

Abstract
The ecotropic viral integration site‐1 (Evi‐1) gene was first identified as a common locus of retroviral integration in murine leukemia models. In humans, EVI‐1 is located on chromosome 3q26, and rearrangements on chromosome 3q26 often activate EVI‐1 expression in hematological malignancies. Overexpression of EVI‐1 also occurs with high frequency in leukemia patients without 3q26 abnormalities, and importantly, high EVI‐1 expression is an independent negative prognostic indicator irrespective of the presence of 3q26 rearrangements. Recent gene targeting studies in mice revealed that Evi‐1 is preferentially expressed in hematopoietic stem cells and plays an essential role in proliferation and maintenance of hematopoietic stem cells. In addition, intense attention has been focused on the EVI‐1 gene complex as retrovirus integration sites because transcription‐activating integrations into the EVI‐1 locus confer survival and self‐renewing ability to hematopoietic cells. The experimental results using animal models suggest that activation of Evi‐1 in hematopoietic cells leads to clonal expansion or dysplastic hematopoiesis, whereas onset of full‐blown leukemia requires cooperative genetic events. EVI‐1 possesses diverse functions as an oncoprotein, including suppression of transforming growth factor‐β‐mediated growth inhibition, upregulation of GATA2, inhibition of the Jun kinase pathway, and stimulation of cell growth via activator protein‐1. In this article, we summarize current knowledge regarding the biochemical properties and biological functions of EVI‐1 in normal and malignant hematopoiesis, with specific focus on its pathogenetic significance in hematological malignancies. (Cancer Sci 2009; 100: 990–995)
Despite the development of multiple new agents, relapse continues to be the most common cause of death for hematological malignancies. In order to improve the rate of cure, it is necessary to clarify the molecular mechanisms underlying therapy‐resistant leukemia. The ecotropic viral integration site‐1 (EVI‐1) is an oncogene that confers a poor prognosis in human hematological malignancies, including acute myeloid leukemia (AML), chronic myeloid leukemia, and myelodysplastic syndrome (MDS). Evi‐1 was first identified as the integration site of the ecotropic retrovirus leading to myeloid leukemia in murine model systems.( 1 , 2 ) Since its identification, numerous studies have shown that EVI‐1 is associated with particularly aggressive forms of human myeloid malignancies. EVI‐1 is a nuclear transcription factor and contains DNA‐binding zinc finger motifs. In addition to its DNA‐binding activity, Evi‐1 has the potential to recruit diverse proteins, such as Smad( 3 ) and C‐terminal Binding Protein (CtBP), thus generating regulatory complexes for transcriptional regulation. Recently, gene targeting studies in mice showed that Evi‐1 is essential for proliferation and maintenance of hematopoietic stem cells (HSC). Furthermore, the EVI‐1 genomic locus attracts much attention as a ‘hotspot’ of retroviral integration sites after gene therapy. The significant function of Evi‐1 in HSC regulation implies that Evi‐1 might participate in the generation of leukemia stem cells (LSC), which has been receiving particular attention as a cause of therapeutic resistance of leukemia. This review summarizes the biological roles and biochemical properties of EVI‐1 in normal and malignant hematopoiesis, and describes future prospects for molecular therapy targeting EVI‐1 in hematological malignancies.
EVI‐1 genomic locus and its gene products
Human EVI‐1 is localized to chromosome 3 band q26,( 3 ) spans 60 kb, and contains 16 exons, with multiple alternative 5′ mRNA variants and several alternative spliced transcripts( 4 ) (Fig. 1a,b). The major EVI‐1 form is a 1051 amino acid protein with an apparent molecular weight of 145 kDa.( 5 , 6 ) EVI‐1 has multiple zinc finger domains that are organized into two sets of seven and three zinc finger domains, respectively. Between the two sets of zinc finger domains, a repression domain has been identified as well as an acidic region at the C‐terminus (Fig. 1c). The Δ324 transcript is an alternative splice variant of EVI‐1 encoding an 88‐kDa protein lacking zinc fingers 6 and 7, and is found at low levels in human and mouse cells.( 7 ) The ‐Rp9 variant lacks nine amino acids in the repression domain and is quite common in human and mouse cells (Fig. 1b,c). EVI‐1 also exists as a longer form, called MDS1–EVI‐1, generated from the in‐frame splicing of the small gene MDS1 to the second exon of EVI‐1.( 8 ) The human MDS1 gene was first identified because it is rearranged in 3;21 translocation in myeloid leukemias.( 9 ) Human MDS1 spans 500 kb, contains three exons, and is located 3 kb telomeric to the first exon of EVI‐1 (Fig. 1a). The MDS1–EVI‐1 protein contains 188 additional amino acids at its N‐terminus, encoding a so‐called ‘PR’ domain (Positive Regulatory‐Domain 1‐Binding Factor 1 (PRD1‐BF1)/Retinoblastoma‐Interacting Zinc‐finger protein 1 (RIZ1) homology), in addition to the entire EVI‐1 sequence( 8 ) (Fig. 1c). The PR domain has homology with the Su(var)3‐9, Enhancer‐of‐zest, Trithorax (SET) domain, which is associated with histone methyltransferase activity. However, to date there is no indication that this activity is functional in MDS1–EVI‐1. Rather, the PR domain in MDS1–EVI‐1 prevents oligomerization, which affects its biochemical functions.( 10 ) Several experimental and clinical observations indicate that EVI‐1 (the PR‐absent form) promotes tumor growth, whereas MDS1–EVI‐1 (the PR‐containing form) contributes to tumor suppression. However, EVI‐1 and MDS1–EVI‐1 cause similar effects in some biological settings (described in more detail below). Therefore, the precise roles of these proteins remain to be elucidated.
