

Abstract
Bone marrow (BM) neovascularization and vascular endothelial growth factor (VEGF) expression in multiple myeloma (MM) correlate with disease progression. Brain derived neurotrophic factor (BDNF) is highly expressed by malignant plasma cells isolated from the majority of MM patients. Recently, BDNF was identified as a potential proangiogenic factor for the promotion of endothelial cell survival, induction of neoangiogenesis in ischemic tissues, and increase of VEGF expression in neuroblastoma. Since tropomyosin receptor kinase B (TrkB), the receptor of BDNF, is expressed by stromal cells within the BM milieu, here we sought to evaluate the involvement of BDNF/TrkB in myeloma–marrow stroma interaction and its effects on BM angiogenesis. TrkB was abundantly expressed by bone marrow stromal cells (BMSCs) isolated from healthy donors. Stimulation of BMSCs with BDNF induced a time‐ and dose‐ dependent increase in VEGF secretion, which was completely abolished by K252α, an inhibitor of TrkB. BDNF triggered activation of signal transducer and activator of transcription 3 (STAT3) and activator protein‐1 (AP‐1), whereas STAT3 was involved in mediating VEGF expression. We further delineated the biological significance of BDNF in MM by using lentiviral short‐interfering RNA (shRNA). When myeloma cells were cocultured with BMSCs in a noncontact Transwell system, VEGF levels in supernatants were significantly decreased when BDNF expression was knocked down. Furthermore, silencing of BDNF expression significantly inhibited xenograft tumor growth and angiogenesis, and prolonged survival in mouse model. Our studies demonstrate that BDNF, as a potential stimulator of angiogenesis, contributes to MM tumorgenesis; it mediates stromal–MM cell interactions via selective activation of specific receptor TrkB and downstream signal transducer STAT3, regulating VEGF secretion.
(Cancer Sci 2010; 101: 1117–1124)
Multiple myeloma (MM) is a B‐cell malignancy characterized by the clonal expansion of malignant plasma cells that reside in the bone marrow (BM) milieu. It has been reported that BM microvessel density is increased in patients with active MM. Angiogenesis is a required step of MM progression and has prognostic potential.( 1 , 2 , 3 ) The pathophysiology of MM‐induced angiogenesis involves both direct production of angiogenic cytokines by plasma cells and their induction within the microenvironment. Consistent with this theory, several studies have demonstrated that bone marrow stromal cells (BMSCs) provide various proangigenic cytokines, such as vascular endothelial growth factor (VEGF), basic fibroblast growth factor (b‐FGF), and interleukin‐6 (IL‐6) via stroma–myeloma cell interaction, and contribute to plasma cell survival, growth, and angiogenesis in the BM milieu.( 4 , 5 , 6 , 7 )
Brain derived neurotrophic factor (BDNF) is highly expressed by malignant plasma cells isolated from the majority of patients with MM. We and others have indicated that BDNF selective activation of tyrosine kinase receptor B pathway promotes myeloma cell growth and survival, and protects cells from chemotherapy.( 8 , 9 ) In addition to its neuropoietic action, BDNF has been recently identified as a mediator of angiogenesis which promotes endothelial cell survival,( 10 , 11 , 12 ) induces neoangiogenesis in ischemic tissues,( 13 ) and increases VEGF expression in neuroblastoma.( 14 ) Recently, we found that MM‐derived BDNF significantly stimulated vessel formation in vitro, and this effect was inhibited by anti‐BDNF mAb. Moreover, humanized anti‐BDNF mAb mediates significant antibody‐dependent cellular cytoxicity (ADCC) against allogeneic and autologous BDNF‐expression MM cells and inhibits tumor cell growth and neovasculariztion in several xenograft models of human MM.( 15 ) These findings indicate that BDNF activation of the TrkB pathway contributes to MM angiogenesis necessary for tumor progression.
Currently, the specific molecular mechanism of BDNF on angiogenesis has not been clarified in MM. Besides its frequent expression by malignant plasma cells, TrkB was found to be expressed by osteoblasts, endothelial cells, and BMSCs in the BM microenvironment.( 16 , 17 , 18 ) Since BDNF and its receptor TrkB expressed by MM cells contribute to plasma cell survival, TrkB expressed by BMSCs may allow stroma–myeloma cell interaction, further influencing BM angiogenesis via secreting angiogenic cytokines. In addition, studies of our group found that the increased serum BDNF levels detected in patients with MM correlated with disease activity and serum VEGF levels.( 5 ) VEGF is a well‐known potential proangiogenic factor and an attractive target for tumor anti‐angiogenic therapies. BMSCs were reported to be a potent resource of VEGF,( 19 , 20 , 21 ) and BMSC release of VEGF is associated with activation of signal transducer and activator of transcription 3 (STAT3).( 21 , 22 , 23 ) Activation of STAT3 in response to a wide array of cytokines and growth factors has been demonstrated as a downstream mediator of Trk signaling and function.( 24 , 25 , 26 , 27 ) Based on these studies, we hypothesized that BDNF activation of the TrkB pathway may regulate VEGF expression by BMSCs through the STAT3 signaling pathway.
In the present study, we characterized the activity of BDNF in MM neoangiogenesis by inhibiting BDNF expression using lentiviral BDNF shRNA vector in BDNF‐expression MM cells and coculturing this BDNF knockdown MM cells with BMSCs in vitro and in vivo. We found that BDNF stimulated VEGF secretion from BMSCs via STAT3 activation, and observed silencing of BDNF down‐regulated VEGF concentration in cocultures of myeloma cells and BMSCs. Importantly, silencing of BDNF blocked in vivo tumor growth and angiogenesis and prolonged survival in a xenograft model of human MM in the presence of BMSCs. These results established a pathophysiological role of BDNF in MM.
Materials and Methods
Cell lines and reagents. Human MM cell lines (HMCLs) RPMI‐8226 and U266 were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in RPMI‐1640 (Hyclone, Logan, UT, USA) with 10% fetal bovine serum (Gibco, Grand Island, NY, USA), 100 units/mL penicillin, and 100 μg/mL streptomycin. Human recombinant BDNF (PeproTech, Princeton, NJ, USA), TrkB‐specific inhibitor K252α (Calbiochem, San Diego, CA, USA), activator protein‐1 (AP‐1) inhibitor curcumin (Sigma‐Aldrich, Deisenhofen, Germany), and the JNK2 inhibitor AG490 (Invitrogen, Carlsbad, CA, USA) were obtained and reconstituted according to the manufacturers’ specifications. Antihuman BDNF, antihuman TrkB, and antihuman STAT3 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti‐phospho STAT3 (Tyr705) and antimouse CD34 were purchased from Cell Signaling (Danvers, MA, USA) and BD Pharmingen (San Diego, CA, USA), respectively.
