number was significantly associated with the melanoma subtypes and locations of metastases. For example, increased TERT gene dosage was abundant in superficial spreading primary melanomas, subcutaneous metastases and malignant effusion‐derived cells, but was completely absent or very rare in cells from primary nodular melanomas and brain, bone and lymph node metastases.( 32 )
Increased TERC copy number in cancer
Like TERT, increased copy number of the TERC gene has been found in many tumor samples and immortalized cell lines by techniques such as FISH and Southern blot analysis using probes containing the TERC sequence (Table 2). Using TERC sequence as the probe in FISH analysis, Soder et al. detected more than 10 TERC DNA signals per nucleus in 4/73 carcinomas of the cervix, head and neck, and lung, and 29/30 (97%) of SCC‐HN and cervical carcinomas had more than two copies of TERC per cell.( 19 ) Southern blot analysis using 17 probes spanning chromosome 3q located an amplicon within band 3q26 which included the TERC gene, supporting TERC as a potential amplification target in tumors of the cervix, ovary and lung.( 24 )
Table 2.
Increased TERC copy number in cancer
| Human cancer type | Number and types of samples | Method of TERC copy number analysis | Definition for TERC gain or amplificaton | Result | References |
|---|---|---|---|---|---|
| Cervix, Head & neck, lung cancer | 33 cervical carcinomas (32 squamous and one adenocarcinoma), 31 head and neck carcinomas (30 squamous, one adenocarcinoma), 9 lung carcinomas (squamous non‐small cell) | FISH probed with a P1 clone containing TERC sequence (interphase) | Amplification definition: >10 TERC hybridization signals per nucleus, high density of signals | TERC Amplification: 4/73 (5%) (2/33 cervix, 1/31 head and neck, 1/9 lung) | ( 19 ) |
| TERC gain definition: TERC signal >2 per cell in >10% of the cells | TERC gain: 29/30 (97%) of tumors investigated | ||||
| Cervix, ovary and lung cancer | 9 ovarian tumor samples, 8 cervical carcinoma cell lines, 7 SCLC cell lines, 1 ovarian carcinoma cell line, 3 NSCLC cell lines, | Southern: used 17 probes spaning 3q including two probes within TERC gene | Not defined | • More than 2 copies of TERC gene: | ( 24 ) |
| FISH: used two 3q26 specific probes | 7/8 ovarian tumor samples, | ||||
| 3/8 cervical carcinoma cell lines, | |||||
| 5/7 SCLC cell lines. | |||||
| • 5/8 (62%) ovarian tumor had more than 4 copies of TERC gene | |||||
| • Increased copy number peaked at 3q26 in the 8 cervical carcinoma cell lines, the 7 SCLC cell lines and the 9 ovarian tumors | |||||
| Melanomas | 48 primary cell cultures from 46 CMM patients | FISH: using a BAC clone containing the TERC locus | Not defined | Frequencies not given, but increase in copy number of chromosome region detected by CGH in 16/50 cell lines | ( 32 ) |
| One culture from a dysplastic naevus | Southern: using primers specific for TERC | ||||
| Melanoma celll line SK‐Mel‐28 | |||||
| Lung cancer | 19 NSCLC cell lines (10 from squamous cell carcinomas and 9 from adenocarcinomas) | FISH using a BAC probe containing TERC gene (metaphase) | Not defined | FISH: 5–16 TERC signals were detected in metaphases of the 5 cell lines examined | ( 37 ) |
| Southern: using a probe containing hTR cDNA sequence | Southern: 47% of the 19 lines showed increased TERC copy number | ||||
| Increased TERC copy number correlates with hTR expression | |||||
| Cervix cancer | 57 thin‐layer cytological specimens | FISH using CEP7, CEP3, and TERC probes | Higher copy numbers of TERC compared to CEP7 | Cells with multiple TERC signals increased with the severity of the cytologic interpretation | ( 40 ) |
| Cervix cancer | 59 pap smears | Same as above | TERC signal >2 per cell in >20% of the cells | increased copy number of TERC was associated with progression of premalignant dysplastic lesions | ( 41 ) |
| Cervix cancer | 12 primary cervical adenocarcinomas | Same as above | Not defined | TERC gain or amplification was found in all cervical adenocarcinomas investigated | ( 39 ) |
| Esophageal carcinomas | 60 primary tumors | qPCR | Not defined | Average TERC copy number: 5.28 (±0.54) | ( 38 ) |
| Leukemia | 29 ALL | FISH using probes for TERC and reference probe of chr3 | Not defined | 2–12 copiesof TERC detected | ( 29 ) |
| 16 ANLL | |||||
| Myeloblastic cell line K562 |
CEP3 and CEP7, probes containing repeat sequences specific for the centromeres of chromosomes 3 (CEP3) and 7 (CEP7);
ALL, acute lympholastic leukemia; ANLL, non‐lymphoblastic leukemia; BAC, bacterial artificial chromosome; CGH, comparative genomic hybridization; CMM, cutaneous malignant melanoma; FISH, fluorescence in situ hybridization; TERC, telomerase RNA component; hTR, human telomerase RNA; NSCLC, non‐small cell lung cancer; qPCR, quantitative polymerase chain reaction; SCLC, small cell lung cancer.