Figure 1.
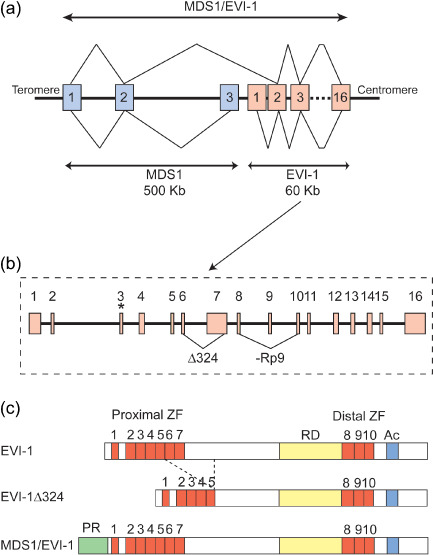
Structure of ecotropic viral integration site‐1 (EVI‐1) and myelodysplastic syndrome (MDS) 1–EVI‐1. (a) Genomic structure of human EVI‐1, MDS1, and MDS1–EVI‐1. Exons are represented by boxes and numbered. Z‐lines represent splicing to produce mRNA. The MDS1 gene has three exons and spans 500 kb. The EVI‐1 gene spans only 60 kb but has 16 exons. MDS1–EVI‐1 is produced by splicing of the second exon of MDS1 and the second exon of EVI‐1. (b) Detailed genomic structure of human EVI‐1. The translation start codon of EVI‐1 in exon 3 is indicated by an asterisk. The alternative splice variants are indicated by triangular lines. (c) Diagrams of EVI‐1, EVI‐1Δ324, and MDS1–EVI‐1 proteins. EVI‐1 has two sets of the zinc finger domains (ZF). Between the two sets of zinc finger domains, a repression domain (RD) has been identified as well as an acidic region (Ac) at the C‐terminus. Structurally in MDS1–EVI‐1, the PR domain is located at the N‐terminus of EVI‐1. Another naturally occurring splice variant, EVI‐1Δ324, has been described that lacks 324 internal amino acids including zinc fingers 6 and 7 of the proximal zinc finger domain.
EVI‐1 and transcriptional regulation
The EVI‐1 protein is located in the nucleus and can bind to specific DNA sequences through both of its zinc finger domains independently.( 11 , 12 , 13 ) The proximal zinc finger domain recognizes a consensus sequence of 15 nucleotides consisting of GA(C/T)AAGA(T/C)AAGATAA, and Evi‐1 was shown to bind directly to the Gata2 promoter through this domain.( 14 , 15 ) In addition, the binding site for this domain has a Gata1 consensus motif, and could potentially compete with Gata1 for DNA binding.( 16 ) Although in vitro studies showed that the distal zinc finger domain recognizes the consensus GAAGATGAG, so far there are no reports of genes that are directly regulated by EVI‐1 through the distal zinc finger domain. EVI‐1 also interacts with several transcription regulators. In particular, interaction with the corepressor CtBP is important for EVI‐1 function.( 17 , 18 ) This interaction relies on amino acids 544–607 on the EVI‐1 protein, a stretch that contains CtBP binding consensus motifs. CtBP increases the transcriptional repression of a reporter gene by EVI‐1, and point mutations in EVI‐1 that abolish the interaction significantly decrease EVI‐1‐mediated transcriptional repression, growth inhibition of Mv1Lu cells in response to transforming growth factor (TGF)‐β, and transformation of Rat‐1 fibroblasts. EVI‐1 also interacts with histone deacetylases directly or through CtBP, and the histone deacetylase inhibitor partially relieves transcriptional repression by EVI‐1.( 17 , 19 ) It was also shown that EVI‐1 binds to the coactivators CREB binding protein (CBP) and P300/CBP‐associated factor (P/CAF), and coexpression of CBP could turn a repressive effect of EVI‐1 on a reporter gene into a moderately activating effect.( 20 ) Furthermore, it was recently shown that EVI‐1 associates with the histone H3 lysine 9‐specific histone methyltransferases SUV39H1 and G9a.( 21 , 22 ) Thus, EVI‐1 forms higher‐order complexes with various transcriptional regulators, and these associations are important for transcriptional regulation by EVI‐1 (Fig. 2).