Isolation and identification of human BMSCs. Anti‐coagulated BM samples were obtained from aspirates of nine healthy donors after the provision of informed consent. Cultures of BMSCs were established and identified according to the method of Pittenger et al. ( 28 ) Additional details are shown in the Supporting Methods.
Lentiviral vector for BDNF small‐hairpin RNA. To directly identify the biological function of BDNF in MM, the small‐hairpin RNA (shRNA) was designed using BDNF Refseq cDNA sequence (GenBank accession number NM_170735) as previously described by our group.( 29 ) A pair of 65‐nucleotide oligonucleotides encoding a 19‐nucleotide BDNF shRNA (5′‐CCGGCCGGCATTGGAACTCCCAGTGTTCAAGACGCACTGGGAGTTCCAATGCCTTTTTTG‐3′, 5′‐AATTCAAAAAAGGCATTGGAACTCCCAGTGCGTCTTGAACACTGGGAGTTCCAATGCCGG‐3′) were chemically synthesized, annealed, and inserted into the expression vector pGCSIL‐eGFP by Genechem (Shanghai, China), and confirmed by sequencing. Lentiviral BDNF shRNA and negative control shRNA were produced in 293t packaging cells and then transduced into MM cell lines.
Stable transfection of shRNA/eGFP constructs in MM Cells. MM cells were transfected with lentiviral BDNF shRNA‐pGCSIL‐eGFP vector or control vector plasmid in the presence of 4 μg/mL polybrene (Sigma‐Aldrich, St. Louis, MO, USA) at multiplicity of infection (MOI) of 10 and centrifuged at 780 g for 30 min. Thereafter, cells were cultured for another 72 h and analyzed for the expression of eGFP by a FACS Calibur flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). For the selection of shRNA‐eGFP‐transfected RPMI‐8226 MM cells, the eGFP bright cells were sorted on a FACS Diva (Becton‐Dickinson, Franklin Lakes, NJ, USA) on three sequential occasions. Down‐regulation of BDNF protein expression was further confirmed by western blotting.
Western blotting. Total cell lysates were subjected to 10% SDS‐PAGE gel electrophoresis and transferred to nitrocellulose membranes (Pierce Biotechnology, Rockford, IL, USA). After blocking for 3 h with 5% nonfat milk in TBS, membranes were incubated with 1:500 diluted BDNF, TrkB, and STAT3 monoclonal antibodies, 1:1000 diluted phosphor‐STAT3 (Tyr705) monoclonal antibody, and 1:2000 diluted β‐actin monoclonal antibody at 4°C overnight. After washing with TBST, membranes were incubated with HRP‐conjugated secondary antibody for 2 h at room temperature. Specific bands were detected using the ECL detection system (Pierce Biotechnology).
VEGF enzyme‐linked immunosorbant assay (ELISA). BMSCs of mouse (3 × 105cells/well) were cultured into a 24‐well plate in serum‐free RPMI‐1640 medium for 6 h and then treat with different concentrations of recombinant human BDNF for 6, 12, 24, 48 h. Cells were treated with 100 ng/mL BDNF for the indicated times with or without pretreatment with TrkB‐inhibitor K252α, JNK‐inhibitor AG490, and AP‐1‐inhibitor curcumin. Next, BMSC‐MM cocultures were performed using a noncontact Transwell system (pore size, 0.4 μm; Corning Costar, Cambridge, MA, USA) with shBDNF or control RPMI‐8226 cells seeded in the inserts at a density of 2 × 106cells/mL and BMSCs growing on the bottom of the plates (3 × 105cells/well) for 48 h. The cocultures of MM‐BMSCs were performed either in the absence or presence of K252α. Thereafter, cells were pelleted and the supernatants were analyzed for VEGF by using the Quantikine human VEGF immunoassay assay kit (R&D Systems, Minneapolis, MN, USA) with a minimum detectable dose of 20 pg/mL.
Nuclear extract and lightshift chemiluminescent electrophoretic mobility shift assay. Confluent BMSCs were starved for 6 h and then were stimulated by BDNF with or without pretreated with K252α; nuclear extracts were prepared as previously described.( 30 ) LightShift Chemiluminescent EMSA kit (Pierce Biotechnology) was used to detect DNA‐protein interaction. The following consensus oligonucleotide was used as probes in EMSA: AP‐1 (5′‐CGC TTG ATG ACT CAG CCG GAA‐3′). The 5′ ends of the oligonucleotides were biotin labeled. Additional details are shown in the Supporting Methods.
Human plasmacytoma xenograft model. All animal studies were conducted according to protocols approved by the Animal Ethics Committee of the Tongji Medical College. The mice were irradiated (200 cGy) using a 60Co resource and were injected with cells the following day under anesthesia. 5 × 106 BDNF shRNA/eGFP RPMI‐8226 MM cells or control shRNA/eGFP RPMI 8226 MM cells which mixed with 1 × 106 BMSC or not inoculated subcutaneously in 100 μL RPMI‐1640 medium. Stable shRNA/eGFP+ transfected MM cells were monitored by noninvasive in‐vivo optical imaging (Kodak Image Station 2000 MM; Eastman Kodak Company, Rochester, NY, USA). Tumor growth and progression were measured weekly by caliper and the tumor volumes were calculated using the formula: [1/2] × a × b 2, where a and b represented the larger and smaller tumor diameters, respectively. Survival was evaluated from the first day of treatment until death. The expression of BDNF and VEGF protein in xenograft tumor was evaluated by western blotting. For histopathological analyses, tumor tissue excised from mice were washed by PBS and fixed with 4% paraformaldehyde and hematoxylin–eosin staining was performed. Tissues were evaluated by immunohistochemical analysis for CD34 expression as previously described.( 15 )
Statistical analysis. In vitro experiments were performed in triplicate, and the results are reported as mean with SE. Statistical significance of differences observed in treatment compared with respective control groups was determined using the Student’s t‐test. The minimal level of significance was P < 0.05. The significance of differences in tumor volume between groups was estimated by a one‐way ANOVA test. Overall survival was assessed using Kaplan–Meier curves and log‐rank analysis.