Other studies that have shown increased TERC copy number in tumors include a FISH analysis that detected 5–16 TERC signals in five non‐small cell lung cancer cell lines (Lc‐1sq, PC‐10, VMRC‐LCP, HUT‐29 and ABC‐1).( 37 ) Southern blot analysis detected increased TERC copy number in these and four additional cell lines (11–18, RERF‐LC‐MS, PC‐14 and Sq‐1) out of a total of 19 lines.( 37 ) PCR analysis of genomic DNA from 60 esophageal carcinomas detected an average of more than five copies of TERC.( 38 ) In other studies, extra copies of TERC were observed in melanomas( 32 ) leukemic cells( 29 ) and 100% (12/12) of primary cervical adenocarcinomas.( 39 )
A potentially important finding is that FISH analysis of TERC gene copy number in routinely prepared Pap smears is able to distinguish normal epithelium and low‐grade dysplasia from high‐grade lesions( 40 ) and to assist in identifying low‐grade lesions with a high progression risk.( 41 ) A probe set consisting of TERC and repeat sequences specific for the centromeres of chromosome 3 (CEP3) and 7 (CEP7) was used to screen 57 thin‐layer slides by FISH.( 40 ) CEP3 was included for evaluating the relative copy number increase of TERC compared to the number of chromosome 3 centromeres whereas CEP7 served as a control for the overall ploidy of the cells. The most frequent increased TERC copy number pattern was 2‐2‐3 (copy number of CEP7‐CEP3‐hTR). One case showed high‐level amplification of TERC (>20 copies) while the two centromere probes were still diploid.( 40 ) Seven of 12 cervical intraepithelial neoplasia (CIN) 1/CIN2 lesions that progressed to CIN3 carried extra copies of TERC (and all matched CIN3 lesions had extra copies of TERC), whereas 0/10 non‐progressing CIN1/CIN2 lesions carried extra copies of TERC.( 41 ) Even more strikingly, extra copies of TERC were detected in 4/12 (33%) cytologically normal Pap smears from women who later developed CIN3 or cervical carcinomas (only 1–3 years after the normal Pap smears).( 41 ) These results suggest that the detection of additional copies of the TERC gene in routinely collected Pap smears might be able to serve as an early and specific marker to identify lesions with a high progression risk.
Although FISH analysis using CEP7‐CEP3‐hTR probes in routinely collected Pap smears facilitated the visualization of extra copies of TERC in a single cell, the threshold for determining whether the TERC copy number increase in the cell population is clinically significant needs careful definition. For example, using a threshold of ≥5% of cells with multiple TERC signals and/or at least one observed cell with six or more TERC signals, no normal Pap smears were positive, but changing the threshold to ≥1% of cells with multiple TERC signals and/or at least one observed cell with five or more TERC signals, resulted in 2/13 (15.4%) cytologically normal Pap smears being positive.( 40 ) The clinical outcomes were not known in this study, and it would be of great interest to determine whether the two women, whose smears were positive when the lower threshold was used, later developed high‐grade lesions.
A correlation between TERC gene copy number and hTR expression level was observed in lung cancer( 37 ) and leukemia( 29 ) but not in melanoma.( 32 ) No other studies have investigated the correlation between TERC gene copy number and hTR expression. Potential explanations for the non‐correlation between TERT gene dosage and hTERT expression discussed before also apply to hTR. Like hTERT expression, hTR expression is regulated at multiple levels (reviewed Cairney et al.( 42 )), so a simple correlation between TERC gene dosage and hTR expression should not be expected.