Figure 2.
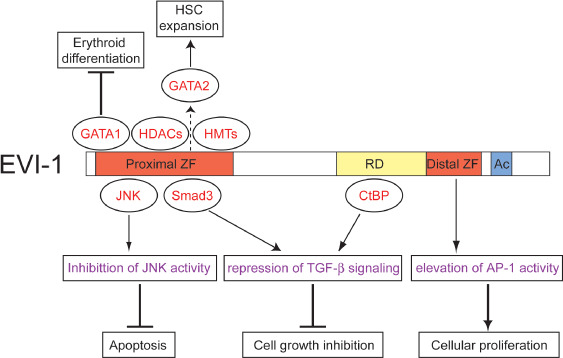
Biochemical properties of ecotropic viral integration site‐1 (EVI‐1). EVI‐1 inhibits c‐Jun N‐terminal kinase (JNK) activity and prevents apoptosis. EVI‐1 interacts with Smad3 and C‐terminal Binding Protein (CtBP), and blocks the growth inhibitory effect of transforming growth factor (TGF)‐β. EVI‐1 raises activator protein (AP)‐1 activity and promotes cell proliferation. EVI‐1 interacts with GATA1 and inhibits erythroid differentiation. EVI‐1 upregulates GATA2 expression and promotes hematopoietic stem cell (HSC) expansion. Interactions with histone deacetylases (HDAC) and histone methyltransferases (HMT) contribute to EVI‐1‐mediated transcriptional regulation. Through these molecular mechanisms, EVI‐1 acts both as a HSC regulator and an oncogenic protein. Proteins that interact with specific regions of EVI‐1 are depicted in corresponding positions. Ac, acidic region; RD, repression domain; ZF, zinc finger domain.
EVI‐1 and signaling pathways
It has been shown that EVI‐1 affects various signaling pathways. Among these, TGF‐β is the best‐characterized pathway that is interfered by EVI‐1. TGF‐β controls proliferation and cellular differentiation of most cell types, and plays an important role in tumor development. EVI‐1 significantly represses TGF‐β‐mediated activation of the p3TP‐Lux reporter plasmid in HepG2 cells, and suppresses TGF‐β‐mediated growth inhibition in Mv1Lu and 32D cells.( 23 , 24 ) Furthermore, EVI‐1 interferes with the induction of endogenous genes by TGF‐β and other TGF‐β family members in Xenopus animal cap explants as well as in C2C12 cells.( 25 ) EVI‐1 inhibits TGF‐β signaling through at least two possible mechanisms: reduction of Smad3 activity by physical interaction and recruitment of the corepressor CtBP.( 17 , 23 ) In contrast to EVI‐1, MDS1–EVI‐1 enhances TGF‐β‐induced growth inhibition in 32D cells( 24 ) and cannot efficiently repress TGF‐β‐mediated activation of p3TP‐Lux in HepG2 cells.( 10 ) The lower repressive activity correlates with a reduced ability of MDS1–EVI‐1, as compared to EVI‐1, to bind to the corepressor CtBP( 10 ) (Fig. 2).
The c‐Jun N‐terminal kinases (JNK) are mitogen‐activated protein kinases that are responsive to various stress stimuli and play an important role in triggering apoptosis. EVI‐1 significantly suppresses the JNK1‐mediated phosphorylation of c‐Jun. Conversely, reduction of EVI‐1 expression using antisense oligonucleotide recovers endogenous JNK1 activity in MOLM‐1 and HEC1B cells. EVI‐1 physically interacts with JNK through the proximal zinc finger domain, and an EVI‐1 mutant lacking this domain fails to suppress JNK1 activity. EVI‐1 also protects cells from stress‐induced cell death with dependence on the ability to inhibit JNK( 26 ) (Fig. 2). In addition to JNK, several mechanisms have been proposed to play a role in the survival function of EVI‐1. EVI‐1 protects murine bone marrow progenitors from apoptosis by activating the Promyelocytic leukemia (Pml) gene.( 27 ) It was also reported that EVI‐1 suppresses TGF‐β or taxol‐mediated apoptosis through a Phosphoinositide 3‐kinase (PI3K)‐Akt dependent mechanism in RIE cells.( 28 )
Activator protein (AP)‐1 is a transcription factor complex consisting of a Fos–Jun heterodimer or Jun–Jun homodimer. It regulates gene expression in response to a variety of stimuli, and controls a number of cellular processes including differentiation, proliferation, and apoptosis. EVI‐1 raises AP‐1 activity and stimulates c‐fos promoter activation with dependence on its distal zinc finger domain in NIH3T3 and P19 cells.( 29 ) Because the distal zinc finger domain is required for EVI‐1‐mediated transformation of Rat‐1 cells, the enhanced AP‐1 activity probably contributes to cell transformation by EVI‐1 (Fig. 2).