Results
Isolation of efficient and stable BDNF knockdown RPMI‐8226 cells. The selected pure BDNF shRNA and control shRNA transfected RPMI‐8826 cells were able to retain eGFP after numerous passages as shown in the in vitro fluorescence microscopic analysis (Fig. 1a). As shown in Figure 1(b), the purity of transfectants was determined by flow cytometric analysis of enhanced GFP reporter protein, and the selected BDNF shRNA and control shRNA RPMI‐8226 cells could reach up to 91.87% and 90.50% when compared with parental RPMI‐8226 cells. The silencing capacity of BDNF shRNA on selected RPMI‐8226 shBDNF cells was further determined by western blotting (Fig. 1c). Transfection of RPMI‐8226 cells with shBDNF‐pGCSIL‐eGFP lentiviral vector resulted in a significantly reduced expression of BDNF protein.
Figure 1.
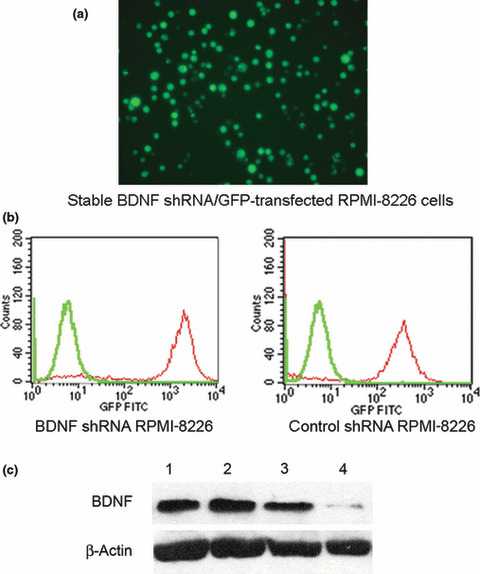
Stable knockdown of brain‐derived neu‐rotrophic factor (BDNF) expression in RPMI‐8226 cells by lentiviral based pGCSIL‐eGFP‐shBDNF vector. (a) Fluorescence microscopic analysis of shBDNF/eGFP+ RPMI‐8226 cells stable transfected with the lentiviral pGCSIL‐eGFP‐shBDNF vector at ×200 magnification. (b) Flow cytometric analysis indicates an approximate 2‐log difference in mean fluorescence intensity of BDNF or control shRNA/eGFP+ transfected RPMI‐8226 cells (red peak) versus parental RPMI‐8226 cells (green peak). (c) Western blot analysis confirmed that both parental U266 (lane 1) and RPMI‐8226 (lane 2) expressed BDNF, and endogenous BDNF was knocked down in BDNF shRNA/eGFP+ RPMI‐8226 (lane 4) compared to control (lane 3) and parental RPMI‐8226 cells (lane 2).
Up‐regulation of VEGF secretion by BDNF‐stimulated BMSCs. BMSCs were isolated from nine healthy donors and expressed CD44 and CD90, but not CD34, CD45, and CD86 (Supporting Fig. S1a). BMSCs were further characterized for differentiation capacity (Supporting Fig. S1b). To investigate whether BDNF might have a paracrine regulation effect on BMSCs, expression of BDNF receptor TrkB was analyzed by western blot. As shown in Figure 2(a), TrkB was consistently expressed by represent BMSC cultures and induced by BDNF stimulation, whereas TrkB expression was inhibited when BMSCs were co‐cultured with BDNF knockdown MM cells compared to control MM cells.
Figure 2.
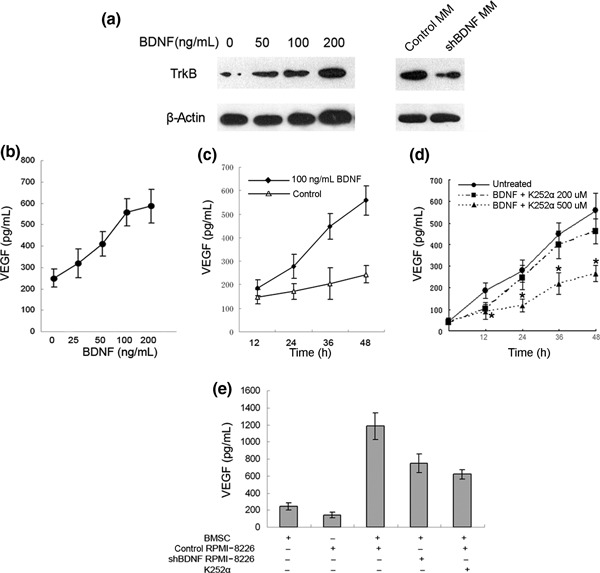
Effect of brain‐derived neurotrophic factor (BDNF) on vascular endothelial growth factor (VEGF) secretion in bone marrow stromal cells. (a) Western blot analysis demonstrating tropomyosin receptor kinase B (TrkB) expression by human bone marrow stromal cells (BMSCs). (b) VEGF concentrations were determined in supernatant of BMSC cultures exposed to various concentrations of BDNF (0–200 ng/mL) for 48 h. (c) VEGF concentrations were determined in supernatants of BMSCs exposed to 100 ng/mL of BDNF at various time points (•), unstimulated controls (△). (d) As a control experiment, stimulation with 100 ng/mL BDNF was also performed with or without K252α pretreatment (200, 500 nm; 30 min) in BMSC cultures. VEGF in supernatants at various time points (0, 12, 24, 48 h) was measured by ELISA. (e) Enhanced VEGF secretion of BMSCs in Transwell cocultures with RPMI‐8226 cells (P < 0.001 vs BMSCs). The addition of K252α results in a significant reduction of VEGF secretion (P < 0.001). Down‐regulation of BDNF expression inhibits VEGF secretion in Transwell cocultures of RPMI‐8226 and BMSCs (P < 0.001).