Interestingly, 5 out of 50 (10%) cell cultures from melonomas displayed increased copy number of both TERT and TERC, although it is not clear whether this can happen in the same cell.( 32 ) Extra copies of TERT and TERC are both observed in leukemic cells, but it cannot be ascertained from the publication whether extra copies of both genes were observed in the same samples.( 29 ) None of the other publications reviewed here appear to have investigated the copy number of both genes, but evidence from separate studies indicates that TERT and TERC are both frequently amplified in lung and cervical carcinomas (1, 2). Given the recent evidence that TERT and TERC gene products are both limiting for telomerase activity, it would be of considerable interest to know how frequently both genes are amplified in the same tumor or cell.
Clonal overgrowth of cells with TERC or TERT amplification
Overgrowth of cells carrying additional copies of TERC appears to occur within cervical carcinomas.( 40 , 41 ) Heselmeyer‐Haddad et al. observed that in many instances cells with extra copies of TERC were located next to each other and this clustering increased with advanced dysplasia.( 40 , 41 ) It is possible that cells carrying extra copies of TERC have a growth advantage, which eventually results in a cervical carcinoma cell population in which the majority of the cells are positive for TERC amplification. It is not clear whether TERC amplification correlates with hTR expression or telomerase activity in this context, and the mechanism of the putative promotion of cell growth by TERC amplification in cervical carcinomas awaits further investigation.
Our data indicate that a clone of cells with TERT amplification can overgrow all other cells in an in vitro cell population. Four independent hTERT‐immortalized mass cultures (B80‐TERT1, 2, 3a and 3b) were obtained by transfecting HMECs with an hTERT expression plasmid.( 43 ) Extensive amplification of the TERT transgene in B80‐TERT1 cells was detected by FISH analysis of metaphase spreads using a probe containing full‐length hTERT cDNA or a 650 bp N‐terminal hTERT probe. A similar pattern of TERT amplification was observed in every metaphase (Fig. 1), indicating that this immortalized cell line has become clonal even though it was originally established as a mass culture. FISH analysis detected a different pattern of TERT amplification in another cell line (B80‐TERT3b), and it had also become clonal because every metaphase had the same pattern.( 33 ) These observations suggest that cells with TERT amplification may have acquired a growth advantage.
Figure 1.
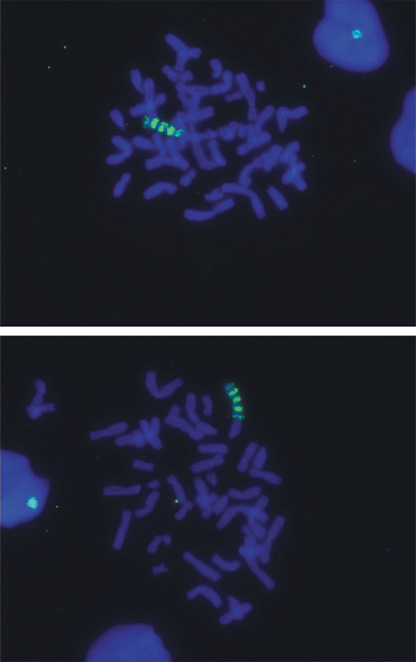
Human telomerase reverse transcriptase (hTERT) amplification in B80‐TERT1 cells detected by fluorescence in situ hybridization analysis using the full length hTERT cDNA as a probe. Fluorescein‐avidin detection of hTERT is shown in green. DAPI staining of the nucleus is shown in blue. Top and bottom panels are two representative images from the whole cell population.
hTERT and hTR roles beyond telomerase activity
As noted above, increased TERT copy number in various tumors or cell lines does not always correlate with telomerase activity. A similar situation may also apply to increased TERC copy number. This could indicate that, at least in some tumors, the increases in copy number are simply random changes that reflect the genetic instability seen in cancers. Another potential explanation is that increased hTERT and hTR expression may promote carcinogenesis through a mechanism independent of the telomere‐lengthening catalytic activity of telomerase.