Roles of Evi‐1 in HSC regulation
Recently, several studies using gene targeting mice revealed an essential role for EVI‐1 in HSC regulation. First, Yuasa et al. showed that Evi‐1 is preferentially expressed in HSC and is downregulated upon differentiation.( 14 ) Furthermore, development of HSC in the para‐aortic splanchnopleural region, from which definitive hematopoiesis originates, was severely impaired in Evi‐1‐knockout embryos.( 14 ) Interestingly, Evi‐1‐mediated para‐aortic splanchnopleural hematopoiesis seems to depend on Gata2 activation and repression of TGF‐β signaling.( 14 , 30 ) These findings suggest that Evi‐1 has a role in the early expansion of HSC during embryogenesis. However, the Evi‐1‐knockout mice used in the study carry a targeted deletion of exon 7, resulting in the expression of a truncated Evi‐1 transcript (Evi‐1Δ324). In addition, the role for Evi‐1 in fetal liver (FL) and bone marrow hematopoiesis was not clarified because these mice die around embryonic day 10.( 31 ) Our group created new Evi‐1 mutant mice in which exon 4 of the Evi‐1 gene can be deleted by the expression of Cre recombinase (Evi‐1‐flox), as well as mice in which the same region was completely deleted (Evi‐1‐KO).( 32 ) Because exon 4 of Evi‐1 exists in all known Evi‐1 transcripts, this strategy resulted in complete deletion of Evi‐1. Interestingly, the new Evi‐1‐KO mice survived slightly longer (they died between embryonic days 13.5 and 16.5) than the prior Evi‐1 mutant mice, which enabled the analysis of FL hematopoiesis. Although the Evi‐1‐KO FL was morphologically indistinguishable from the wild‐type FL, the population of lineage−, c‐Kit+, Sca‐1+ cells (LSK cells), which enriches hematopoietic stem and progenitor cells, was severely reduced in Evi‐1‐KO mice. The total number of colony‐forming cells, especially the number of mixed colonies, in Evi‐1‐KO FL cells was also severely decreased. Furthermore, Evi‐1‐KO FL cells did not reconstitute hematopoiesis of the irradiated recipient mice. Thus, Evi‐1 deletion causes severe reduction in HSC in FL hematopoiesis.( 32 ) The roles of Evi‐1 in adult hematopoiesis were assessed by crossing Evi‐1‐flox mice with Mx‐Cre transgenic mice, in which a high level of Cre recombinase is produced by treatment with the interferon inducer pI‐pC, leading to recombination in hematopoietic cells of in all lineages. By 4 weeks after pI‐pC injection, Evi‐1‐excised mice exhibited a significant decrease in the frequency of HSC compared with control mice. Moreover, by 12 weeks after pI‐pC injection, Evi‐1‐deleted hematopoietic cells were outcompeted by the cells that escaped Cre‐mediated Evi‐1 excision. Thus, Evi‐1 is also required for proliferation and maintenance of adult HSC.( 32 ) In addition, Evi‐1+/– mice exhibited the intermediate phenotype as for the number of HSC, as well as hematopoietic reconstitution activity of the bone marrow cells, suggesting a gene dosage requirement for Evi‐1 in the regulation of HSC( 32 ) (Fig. 3).
Figure 3.
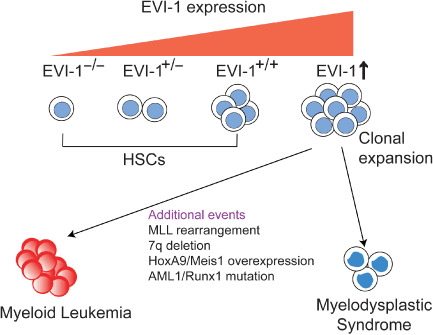
Gene dosage effects of ecotropic viral integration site‐1 (EVI‐1) in hematopoietic stem cells (HSC) and hematological malignancies. EVI‐1 regulates proliferation and maintenance of HSC in a dose‐dependent manner. Activation of EVI‐1 in hematopoietic cells leads to clonal expansion or dysplastic hematopoiesis, whereas onset of full‐blown leukemia requires cooperative genetic events. Hox, Homeobox; Meis, Myeloid ecotropic viral integration site; MLL, Mixed‐Lineage leukemia; Runx, Runt‐related transcription factor.