We therefore examined the direct incentive effect of BDNF on baseline VEGF secretion from BMSCs using ELISA. BMSCs constitutively produced VEGF (249.69 ± 41.69 pg/mL per 1 × 105 cells). Stimulation of BMSCs with BDNF induced a time‐ and dose‐ dependent increase in VEGF secretion (2.29‐fold over unstimulated controls at 100 ng/mL of BDNF after 48 h; P < 0.001; Fig. 2a,b). K252α abrogated VEGF protein expression at a concentration of 500 nM at 12 to 48 h (P < 0.001, Fig. 2c).
Silencing of BDNF expression down‐regulating VEGF secretion in Transwell cocultures of myeloma and marrow stroma cells. After having established that BDNF is able to induce VEGF secretion in BMSCs, we examined the effect of myeloma‐derived BDNF on VEGF secretion in a Transwell coculture system to preclude direct myeloma‐stroma cell contacts. When RPMI‐8226 cells were cultured with BMSCs, VEGF secretion was up‐regulated 3.1‐fold over the sum of basal concentrations in the monoculture controls. This up‐regulation of VEGF secretion is partially abrogated by K252α (47.78%, Fig. 2c). We next examined the effect of silencing the expression of myeloma‐derived BDNF on VEGF secretion by BMSCs. VEGF levels in the coculture supernatants decreased 36.89% when BDNF shRNA RPMI‐8226 cells were cocultured with BMSCs (Fig. 2c). Silencing of myeloma‐derived BDNF protein inhibited VEGF secretion by co‐cultured BMSCs.
STAT3 and AP‐1 mediate BDNF‐induced VEGF expression. Since AP‐1 is involved in regulation of hypoxia‐inducible factor‐1α (HIF‐1α) which is identified as a crucial factor modulating BDNF‐induced VEGF secretion in neuroblastoma.( 14 ) STAT3 has been shown to mediate VEGF production by BMSCs( 20 ) and works as a downstream mediator of TrkB signaling and function 23 . Therefore, we investigated whether BDNF affects VEGF expression via activatiion of transcription factor STAT3 and/or AP‐1. We first examined AP‐1 activity in BMSCs stimulated with 100 ng/mL BDNF by EMSA using AP‐1–biotin labeled consensus oligonucleotides. AP‐1 DNA binding activity was significantly increased and reached a peak at 1 h after BDNF stimulation, and pre‐incubation of BMSCs with K252α efficiently inhibited BDNF‐induced increase (P < 0.05, Fig. 3a,b). Increased phosphorylation of STAT3 was also detected in BDNF‐treated BMSCs by western blotting and the peak was seen at 30 min. When the BMSCs were pre‐incubated with K252α and then stimulated with BDNF, BDNF‐induced STAT3 activation was significantly blocked (P < 0.001, Fig. 3c,d). Furthermore, to determine whether BDNF induces AP‐1 activation is involved in the increased VEGF secretion, BMSCs were pretreated with the pharmacologic inhibitors of AP‐1 (12.5 μm, Curcumin) or STAT3 (100 μm, AG490) following stimulation with 100 ng/mL BDNF for 48 h. AG490 significantly blocked the increased VEGF secretion of BMSCs induced by BDNF, but the levels were still higher than the basal level of VEGF (Fig. 3e). Treatment of these cells with 12.5 μm curcumin partially inhibited VEGF secretion induced by BDNF stimulation with no statistical significance. These findings indicate that STAT3 is mainly the mediator of BDNF‐induced VEGF secretion in BMSCs.
Figure 3.
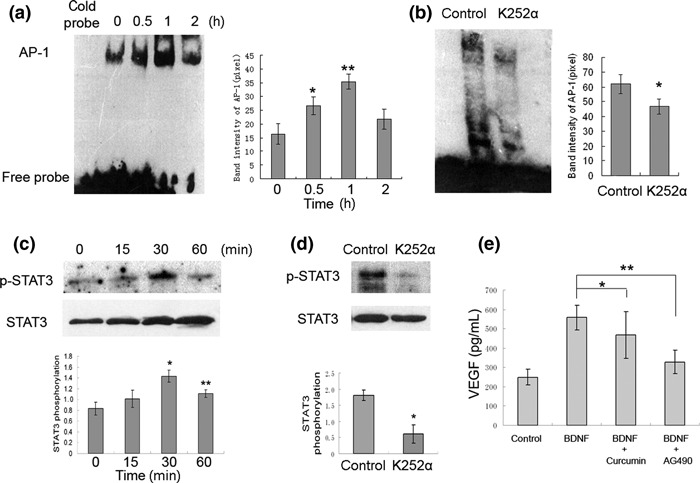
Brain‐derived neurotrophic factor (BDNF) stimulates activator protein‐1 (AP‐1) translocation and signal transducer and activator of transcription 3 (STAT3) phosphorylation, and STAT3 is involved in BDNF‐induced VEGF secretion in bone marrow stromal cells (BMSCs). (a) BMSCs treated with 100 ng/mL BDNF for 0.5, 1, and 2 h, AP‐1 DNA binding activity in the nuclear extracts was measured by EMSA. (*P < 0.05 vs control; **P < 0.005 vs control). (b) The DNA binding activity of AP‐1 in cultured BMSC stimulated by 100 ng/mL BDNF for 1 h in the absence or presence of tropomyosin receptor kinase B (TrkB) inhibitor K252α (*P < 0.05). (c) BMSCs were stimulated with 100 ng/mL BDNF for 0, 15, 30, 60 min; cell lysates were analyzed by western blotting with anti‐pospho (Tyr 705) STAT3. Anti‐STAT3 antibodies were used as loading control (100 ng/mL BDNF vs control, *P < 0.01 at 30 min, **P < 0.05 at 60 min). (d) BMSCs were pretreated with or without K252α and then stimulated with 100 ng/mL BDNF for 30 min (*P < 0.001). (e) Inhibition of BDNF induced AP‐1 and STAT3 activity by curcumin and AG490, and then incubation with BDNF for 48 h and VEGF protein was measured by ELISA. (BDNF + curcumin vs BDNF, *P = 0.064; BDNF + AG490 vs BDNF, **P < 0.001).