Evidence that telomerase may promote tumorigenesis in ways beyond telomere length maintenance has been reviewed elsewhere (for example, see reference,( 44 )). Some of the evidence suggesting that specific telomerase components can have pro‐oncogenic functions beyond their contribution to telomerase enzyme activity is as follows. hTERT has been shown to promote cell survival and proliferation in human breast cancer PMC42 cells; this effect was independent of its telomere‐lengthening catalytic activity because an hTERT mutant (D788N) that lacks telomerase catalytic activity had a similar pro‐survival effect.( 45 ) An independent study showed that a catalytically inactive dominant‐negative mutant of hTERT antagonized p53‐induced apoptosis as efficiently as wild‐type hTERT.( 46 ) A third study has shown that hTERT protects against Bcl‐2–dependent apoptosis, and that a dominant negative catalytically inactive mutant of hTERT had a similar antiapoptotic effect.( 47 ) This antiapoptotic effect of hTERT that is independent of its catalytic activity was observed in three different human cancer cell lines: MCF7 breast cancer cells, M14 melanoma cells, and HCT116 colon cancer cells. Furthermore, hTERT protected against Bcl‐2–dependent apoptosis independently of p53, because overexpression of hTERT antagonized apoptosis induced by a Bcl‐2 inhibitor both in HCT116 p53+/+ and HCT116 p53–/– cells.( 47 )
A role for TERT independent of telomerase activity has also been demonstrated in mouse stem cells.( 14 ) Conditional expression of mTERT in mouse skin epithelium caused activation of quiescent stem cells in the hair follicle and a rapid transition from the resting phase (telogen) to the active phase of the hair follicle cycle (anagen).( 14 ) Induction of anagen by mTERT overexpression facilitated robust hair growth, regardless of whether mTR was expressed or not.( 14 ) Furthermore, a catalytically inactive mTERT mutant (D702A) had a similar hair growth promoting effect compared to wild‐type mTERT.( 48 )
It is also possible that hTR may have a function independent of its role as the template for telomere lengthening. Kedde et al. showed that inhibition of hTR expression in a number of different human cells triggered a rapid growth arrest which was associated with p53 and CHK1 activation.( 49 ) Moreover, the rapid growth arrest resulting from hTR inhibition was independent of hTERT because a similar growth arrest and ATR activation was observed in a cell line (GM847) expressing no hTERT.( 49 )
Both hTERT and hTR can be limiting for telomerase activity
Following the cloning of hTERT in 1997( 3 , 50 , 51 , 52 ), it was shown that hTERT levels can be limiting for telomerase activity. hTERT expression is often undetectable in normal telomerase‐negative cells, whereas hTR is expressed at detectable levels. It was shown that introduction of hTERT expression constructs into normal human cells induced telomerase activity, resulting in telomere length maintenance, escape from senescence, and extension of proliferative lifespan.( 9 , 10 , 53 ) It was therefore deduced that hTERT levels are limiting, and that the other telomerase components must be expressed at sufficient levels. Studies in mice also supported the limiting role of TERT. For example, forced expression of mTERT in cardiac muscle in mice was sufficient to induce telomerase activity, resulting in hyperplasia and hypertrophy of cardiac myocytes.( 54 )
It has recently become clear that hTR is also limiting for telomerase activity and telomere maintenance (reviewed in Cairney et al.( 42 )). Early clues included the observation that hTR expression is upregulated in telomerase‐positive immortal cell lines in comparison to telomerase‐negative mortal cell strains.( 3 , 4 ) Moreover, it has been observed that hTR levels are substantially elevated in a wide variety of human tumors.( 55 , 56 , 57 , 58 , 59 ) Evidence that telomerase RNA levels may also be limiting in chicken cells is provided by the observation that oncogenic strains of Marek's disease virus (MDV) but not non‐oncogenic strains encode a viral form of telomerase RNA (vTR), that shares 88% homology to chicken telomerase RNA.( 60 ) MDV carrying both copies of vTR promoted malignant T cell lymphomagenesis in chickens, whereas mutants of the oncogenic strain of MDV lacking one or both copies of vTR were impaired in their ability to induce T cell lymphomas.( 61 )