Effects of EVI‐1 overexpression on hematopoietic differentiation
In Evi‐1‐excised mice, the frequencies of mature myeloid cells, lymphoid cells, and erythroid cells were not affected in spite of the efficient excision of Evi‐1 alleles.( 32 ) Therefore, it is likely that Evi‐1 is physiologically dispensable for lineage commitment in normal hematopoiesis. However, a large body of evidence suggests that aberrant expression of EVI‐1 affects hematopoietic differentiation in various lineages. Ectopic expression of EVI‐1 blocks granulocytic differentiation of interleukin‐3‐dependent 32D cells when stimulated with granulocyte‐colony stimulating factor (G‐CSF)( 33 ) whereas MDS1–EVI‐1 had no such effect.( 24 ) In addition, retrovirally expressed EVI‐1 delayed myeloid differentiation of murine bone marrow cells and induce the enhanced replating of myeloid progenitors in methylcellulose medium.( 34 ) These results indicate that EVI‐1 overexpression interferes with myeloid differentiation. EVI‐1 might also block erythroid differentiation. Ectopic expression of EVI‐1 into murine bone marrow cells drastically reduced the capacity to produce erythroid colonies or Ter119+ immature erythroid cells both in vitro and in vivo.( 16 , 35 , 36 ) The EVI‐1‐mediated block of erythroid differentiation may be due to the physical interaction and competition for DNA binding with GATA1, a crucial transcription factor required for erythropoiesis( 16 , 36 ) (Fig. 2). Finally, several experiments suggest that EVI‐1 promotes megakaryopoiesis. Using an experimental protocol to differentiate embryonic stem (ES) cells to hematopoietic cells, Sitailo et al. showed that Evi‐1, but not Mds1–Evi‐1, promotes megakaryocytic differentiation.( 37 ) Similarly, EVI‐1 induced megakaryocytic differentiation in UT7‐GM cells.( 38 ) These findings are interesting because leukemias with EVI‐1 rearrangements typically show elevated platelet counts.
Effects of retroviral insertions into the EVI‐1 and MDS1–EVI‐1 loci
The EVI‐1 and MDS1–EVI‐1 genomic loci have been defined as ‘hotspots’ for retroviral integration in both clinical and experimental models. The vast majority of the insertions are located in the introns of MDS1, between the last MDS1 and the first EVI‐1 exons, or in the first two introns of EVI‐1.( 39 ) In a gene therapy trial for chronic granulomatous disease (CGD), activating insertions of the therapeutic vector into the EVI‐1 locus lead to preferential expansion of the affected cells.( 40 ) Two young adult CGD patients were reinfused with autologous CD34+ cells transduced with a retroviral vector containing an intact copy of the gp91phox gene, whose mutation causes CGD. Both patients showed significant clinical improvement, and analysis of vector insertion sites revealed that cell clones containing insertions in the MDS1–EVI‐1 locus increasingly dominated hematopoiesis after transplantation. Despite their marked clonal dominance, no evidence for the development of leukemia was found in either patient at up to 2 years. Similar to the findings in the human CGD trial, a study using rhesus macaques showed significant overrepresentation of vector insertions in the MDS1–EVI‐1 locus in granulocytes long‐term after transplantation.( 41 ) These MDS1–EVI‐1 clones became apparent 3–6 months after transplantation and then contributed continuously with no signs of leukemia. Furthermore, the in vivo analysis of mice using serial transplantation showed that retroviral integrations at the Evi‐1 or Mds1–Evi‐1 locus can be related to long‐term non‐malignant clonal expansion in healthy C57Bl/6 recipient mice.( 42 ) In this experiment, a relatively small number of clones always became dominant in recipient mice, and the dominant clones always had insertions near genes with a role in self‐renewal or survival of HSC. Thus, transcription‐activating retroviral integrations into the EVI‐1 and MDS1–EVI‐1 loci confer survival and self‐renewing ability to hematopoietic cells.
Experimental models of EVI‐1‐related hematological malignancies
Several mouse models of EVI‐1‐related hematological malignancies have been generated. Transgenic mice expressing Evi‐1 under the Sca‐1 promoter, which is active in HSC and progenitor cells, developed normally and did not show clear hematological abnormalities. However, these mice were more susceptible to retrovirally induced leukemia than controls, supporting an oncogenic role for Evi‐1.( 43 ) Other groups used bone marrow infection and transplantation to establish the mouse model for Evi‐1 overexpression. Cuenco and Ren used Balb/c mice as bone marrow donors and recipients, and found that some Evi‐1‐positive mice developed lymphoma with a block in early B‐cell development. In contrast, Mds1–Evi‐1‐positive cells did not develop any disease.( 44 ) Buonamici et al. used C57BL/6 mice and found that all Evi‐1‐positive mice displayed a lethal condition resembling human MDS, including hyperproliferation of bone marrow and progressive pancytopenia that resulted in their death.( 35 ) Jin et al. also reported the emergence of a MDS‐like disease in C57BL/6 mice transplanted with Evi‐1‐transduced bone marrow cells. In addition, they showed that coexpression of Evi‐1 with Homeobox A9 (HoxA9) and Myeloid ecotropic viral integration site 1 (Meis1) significantly accelerated the onset of HoxA9/Meis1‐induced AML.( 45 ) It was also shown that Evi‐1 collaborated with AML1/RUNX1 mutants to induce AML in recipient mice.( 46 ) These results suggest that activation of Evi‐1 in hematopoietic cells primarily promotes clonal expansion or myeloid dysplasia, whereas leukemogenesis requires additional genetic events. The extent of Evi‐1 expression, genetic background of the host, and the cooperative genetic events will determine the fate of Evi‐1‐positive hematopoietic cells (Fig. 3).