Stable knockdown of BDNF inhibits tumor growth and angiogenesis in vivo. We investigated whether knockdown of myeloma‐derived BDNF inhibits tumor growth and angiogenesis in myeloma xenograft mice. Therefore, BDNF/control shRNA RPMI‐8226 cells were subcutaneously injected into BALB/cA nude mice to compare tumor growth and microvessel density. Mice were monitored serially by real‐time fluorescence imaging as an indicator of BDNF knockdown maintenance and in vivo MM tumor growth (Fig. 4a). A significant decrease in tumor volume was observed in the BDNF shRNA RPMI‐8226 cell‐treated group (n = 12) when compared to control shRNA (n = 12) or the parental RPMI‐8226 cell‐treated group (n = 12) (Fig. 4b). Moreover, we found BMSCs accelerated the growth of tumors in mice inoculated with BMSCs and MM cells, and silencing of BDNF expression in MM cells also significantly blocked tumor growth in the present of BMSCs (Fig. 4b). Kaplan–Meier curves and log‐rank analysis showed statistically significant prolongations in mean overall survival compared with control mice were observed in BDNF knockdown MM cell‐treated mice (control RPMI‐8226 vs shBDNF RPMI‐8226, P = 0.001. BMSC + control RPMI‐8226 vs BMSC + shBDNF RPMI 8226, P < 0.001) (Fig. 4c). We observed that tumors from animals treated with BMSCs had a greater proportion of tumor stroma compared with tumors from untreated animals (Fig. 5a). Western blotting analysis indicated that BDNF shRNA significantly inhibits BDNF expression by myeloma cells and downregulates VEGF secretion by tumor tissues in vivo (Fig. 5b). Moreover, decreased microvessel density, which was assessed by immunohistochemistry staining using rat antimouse CD34 mAb, was seen in tumors from mice inoculated with BDNF shRNA MM cells with or without treatment of BMSCs (control vs shBDNF, *P < 0.001; control + BMSCs vs shBDNF + BMSCs, **P < 0.001; Fig. 6). The results indicated that BDNF shRNA suppresses MM growth in vivo, which accompanied with decreased microvessel density and prolonged mean overall survival.
Figure 4.
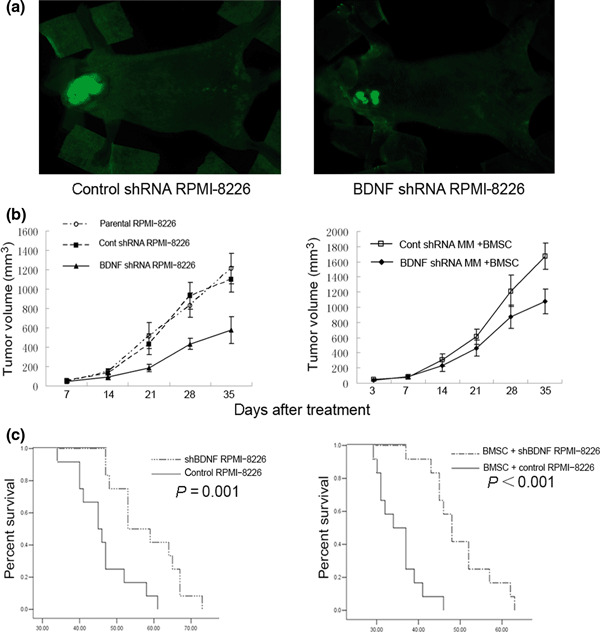
Knockdown of brain‐derived neurotrophic factor (BDNF) protein by lentiviral BDNF shRNA inhibits human multiple myeloma (MM) cell growth and prolonged overall survival in vivo. (a) Representative bright fluorescence images of mice bearing the myeloma xenograft tumors; tumors are indicated by arrows. (b) Tumor volume measurements of BDNF/control shRNA RPMI‐8226 cells injected subcutaneously into nude mice with or without bone marrow stromal cells (BMSCs). (c) The effect of BDNF knockdown on overall survival in the absence or presence of BMSCs were analyzed by Kaplan–Meier curve and long‐rank test.
Figure 5.
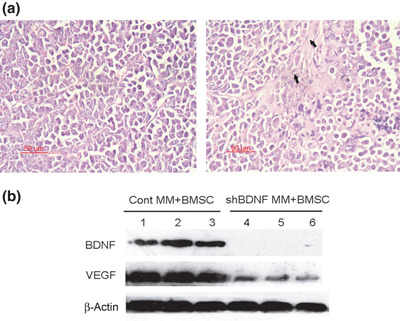
Brain‐derived neurotrophic factor (BDNF) shRNA knockdown of BDNF expression and inhibition of VEGF secretion in myeloma xenografts. (a) Representative haematoxylin–eosin‐stained sections of tumor of mice inoculated with multiple myeloma (MM) cells (left panel) or BMSC + MM cells (right panel). (b) The expression of BDNF and vascular endothelial growth factor (VEGF) protein in xenograft tumors was evaluated by western blotting analysis. BDNF shRNA significantly knocked down of BDNF expression and down‐regulated VEGF secretion in vivo. Lanes 1–3, control MM + BMSCs xenograft; lanes 4–6, shBDNF MM + BMSCs xenograft.
Figure 6.
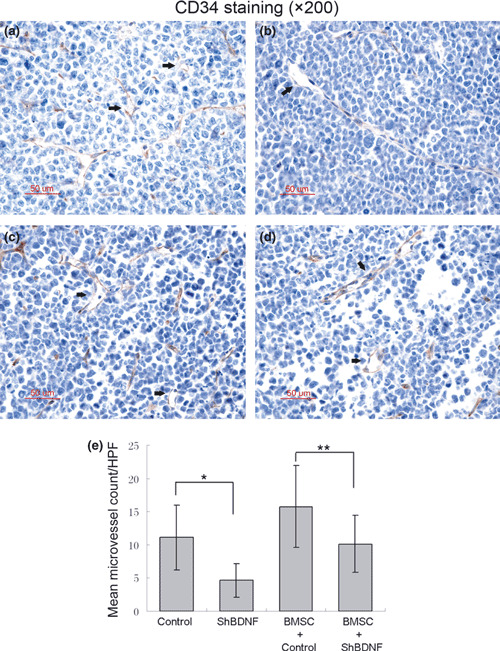
Effect of brain‐derived neurotrophic factor (BDNF) shRNA on angiogenesis in vivo. Microvessel density (MVD) was evaluated by immuno‐histochemical analysis for CD34 expression on formalin‐fixed paraffin‐embedded myeloma xeno‐graft tissue sections, and was determined by microscopic counting of CD‐34 stained microvessels at ×200 magnification. Data represent means ± SEM of five separated high‐power fields (HPFs). Representative vessels indicated with arrows. A significant decrease of MVD was observed in BDNF shRNA RPMI‐8226 cell‐treated mice compared with control RPMI‐8226 cell‐treated mice with or without bone marrow stromal cells (BMSC) combined treatment. (a) Control RPMI‐8226 cells; (b) BDNF shRNA RPMI‐8226 cells; (c) control RPMI‐8226 cells + BMSCs; (d) BDNF shRNA RPMI‐8226 cells + BMSCs. (e) Quantitative analysis of MVD.