Direct evidence that hTR levels are limiting for telomerase activity was published recently by Lingner and colleagues, who showed that concomitant overexpression of hTERT and hTR in several human cell lines resulted in higher levels of telomerase activity compared to hTERT or hTR overexpression alone.( 16 ) It can therefore be concluded that hTERT and hTR levels both limit telomerase activity in these cell lines. hTR levels often become upregulated following transduction of normal cells with an hTERT expression construct( 33 , 62 ), which probably explains why TERT levels alone were previously regarded as limiting. We also identified an hTERT‐immortalized cell line in which hTR up‐regulation was minimal.( 33 ) In this latter cell line, transduction by an hTR expression plasmid resulted in a large increase in telomerase activity and telomere lengthening. The reason that hTR levels are upregulated in response to hTERT overexpression most likely includes stabilization of hTR by binding to TERT( 62 ), but there are other aspects of the mechanism that are currently unexplained.( 33 )
Further evidence that hTR is limiting for telomerase activity comes from the observation that TERC is haploinsufficient in both humans and mice. Dyskeratosis congenita is a human syndrome characterized by abnormally short telomeres and premature proliferative exhaustion in tissues such as the bone marrow, and may be associated with mutations in various genes including TERC (reviewed in Kirwan et al. ( 6 )). Although telomerase levels are insufficient to completely prevent telomere shortening in normal human cells, highly proliferative tissues such as the bone marrow require normal levels of telomerase activity to maintain an adequate proliferative capacity. It appears that the TERC mutations associated with dyskeratosis congenita reduce telomerase activity via haploinsufficiency rather than by a dominant negative mechanism.( 63 ) Studies in mouse knockout models also suggest that this gene is haploinsufficient.( 64 , 65 )
Conclusions
On the basis of the evidence that the levels of both TERT and TERC are limiting for telomerase activity and that the copy number of these genes is frequently increased in cancers by chromosomal gains or by amplification, we propose that increased TERT and/or TERC gene dosage is an important mechanism for upregulation of telomerase activity in human cancer. Interestingly, transduction of normal cells with hTERT expression constructs may result in upregulation of endogenous hTR expression, by mechanisms which are incompletely understood. Very little is currently known about changes in expression of the other known telomerase subunit, dyskerin. More extensive surveys will be required to determine how common increased dosage of TERT and TERC is across a wider range of tumors, and further studies are also required to determine whether dosage of both genes is frequently increased in the same tumors. It is also possible that the increased copy number of genes encoding telomerase components has prooncogenic effects in addition to the ability of telomerase to synthesize telomeric repeats, prevent telomere shortening, and permit cells to escape from senescence. Detection of TERT and/or TERC amplification may have useful applications in cancer diagnosis and prognosis. Realization of this potential will require robust definition of what constitutes a biologically significant increase in TERT or TERC copy number, and more extensive studies of the clinical outcome in patient cohorts.
Acknowledgments
We gratefully acknowledge Axel Neumann for help with FISH analysis and the following grant support: National Health and Medical Research Council (NHMRC) Peter Doherty Postdoctoral Fellowship for YC (228413), NHMRC Senior Principal Research Fellowship (272503) and Healthy Aging Research Grant (219306) for RRR and a Wellcome Trust Senior Research Fellowship for TMB (GRO66727MA).
References
- 1. Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985; 43: 405–13. [DOI] [PubMed] [Google Scholar]
- 2. Cohen SB, Graham ME, Lovrecz GO, Bache N, Robinson PJ, Reddel RR. Protein composition of catalytically active human telomerase from immortal cells. Science 2007; 315: 1850–3. [DOI] [PubMed] [Google Scholar]