Recently, our group found that complete loss of Evi‐1 attenuates proliferative activity in a wide variety of leukemic cells.( 32 ) Disruption of Evi‐1 in MLL/ENL‐transformed or E2A/HLF‐transformed progenitors in methylcellulose medium caused a significant reduction in colony numbers to approximately 30 or 60% of the control, respectively. Furthermore, Cre‐mediated Evi‐1 deletion in cMyc‐bcl2‐transduced bone marrow cells delayed the onset of leukemia in vivo. In particular, Evi‐1 deletion led to a large decrease in colony numbers in MLL/ENL‐transformed cells, indicating a crucial role for Evi‐1 in MLL leukemias.
Clinical aspects of EVI‐1‐related hematological malignancies
The EVI‐1 overexpression often occurs as a consequence of chromosomal rearrangements involving 3q26, where EVI‐1 is mapped. Of these, the translocations t(3;21)(q26;q22) and t(3;12)(q26;p13) lead to the formation of the AML1–MDS1–EVI‐1 and Translocation Ets Leukemia (TEL)–(MDS1)–EVI‐1 fusion transcripts, respectively.( 9 , 47 , 48 ) Meanwhile, inv(3)(q21q26), t(3;3)(q21;q26), and other 3q26 rearrangements lead to overexpression of intact EVI‐1 mRNA.( 49 , 50 ) These cases have elevated platelet counts, marked hyperplasia with dysplastic megakaryocytes, and poor prognosis, which are characterized as 3q21q26 syndrome.( 51 ) EVI‐1 is also highly expressed in a subgroup of AML without 3q26 rearrangements, indicating the presence of other mechanisms for EVI‐1 activation. A study using gene expression profiles of 285 patients with AML revealed that EVI‐1 expression could define a new subtype of AML. These samples have overall expression patterns closest to normal CD34+ cells, suggesting a stem cell phenotype for EVI‐1‐positive leukemias, and these patients have a very poor prognosis.( 52 ) Although previous studies could not distinguish between EVI‐1 and MDS1–EVI‐1 expression, recently, several groups developed real‐time quantitative PCR assays to measure specific expression of both EVI‐1 and MDS1–EVI‐1 transcripts. Interestingly, studies involving a large number of de novo AML patients without 3q26 rearrangements found a prognostic significance of EVI‐1, but not of MDS1–EVI‐1, overexpression in this patient group.( 53 , 54 ) However, another group reported that MDS1–EVI‐1 overexpression also predicts short remission duration in AML patients.( 55 ) Therefore, further study will be necessary to clarify the significance of EVI‐1 versus MDS1–EVI‐1 expression in AML. The clinical studies also revealed that high EVI‐1 expression correlates with the presence of 7q‐deletions and translocations involving 11q23 (MLL‐rearrangements).( 54 ) The relationship between EVI‐1 and these genetic abnormalities should be investigated in the future.
Conclusions and future directions
Recently, EVI‐1 has received much attention as a crucial gene for HSC regulation as well as an oncogene whose expression predicts poor patient survival in hematological malignancies. Current evidence suggests that leukemias are maintained by rare stem cells (LSC), and elimination of LSC is necessary and potentially sufficient for cure of the leukemia. It is tempting to speculate that activation of EVI‐1 enhances proliferation and/or survival of LSC and, thus, confers drug resistance on various types of leukemia. Therefore, therapies designed to target EVI‐1 will be an attractive option in treatment for hematopoietic malignancies. Significant in this regard is the report that arsenic trioxide (ATO) selectively degrades EVI‐1 protein.( 56 ) Furthermore, ATO and thalidomide combination therapy produces multilineage hematological responses in MDS patients, particularly in those with high EVI‐1 expression.( 57 ) Thus, ATO may be used as part of a target therapy for patients with EVI‐1‐positive MDS. Further elucidation of the mechanisms regulating EVI‐1 expression (i.e. upstream signaling pathways, regulation of mRNA translation, and regulation of protein stability of EVI‐1) will be helpful to develop novel therapies targeting EVI‐1. In order to determine the ‘true’ therapeutic target, the precise roles of EVI‐1 variants should be clarified. In particular, the relationship between EVI‐1 and MDS1–EVI‐1 in normal and malignant hematopoiesis is important because they act either in a similar or opposite manner depending on the context. Furthermore, identification of downstream target genes as well as interacting proteins of EVI‐1 will provide essential information on EVI‐1‐mediated hematopoiesis and leukemogenesis. Because EVI‐1 is preferentially expressed in the stem cell compartment, marking of EVI‐1 expression might enable the in vivo visualization of HSC and LSC. Thus, future investigations regarding EVI‐1 will shed light on the basic mechanisms underlying stem cell behavior and leukemia development, and then improve the current treatment strategies for hematological malignancies.