Discussion
A potential role of BDNF in MM pathophysiology was suggested by its over‐expression in malignant plasma cells isolated from MM patients as well as in most HMCLs, and its effects on supporting myeloma cells survival.( 8 , 9 ) Previously, we identified that BDNF was a crucial proangiogeneic factor modulating vessel formation, and its serum level parallels serum VEGF level in MM patients.( 15 ) In the present study, we sought to elucidate the molecular mechanism of BDNF‐mediated angiogenesis in the BM microenvironment of MM. Our findings corroborate previous reports demonstrating that BMSCs isolated from healthy donors express BDNF high affinity receptor TrkB, indicating that paracrine BDNF signaling in the marrow microenvironment may occur.( 16 , 17 , 18 ) The data suggested that the addition of BDNF can stimulate VEGF secretion by BMSC, and this effect can be blocked by K252α, which is an specific inhibitor of TrkB. We further detected VEGF secretion by MM cell lines and BMSCs isolated from healthy donors. Importantly, VEGF concentration is highly increased in the supernatants of BMSC and MM transwell cocultures, which suggests that VEGF secretion in cocultures is triggered by diffusible factor. Moreover, we used potential lentiviral shRNA‐targeting BDNF gene expression on BDNF‐expressing MM cells. This approach has proved to be a powerful tool in the analysis of gene function in vitro as well as in vivo. It can result in prolonged selective inhibition of target gene expression even in eukaryotic cells.( 31 , 32 ) Our studies show that silencing BDNF expression in MM cells down‐regulates VEGF secretion from BMSCs in a Transwell coculture system with no concern for cell–cell contact stimulus. Taken together, in addition to its previously described effects on myeloma cell survival, growth, and migration,( 8 , 9 ) BDNF stimulates VEGF secretion in the stromal cells in the BM milieu. Because VEGF is a potent proangiogenic cytokine whose high expression in MM is a marker of poor prognosis, BDNF may be involved in the regulating of MM neovascularization.
The JAK/STAT pathway, initially defined as a downstream signaling of the interferon, is responsive to a wide array of cytokines and growth factors. STAT3 plays a central role in mediating cell growth, differentiation, and survival signals.( 33 ) Recent evidence suggests that the STAT3 pathway has been implicated in regulating VEGF secretion in BMSCs,( 21 , 22 , 23 ) and neurotrophin‐induced activation of STAT3 is also required for several downstream functions of neutrophin signaling.( 24 ) In this study, we observed BDNF activation of TrkB‐induced STAT3 phosphorylation. We further confirmed that BDNF induction of STAT regulated the expression of VEGF in TrkB‐expressing BMSCs by using JAK/STAT3 inhibitor AG490. The VEGF promoter also contains three AP‐1 sites.( 34 , 35 ) In addition, BDNF can stimulate AP‐1 transactivation in cultured cerebellar neurons.( 36 , 37 ) In the present study, we found that BDNF stimulation can increase AP‐1 DNA binding in BMSCs. However, a direct relationship between AP‐1 translocation and VEGF up‐regulation was not determined in this experiment.
Our further studies examined the effect of blocking endogenous BDNF on tumor growth and BM microenvironment angiogenesis which is associated with disease progression. MM cells co‐transfected with reporter gene enhanced green fluorescent protein and BDNF/control shRNA were selected by FACS for the creation of stable cell lines and applied to in vivo experiment. The eGFP MM xenograft tumor murine model provides us with a valuable experimental setting to evaluate whether silencing of BDNF affects MM cells proliferation in vivo. Here we observed that BMSCs enhanced MM tumor growth in vivo, and tumors from mice treated with BMSCs had a greater proportion of tumor stroma and increased microvessel density. The increased angiogenesis of tumors from BMSCs‐treated mice may represent more angiogenic cytokine secretion induced by myeloma‐BMSC interaction. We next demonstrated that blocking BDNF secretion dramatically inhibits tumor growth and prolongs survival of mice inoculated with MM cell. In addition, we confirmed the inhibitory effect of silencing BDNF on VEGF secretion by western blotting and microvessel density by immunohistochemical analysis in vivo. In in vitro coculture experiment, BDNF secreted by MM cells is a stimulus of VEGF production by BMSCs. On the other hand, BDNF activation of TrkB stimulates VEGF expression also relevant to promote angiogenesis in neuroblastoma. Therefore, it is possible that within the tumor microenvironment, silencing of BDNF expression in MM cells inhibits angiogenesis through down‐regulation of VEGF secretion in vivo.
Neurotrophins are structurally and functionally related growth factors, including nerve growth factor (NGF), NT‐3, NT‐4/5, and BDNF, which contribute to neural cell survival, differentiation, and function via selective activation of tyrosine kinase receptor.( 38 , 39 ) Numerous studies have uncovered the critical role for neurotrophins and their receptors on neuronal and non‐neuronal tumor pathology, such as neuroblastoma, adenocarcinoma of the thyroid, pancreatic ductal adenocarcinoma, prostate cancer, and hematological malignancies of myeloid leukemia and multiple myeloma.( 6 , 14 , 40 , 41 , 42 , 43 , 44 , 45 ) In this study, we demonstrated that BDNF activation of TrkB mediates myeloma–BMSC interaction in the BM milieu, and promotes angiogenesis by elevating VEGF secretion in BMSCs. This is analogous to the effect of the BDNF/TrkB pathway in neuroblastoma.( 14 ) Therefore, these findings provide a possible mechanism for the effect of the BDNF/TrkB pathway on survival and angiogenesis in MM tumorgenesis.