- 3. Nakamura TM, Morin GB, Chapman KB et al . Telomerase catalytic subunit homologs from fission yeast and human. Science 1997; 277: 955–9. [DOI] [PubMed] [Google Scholar]
- 4. Feng J, Funk WD, Wang SS et al . The RNA component of human telomerase. Science 1995; 269: 1236–41. [DOI] [PubMed] [Google Scholar]
- 5. Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature 1999; 402: 551–5. [DOI] [PubMed] [Google Scholar]
- 6. Kirwan M, Dokal I. Dyskeratosis congenita: a genetic disorder of many faces. Clin Genet 2008; 73: 103–12. [DOI] [PubMed] [Google Scholar]
- 7. Kim NW, Piatyszek MA, Prowse KR et al . Specific association of human telomerase activity with immortal cells and cancer. Science 1994; 266: 2011–5. [DOI] [PubMed] [Google Scholar]
- 8. Colgin LM, Reddel RR. Telomere maintenance mechanisms and cellular immortalization. Curr Opin Genet Dev 1999; 9: 97–103. [DOI] [PubMed] [Google Scholar]
- 9. Bodnar AG, Ouellette M, Frolkis M et al . Extension of life‐span by introduction of telomerase into normal human cells. Science 1998; 279: 349–52. [DOI] [PubMed] [Google Scholar]
- 10. Vaziri H, Benchimol S. Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol 1998; 8: 279–82. [DOI] [PubMed] [Google Scholar]
- 11. Hahn WC, Stewart SA, Brooks MW et al . Inhibition of telomerase limits the growth of human cancer cells. Nat Med 1999; 5: 1164–70. [DOI] [PubMed] [Google Scholar]
- 12. Zhang X, Mar V, Zhou W, Harrington L, Robinson MO. Telomere shortening and apoptosis in telomerase‐inhibited human tumor cells. Genes Dev 1999; 13: 2388–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Herbert BS, Pitts AE, Baker SI et al . Inhibition of human telomerase in immortal human cells leads to progressive telomere shortening and cell death. Proc Natl Acad Sci USA 1999; 96: 14276–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sarin KY, Cheung P, Gilison D et al . Conditional telomerase induction causes proliferation of hair follicle stem cells. Nature 2005; 436: 1048–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Avilion AA, Piatyszek MA, Gupta J, Shay JW, Bacchetti S, Greider CW. Human telomerase RNA and telomerase activity in immortal cell lines and tumor tissues. Cancer Res 1996; 56: 645–50. [PubMed] [Google Scholar]
- 16. Cristofari G, Lingner J. Telomere length homeostasis requires that telomerase levels are limiting. EMBO J 2006; 25: 565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rooney PH, Murray GI, Stevenson DA, Haites NE, Cassidy J, McLeod HL. Comparative genomic hybridization and chromosomal instability in solid tumours. Br J Cancer 1999; 80: 862–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bryce LA, Morrison N, Hoare SF, Muir S, Keith WN. Mapping of the gene for the human telomerase reverse transcriptase, hTERT, to chromosome 5p15.33 by fluorescence in situ hybridization. Neoplasia 2000; 2: 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Soder AI, Hoare SF, Muir S, Going JJ, Parkinson EK, Keith WN. Amplification, increased dosage and in situ expression of the telomerase RNA gene in human cancer. Oncogene 1997; 14: 1013–21. [DOI] [PubMed] [Google Scholar]
- 20. Knuutila S, Bjorkqvist AM, Autio K et al . DNA copy number amplifications in human neoplasms: review of comparative genomic hybridization studies. Am J Pathol 1998; 152: 1107–23. [PMC free article] [PubMed] [Google Scholar]
- 21. Albertson DG. Gene amplification in cancer. Trends Genet 2006; 22: 447–55. [DOI] [PubMed] [Google Scholar]
- 22. Mosse YP, Greshock J, Margolin A et al . High‐resolution detection and mapping of genomic DNA alterations in neuroblastoma. Genes Chromosomes Cancer 2005; 43: 390–403. [DOI] [PubMed] [Google Scholar]
- 23. Coe BP, Henderson LJ, Garnis C et al . High‐resolution chromosome arm 5p array CGH analysis of small cell lung carcinoma cell lines. Genes Chromosomes Cancer 2005; 42: 308–13. [DOI] [PubMed] [Google Scholar]
- 24. Sugita M, Tanaka N, Davidson S et al . Molecular definition of a small amplification domain within 3q26 in tumors of cervix, ovary, and lung. Cancer Genet Cytogenet 2000; 117: 9–18. [DOI] [PubMed] [Google Scholar]
- 25. Zhang A, Zheng C, Lindvall C et al . Frequent amplification of the telomerase reverse transcriptase gene in human tumors. Cancer Res 2000; 60: 6230–5. [PubMed] [Google Scholar]