Acknowledgments
This work was supported in part by a Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science, and by Health and Labour Sciences Research grants from the Ministry of Health, Labour, and Welfare.
References
- 1. Morishita K, Parker DS, Mucenski ML, Jenkins NA, Copeland NG, Ihle JN. Retroviral activation of a novel gene encoding a zinc finger protein in IL‐3‐dependent myeloid leukemia cell lines. Cell 1988; 54: 831–40. [DOI] [PubMed] [Google Scholar]
- 2. Mucenski ML, Taylor BA, Ihle JN et al . Identification of a common ecotropic viral integration site, Evi‐1, in the DNA of AKXD murine myeloid tumors. Mol Cell Biol 1988; 8: 301–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morishita K, Parganas E, Bartholomew C et al . The human Evi‐1 gene is located on chromosome 3q24‐q28 but is not rearranged in three cases of acute nonlymphocytic leukemias containing t(3;5)(q25;q34) translocations. Oncogene Res 1990; 5: 221–31. [PubMed] [Google Scholar]
- 4. Wieser R. The oncogene and developmental regulator EVI1: expression, biochemical properties, and biological functions. Gene 2007; 396: 346–57. [DOI] [PubMed] [Google Scholar]
- 5. Morishita K, Parganas E, Douglass EC, Ihle JN. Unique expression of the human Evi‐1 gene in an endometrial carcinoma cell line: sequence of cDNAs and structure of alternatively spliced transcripts. Oncogene 1990; 5: 963–71. [PubMed] [Google Scholar]
- 6. Matsugi T, Morishita K, Ihle JN. Identification, nuclear localization, and DNA‐binding activity of the zinc finger protein encoded by the Evi‐1 myeloid transforming gene. Mol Cell Biol 1990; 10: 1259–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bordereaux D, Fichelson S, Tambourin P, Gisselbrecht S. Alternative splicing of the Evi‐1 zinc finger gene generates mRNAs which differ by the number of zinc finger motifs. Oncogene 1990; 5: 925–7. [PubMed] [Google Scholar]
- 8. Fears S, Mathieu C, Zeleznik‐Le N, Huang S, Rowley JD, Nucifora G. Intergenic splicing of MDS1 and EVI1 occurs in normal tissues as well as in myeloid leukemia and produces a new member of the PR domain family. Proc Natl Acad Sci USA 1996; 93: 1642–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mitani K, Ogawa S, Tanaka T et al . Generation of the AML1‐EVI‐1 fusion gene in the t(3;21)(q26;q22) causes blastic crisis in chronic myelocytic leukemia. EMBO J 1994; 13: 504–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nitta E, Izutsu K, Yamaguchi Y et al . Oligomerization of Evi‐1 regulated by the PR domain contributes to recruitment of corepressor CtBP 2005.. Oncogene 2005; 24: 6165–73. [DOI] [PubMed] [Google Scholar]
- 11. Perkins AS, Fishel R, Jenkins NA, Copeland NG. Evi‐1, a murine zinc finger proto‐oncogene, encodes a sequence‐specific DNA‐binding protein. Mol Cell Biol 1991; 11: 2665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Delwel R, Funabiki T, Kreider BL, Morishita K, Ihle JN. Four of the seven zinc fingers of the Evi‐1 myeloid‐transforming gene are required for sequence‐specific binding to GA(C/T)AAGA(T/C)AAGATAA. Mol Cell Biol 1993; 13: 4291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morishita K, Suzukawa K, Taki T, Ihle JN, Yokota J. EVI‐1 zinc finger protein works as a transcriptional activator via binding to a consensus sequence of GACAAGATAAGATAAN1‐28 CTCATCTTC. Oncogene 1995; 10: 1961–7. [PubMed] [Google Scholar]
- 14. Yuasa H, Oike Y, Iwama A et al . Oncogenic transcription factor Evi1 regulates hematopoietic stem cell proliferation through GATA‐2 expression. EMBO J 2005; 24: 1976–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yatsula B, Lin S, Read AJ et al . Identification of binding sites of EVI1 in mammalian cells. J Biol Chem 2005; 280: 30 712–22. [DOI] [PubMed] [Google Scholar]
- 16. Kreider BL, Orkin SH, Ihle JN. Loss of erythropoietin responsiveness in erythroid progenitors due to expression of the Evi‐1 myeloid‐transforming gene. Proc Natl Acad Sci USA 1993; 90: 6454–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Izutsu K, Kurokawa M, Imai Y, Maki K, Mitani K, Hirai H. The corepressor CtBP interacts with Evi‐1 to repress transforming growth factor beta signaling. Blood 2001; 97: 2815–22. [DOI] [PubMed] [Google Scholar]