In conclusion, our data demonstrated that myeloma cell‐derived BDNF can stimulate VEGF secretion from BMSCs in the BM milieu via induction of STAT3, and that silencing of endogenous BDNF also inhibits MM tumor growth and angogensis in vivo. Since we previously found that serum BDNF levels correlate with BM microvessel density and parallel serum VEGF levels, it is strongly suggested that BDNF acts as a proangiogenic cytokine in paracrine interactions between myeloma and stroma cells besides its direct effects on MM survival and migration. These findings support targeting BDNF as a new therapeutic strategy for BM neovaculariztion to improve patient outcome in MM.
Supporting information
Supporting Methods.
Fig. S1. Isolation and identification of human BMSCs.
Fig. S2. Representative bright fluorescence images of mice bearing the myeloma xenograft tumors.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
We thank W.N. Wei, Y. Tian, and J.H. Zhang for excellent laboratory management, and J. Li and J. Cui for assistance with animal studies. This work was supported by the National Natural Science Foundation for Distinguished Young Scholars of China (grant no. 30825018).
References
- 1. Vacca A, Ribatti D, Roncali L et al. Bone marrow angiogenesis and progression in multiple myeloma. Br J Haematol 1994; 87: 503–8. [DOI] [PubMed] [Google Scholar]
- 2. Vacca A, Ribatti D, Presta M et al. Bone marrow neovascularization, plasma cell angiogenic potential and matrix metalloproteinase‐2 secretion parallel progression of human multiple myeloma. Blood 1999; 93: 3064–73. [PubMed] [Google Scholar]
- 3. Munshi NC, Wilson C. Increased bone marrow microvessel density in newly diagnosed multiple myeloma carries a poor prognosis. Semin Oncol 2001; 28: 565–9. [DOI] [PubMed] [Google Scholar]
- 4. Vacca A, Ria R, Ribatti D et al. A paracrine loop in the vascular endothelial growth factor pathway triggers tumor angiogenesis and growth in multiple myeloma. Haematologica 2003; 88: 176–85. [PubMed] [Google Scholar]
- 5. Loı¨c V, Jin DK, Karajannis MA et al. Fetal stromal‐dependent paracrine and intracrine vascular endothelial growth factor‐a/vascular endothelial growth factor receptor‐1 signaling promotes proliferation and motility of human primary myeloma cells. Cancer Res 2005; 65: 3185–92. [DOI] [PubMed] [Google Scholar]
- 6. Di Raimondo F, Azzaro MP, Palumbo GA et al. Angiogenic factors in multiple myeloma: higher levels in bone marrow than in peripheral blood. Haematologica 2000; 85: 800–5. [PubMed] [Google Scholar]
- 7. Vacca A, Ribatti D, Roccaro AM, Frigeri A, Dammacco F. Bone marrow angiogenesis in patients with active multiple myeloma. Semin Oncol 2001; 28: 543–50. [DOI] [PubMed] [Google Scholar]
- 8. Pearse RN, Swendeman SL, Li Y et al. A neurotrophin axis in myeloma: TrkB and BDNF promote tumor‐cell survival. Blood 2005; 105: 4429–36. [DOI] [PubMed] [Google Scholar]
- 9. Hu Y, Sun CY, Wang HF et al. Brain‐derived neurotrophic factor (BDNF) promotes growth and migration of multiple myeloma (MM) cells. Cancer Genet Cytogenet 2006; 169: 12–20. [DOI] [PubMed] [Google Scholar]
- 10. Turrini P, Gaetano C, Antonelli A, Capogrossi MC, Aloe L. Nerve growth factor induces angiogenic activity in a mouse model of hindlimb ischemia. Neurosci Lett 2002; 323: 102–12. [DOI] [PubMed] [Google Scholar]
- 11. Donavan MJ, Lin MI, Wiegn P et al. Brain derived neorotrophic factor is an endothelial cell survival factor required for intamyocardial vessel stabilization. Development 2000; 127: 4531–40. [DOI] [PubMed] [Google Scholar]
- 12. Kermani P, Rafii D, Jin DK et al. Neurotrophins promote revascularization by local recruitment of TrkB+ endothelial cells and systemic mobilization of hematopoietic progenitors. J Clin Invest 2005; 115: 653–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim H, Li Q, Hempstead BL, Madri JA. Paracrine and autocrine functions of brain‐derived neurotrophic factor and nerve growth factor (NGF) in brain‐derived endothelial cells. J Biol Chem 2004; 279: 33538–46. [DOI] [PubMed] [Google Scholar]
- 14. Nakamura K, Martin KC, Jackson JK, Beppu K, Woo CW, Thiele CJ. Brain‐derived neurotrophic factor activation of TrkB induces vascular endothelial growth factor expression via hypoxia‐inducible factor‐1alpha in neuroblastoma cells. Cancer Res 2006; 66: 4249–55. [DOI] [PubMed] [Google Scholar]
- 15. Hu Y, Wang HF, Guo T et al. Identification of BDNF as a novel angiogenic protein in multiple myeloma. Cancer Genet Cytogenet 2007; 178: 1–10. [DOI] [PubMed] [Google Scholar]
- 16. Cattoretti G, Schiro R, Orazi A, Soligo D, Colombo MP. Bone marrow stroma in humans: anti‐nerve growth factor receptor antibodies selectively stain reticular cells in vivo and in vitro. Blood 1993; 81: 1726–38. [PubMed] [Google Scholar]
- 17. Labouyrie E, Dubus P, Groppi A et al. Expression of neurotrophins and their receptors in human bone marrow. Am J Pathol 1999; 154: 405–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yaghoobia MM, Mowla SJ. Differential gene expression pattern of neurotrophins and their receptors during neuronal differentiation of rat bone marrow stromal cells. Neurosci Lett 2006; 397: 149–54. [DOI] [PubMed] [Google Scholar]
- 19. Wang M, Crisostomo PR, Herring C, Meldrum KK, Meldrum DR. Human progenitor cells from bone marrow or adipose tissue produce VEGF, HGF, and IGF‐I in response to TNF by a p38 MAPK‐dependent mechanism. Am J Physiol Regul Integr Comp Physiol 2006; 291: R880–4. [DOI] [PubMed] [Google Scholar]