- 26. Fan X, Wang Y, Kratz J et al . hTERT gene amplification and increased mRNA expression in central nervous system embryonal tumors. Am J Pathol 2003; 162: 1763–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang A, Zheng C, Hou M et al . Amplification of the telomerase reverse transcriptase (hTERT) gene in cervical carcinomas. Genes Chromosomes Cancer 2002; 34: 269–75. [DOI] [PubMed] [Google Scholar]
- 28. Saretzki G, Petersen S, Petersen I, Kolble K, Von Zglinicki T. hTERT gene dosage correlates with telomerase activity in human lung cancer cell lines. Cancer Lett 2002; 176: 81–91. [DOI] [PubMed] [Google Scholar]
- 29. Nowak T, Januszkiewicz D, Zawada M et al . Amplification of hTERT and hTERC genes in leukemic cells with high expression and activity of telomerase. Oncol Rep 2006; 16: 301–5. [PubMed] [Google Scholar]
- 30. Palmqvist R, Zhang A, Xu D et al . hTERT gene copy number is not associated with hTERT RNA expression or telomerase activity in colorectal cancer. Int J Cancer 2005; 116: 395–400. [DOI] [PubMed] [Google Scholar]
- 31. Takuma Y, Nouso K, Kobayashi Y et al . Telomerase reverse transcriptase gene amplification in hepatocellular carcinoma. J Gastroenterol Hepatol 2004; 19: 1300–4. [DOI] [PubMed] [Google Scholar]
- 32. Pirker C, Holzmann K, Spiegl‐Kreinecker S et al . Chromosomal imbalances in primary and metastatic melanomas: over‐representation of essential telomerase genes. Melanoma Res 2003; 13: 483–92. [DOI] [PubMed] [Google Scholar]
- 33. Cao Y, Huschtscha LI, Nouwens AS et al . Amplification of hTERT in human mammary epithelial cells with limiting hTR expression levels. Cancer Res 2008. (In Press). [DOI] [PubMed] [Google Scholar]
- 34. Horikawa I, Barrett JC. Transcriptional regulation of the telomerase hTERT gene as a target for cellular and viral oncogenic mechanisms. Carcinogenesis 2003; 24: 1167–76. [DOI] [PubMed] [Google Scholar]
- 35. Ducrest AL, Szutorisz H, Lingner J, Nabholz M. Regulation of the human telomerase reverse transcriptase gene. Oncogene 2002; 21: 541–52. [DOI] [PubMed] [Google Scholar]
- 36. Zhu CQ, Cutz JC, Liu N et al . Amplification of telomerase (hTERT) gene is a poor prognostic marker in non‐small‐cell lung cancer. Br J Cancer 2006; 94: 1452–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yokoi S, Yasui K, Iizasa T, Imoto I, Fujisawa T, Inazawa J. TERC identified as a probable target within the 3q26 amplicon that is detected frequently in non‐small cell lung cancers. Clin Cancer Res 2003; 9: 4705–13. [PubMed] [Google Scholar]
- 38. Yen CC, Chen YJ, Pan CC et al . Copy number changes of target genes in chromosome 3q25.3‐qter of esophageal squamous cell carcinoma: TP63 is amplified in early carcinogenesis but down‐regulated as disease progressed. World J Gastroenterol 2005; 11: 1267–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Andersson S, Wallin KL, Hellstrom AC et al . Frequent gain of the human telomerase gene TERC at 3q26 in cervical adenocarcinomas. Br J Cancer 2006; 95: 331–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heselmeyer‐Haddad K, Janz V, Castle PE et al . Detection of genomic amplification of the human telomerase gene (TERC) in cytologic specimens as a genetic test for the diagnosis of cervical dysplasia. Am J Pathol 2003; 163: 1405–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Heselmeyer‐Haddad K, Sommerfeld K, White NM et al . Genomic amplification of the human telomerase gene (TERC) in Pap smears predicts the development of cervical cancer. Am J Pathol 2005; 166: 1229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cairney CJ, Keith WN. Telomerase redefined. integrated regulation of hTR and hTERT for telomere maintenance and telomerase activity. Biochimie 2008; 90: 13–23. [DOI] [PubMed] [Google Scholar]
- 43. Toouli CD, Huschtscha LI, Neumann AA et al . Comparison of human mammary epithelial cells immortalized by simian virus 40 T‐Antigen or by the telomerase catalytic subunit. Oncogene 2002; 21: 128–39. [DOI] [PubMed] [Google Scholar]
- 44. Blasco MA. Telomerase beyond telomeres. Nat Rev Cancer 2002; 2: 627–33. [DOI] [PubMed] [Google Scholar]
- 45. Cao Y, Li H, Deb S, Liu JP. TERT regulates cell survival independent of telomerase enzymatic activity. Oncogene 2002; 21: 3130–8. [DOI] [PubMed] [Google Scholar]
- 46. Rahman R, Latonen L, Wiman KG. hTERT antagonizes p53‐induced apoptosis independently of telomerase activity. Oncogene 2005; 24: 1320–7. [DOI] [PubMed] [Google Scholar]
- 47. Del Bufalo D, Rizzo A, Trisciuoglio D et al . Involvement of hTERT in apoptosis induced by interference with Bcl‐2 expression and function. Cell Death Differ 2005; 12: 1429–38. [DOI] [PubMed] [Google Scholar]
- 48. Choi J, Southworth LK, Sarin KY et al . TERT promotes epithelial proliferation through transcriptional control of a Myc‐ and Wnt‐related developmental program. PLoS Genet 2008; 4: e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kedde M, Sage CL, Duursma A et al . Telomerase independent regulation of ATR by human telomerase RNA. J Biol Chem 2006; 281: 40503–14. [DOI] [PubMed] [Google Scholar]
- 50. Meyerson M, Counter CM, Eaton EN et al . hEST2, the putative human telomerase catalytic subunit gene, is up‐regulated in tumor cells and during immortalization. Cell 1997; 90: 785–95. [DOI] [PubMed] [Google Scholar]
- 51. Kilian A, Bowtell DD, Abud HE et al . Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum Mol Genet 1997; 6: 2011–19. [DOI] [PubMed] [Google Scholar]
- 52. Harrington L, Zhou W, McPhail T et al . Human telomerase contains evolutionarily conserved catalytic and structural subunits. Genes Dev 1997; 11: 3109–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nakayama J, Tahara H, Tahara E et al . Telomerase activation by hTRT in human normal fibroblasts and hepatocellular carcinomas. Nat Genet 1998; 18: 65–8. [DOI] [PubMed] [Google Scholar]
- 54. Oh H, Taffet GE, Youker KA et al . Telomerase reverse transcriptase promotes cardiac muscle cell proliferation, hypertrophy, and survival. Proc Natl Acad Sci USA 2001; 98: 10308–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Soder AI, Going JJ, Kaye SB, Keith WN. Tumour specific regulation of telomerase RNA gene expression visualized by in situ hybridization. Oncogene 1998; 16: 979–83. [DOI] [PubMed] [Google Scholar]
- 56. Heine B, Hummel M, Demel G, Stein H. Demonstration of constant upregulation of the telomerase RNA component in human gastric carcinomas using in situ hybridization. J Pathol 1998; 185: 139–44. [DOI] [PubMed] [Google Scholar]
- 57. Paradis V, Dargere D, Laurendeau I et al . Expression of the RNA component of human telomerase (hTR) in prostate cancer, prostatic intraepithelial neoplasia, and normal prostate tissue. J Pathol 1999; 189: 213–18. [DOI] [PubMed] [Google Scholar]
- 58. Yashima K, Litzky LA, Kaiser L et al . Telomerase expression in respiratory epithelium during the multistage pathogenesis of lung carcinomas. Cancer Res 1997; 57: 2373–7. [PubMed] [Google Scholar]
- 59. Morales CP, Lee EL, Shay JW. In situ hybridization for the detection of telomerase RNA in the progression from Barrett's esophagus to esophageal adenocarcinoma. Cancer 1998; 83: 652–9. [PubMed] [Google Scholar]
- 60. Fragnet L, Blasco MA, Klapper W, Rasschaert D. The RNA subunit of telomerase is encoded by Marek's disease virus. J Virol 2003; 77: 5985–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Trapp S, Parcells MS, Kamil JP et al . A virus‐encoded telomerase RNA promotes malignant T cell lymphomagenesis. J Exp Med 2006; 203: 1307–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yi X, Tesmer VM, Savre‐Train I, Shay JW, Wright WE. Both transcriptional and posttranscriptional mechanisms regulate human telomerase template RNA levels. Mol Cell Biol 1999; 19: 3989–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Marrone A, Stevens D, Vulliamy T, Dokal I, Mason PJ. Heterozygous telomerase RNA mutations found in dyskeratosis congenita and aplastic anemia reduce telomerase activity via haploinsufficiency. Blood 2004; 104: 3936–42. [DOI] [PubMed] [Google Scholar]
- 64. Hathcock KS, Hemann MT, Opperman KK, Strong MA, Greider CW, Hodes RJ. Haploinsufficiency of mTR results in defects in telomere elongation. Proc Natl Acad Sci USA 2002; 99: 3591–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chiang YJ, Hemann MT, Hathcock KS et al . Expression of telomerase RNA template, but not telomerase reverse transcriptase, is limiting for telomere length maintenance in vivo. Mol Cell Biol 2004; 24: 7024–31. [DOI] [PMC free article] [PubMed] [Google Scholar]