- 20. Uemura R, Xu M, Ahmad N, Ashraf M. Bone marrow stem cells prevent left ventricular remodeling of ischemic heart through paracrine signaling. Circ Res 2006; 98: 1414–21. [DOI] [PubMed] [Google Scholar]
- 21. Wang M, Zhang W, Crisostomo P et al. STAT3 mediates bone marrow mesenchymal stem cell VEGF production. J Mol Cell Cardiol 2007; 42: 1009–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu Q, Briggs J, Park S et al. Targeting Stat3 blocks both HIF‐1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 2005; 24: 5552–60. [DOI] [PubMed] [Google Scholar]
- 23. Jung JE, Lee HG, Cho IH et al. STAT3 is a potential modulator of HIF‐1‐mediated VEGF expression in human renal carcinoma cells. FASEB J 2005; 19: 1296–8. [DOI] [PubMed] [Google Scholar]
- 24. Nq YP, Cheung ZH, Ip NY. STAT3 as a downstream mediator of Trk signaling and functions. J Biol Chem 2006; 281: 15636–44. [DOI] [PubMed] [Google Scholar]
- 25. Lin G, Bella AJ, Lue TF, Lin CS. Brain–derived neurotrophic factor acts primarily via the JAK/STAT pathway to promote neurite growth in the major pelvic ganglion of the rat. J Sex Med 2006; 3: 821–7. [DOI] [PubMed] [Google Scholar]
- 26. Lund IV, Hu Y, Raol YH et al. BDNF selectively regulates GABAA receptor transcription by activation of the JAK/STAT pathway. Sci Signal 2008; 1: ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Islam O, Loo TX, Heese K. Brain‐derived neurotrophic factor (BDNF) has proliferative effects on neural stem cells through the truncated TRK‐B receptor, MAP Kinase, AKT, and STAT‐3 signaling pathways. Curr Neurovasc Res 2009; 6: 42–53. [DOI] [PubMed] [Google Scholar]
- 28. Pittenger MF, Mackay AM, Beck SC et al. Multilineage potential of adult human mesenchymal stem cells. Science 1999; 284: 143–7. [DOI] [PubMed] [Google Scholar]
- 29. Huang J, Hu Y, Sun CY et al. Effect of interference targeting BDNF gene on Hela cell proliferation and apoptosis. Zhonghua Bing Li Xue Za Zhi 2009; 38: 686–90. [PubMed] [Google Scholar]
- 30. Zhang L, Hu Y, Sun CY, Huang J, Chu ZB. Brian‐derived neurotrophic factor promotes the secretion of MMP‐9 in human myeloma cell through modulation of nucleus factor‐kappaB. Zhonghua Xue Ye Xue Za Zhi 2008; 29: 243–6. [PubMed] [Google Scholar]
- 31. An DS, Xie Y, Mao SH, Morizono K, Kung SK, Chen IS. Efficient lentiviral vectors for short hairpin RNA delivery into human cells. Hum Gene Ther 2003; 14: 1207–12. [DOI] [PubMed] [Google Scholar]
- 32. Scherr M, Battmer K, Dallmann I, Ganser A, Eder M. Inhibition of GM‐CSF receptor function by stable RNA interference in a NOD/SCID mouse hematopoietic stem cell transplantation model. Oligonucleotides 2003; 13: 353–63. [DOI] [PubMed] [Google Scholar]
- 33. Aaronson DS, Horvath CM. A road map for those who don’t know JAK‐STAT. Science 2002; 296: 1653–5. [DOI] [PubMed] [Google Scholar]
- 34. Shi Q, Le X, Abbruzzese JL et al. Constitutive Sp1 activity is essential for differential constitutive expression of vascular endothelial growth factor in human pancreatic adenocarcinoma. Cancer Res 2001; 61: 4143–54. [PubMed] [Google Scholar]
- 35. Tischer E, Mitchell R, Hartman T et al. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J Biol Chem 1991; 266: 11947–54. [PubMed] [Google Scholar]
- 36. Li Z, Ding M, Thiele CJ, Luo J. Ethanol inhibits brain‐derived neurotrophic factor‐mediated intracellular signaling and activator protein‐1 activation in cerebellar granule neurons. Neuroscience 2004; 126: 149–62. [DOI] [PubMed] [Google Scholar]
- 37. Gaiddon C, Loeffler JP, Larmet Y. Brain‐derived neurotrophic factor stimulates AP‐1 and cyclic AMP‐responsive element dependent transcriptional activity in central nervous system neurons. J Neurochem 1996; 66: 2279–86. [DOI] [PubMed] [Google Scholar]
- 38. Persson H, Ibanez CF. Role and expression of neurotrophins and trk family of tyrosine kinase receptors in neural growth and rescue after injury. Curr Opin Neurol Neurosurg 1993; 6: 11–8. [PubMed] [Google Scholar]
- 39. Tessarollo L. Pleiotropic functions of neurotrophins in development. Cytokine Growth Factor Rev 1998; 9: 125–37. [DOI] [PubMed] [Google Scholar]
- 40. Mitra G, Martin‐Zanca D, Barbacid M. Identification and biochemical characterization of p70TRK, product of the human TRK oncogene. Proc Natl Acad Sci U S A 1987; 84: 6707–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eguchi M, Eguchi‐Ishimae M, Tojo A et al. Fusion of ETV6 to neurotrophin‐3 receptor TRKC in acute myeloid leukemia with t(12;15)(p13;q25). Blood 1999; 93: 1355–63. [PubMed] [Google Scholar]
- 42. Miknyoczki SJ, Wan W, Chang H et al. The neurotrophin‐trk receptor axes are critical for the growth and progression of human prostatic carcinoma and pancreatic ductal adenocarcinoma xenografts in nude mice. Clin Cancer Res 2002; 8: 1924–31. [PubMed] [Google Scholar]
- 43. Lamballe F, Klein R, Barbacid M. The trk family of oncogenes and neurotrophin receptors. Princess Takamatsu Symp 1991; 22: 153–70. [PubMed] [Google Scholar]
- 44. Satoh F, Mimata H, Nomura T. Autocrine expression of neurotrophins and their receptors in prostate cancer. Int J Urol 2001; 8: S28–34. [DOI] [PubMed] [Google Scholar]
- 45. Ohta T, Numata M, Tsukioka Y et al. Neurotrophin‐3 expression in human pancreatic cancers. J Pathol 1997; 181: 405–12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Methods.
Fig. S1. Isolation and identification of human BMSCs.
Fig. S2. Representative bright fluorescence images of mice bearing the myeloma xenograft tumors.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item