- 18. Palmer S, Brouillet JP, Kilbey A et al . Evi‐1 transforming and repressor activities are mediated by CtBP co‐repressor proteins. J Biol Chem 2001; 276: 25 834–40. [DOI] [PubMed] [Google Scholar]
- 19. Vinatzer U, Taplick J, Seiser C, Fonatsch C, Wieser R. The leukaemia‐associated transcription factors EVI‐1 and MDS1/EVI1 repress transcription and interact with histone deacetylase. Br J Haematol 2001; 114: 566–73. [DOI] [PubMed] [Google Scholar]
- 20. Chakraborty S, Senyuk V, Sitailo S, Chi Y, Nucifora G. Interaction of EVI1 with cAMP‐responsive element‐binding protein‐binding protein (CBP) and p300/CBP‐associated factor (P/CAF) results in reversible acetylation of EVI1 and in co‐localization in nuclear speckles. J Biol Chem 2001; 276: 44 936–43. [DOI] [PubMed] [Google Scholar]
- 21. Spensberger D, Delwel R. A novel interaction between the proto‐oncogene Evi1 and histone methyltransferases, SUV39H1 and G9a. FEBS Lett 2008; 582: 2761–7. [DOI] [PubMed] [Google Scholar]
- 22. Cattaneo F, Nucifora G. EVI1 recruits the histone methyltransferase SUV39H1 for transcription repression. J Cell Biochem 2008; 105: 344–52. [DOI] [PubMed] [Google Scholar]
- 23. Kurokawa M, Mitani K, Irie K et al . The oncoprotein Evi‐1 represses TGF‐β signalling by inhibiting Smad3. Nature 1998; 394: 92–6. [DOI] [PubMed] [Google Scholar]
- 24. Sood R, Talwar‐Trikha A, Chakrabarti SR, Nucifora G. MDS1/EVI1 enhances TGF‐β1 signaling and strengthens its growth‐inhibitory effect but the leukemia‐associated fusion protein AML1/MDS1/EVI1, product of the t(3;21), abrogates growth‐inhibition in response to TGF‐β1. Leukemia 1999; 13: 348–57. [DOI] [PubMed] [Google Scholar]
- 25. Alliston T, Ko TC, Cao Y et al . Repression of BMP and activin‐inducible transcription by Evi‐1. J Biol Chem 2005; 280: 24227–37. [DOI] [PubMed] [Google Scholar]
- 26. Kurokawa M, Mitani K, Yamagata T et al . The evi‐1 oncoprotein inhibits c‐Jun N‐terminal kinase and prevents stress‐induced cell death. EMBO J 2000; 19: 2958–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Buonamici S, Li D, Mikhail FM et al . EVI1 abrogates interferon‐α response by selectively blocking PML induction. J Biol Chem 2005; 280: 428–36. [DOI] [PubMed] [Google Scholar]
- 28. Liu Y, Chen L, Ko TC, Fields AP, Thompson EA. Evi1 is a survival factor which conveys resistance to both TGFβ‐ and taxol‐mediated cell death via PI3K/AKT. Oncogene 2006; 25: 3565–75. [DOI] [PubMed] [Google Scholar]
- 29. Tanaka T, Nishida J, Mitani K, Ogawa S, Yazaki Y, Hirai H. Evi‐1 raises AP‐1 activity and stimulates c‐fos promoter transactivation with dependence on the second zinc finger domain. J Biol Chem 1994; 269: 24 020–6. [PubMed] [Google Scholar]
- 30. Sato T, Goyama S, Nitta E et al . Evi‐1 promotes para‐aortic splanchnopleural hematopoiesis through upregulation of GATA‐2 and repression of TGF‐β signaling. Cancer Sci 2008; 99: 1407–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hoyt PR, Bartholomew C, Davis AJ et al . The Evi1 proto‐oncogene is required at midgestation for neural, heart, and paraxial mesenchyme development. Mech Dev 1997; 65: 55–70. [DOI] [PubMed] [Google Scholar]
- 32. Goyama S, Yamamoto G, Shimabe M et al . Evi‐1 is a critical regulator for hematopoietic stem cells and transformed leukemic cells. Cell Stem Cell 2008; 3: 207–20. [DOI] [PubMed] [Google Scholar]
- 33. Morishita K, Parganas E, Matsugi T, Ihle JN. Expression of the Evi‐1 zinc finger gene in 32Dc13 myeloid cells blocks granulocytic differentiation in response to granulocyte colony‐stimulating factor. Mol Cell Biol 1992; 12: 183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Laricchia‐Robbio L, Nucifora G. Significant increase of self‐renewal in hematopoietic cells after forced expression of EVI1. Blood Cells Mol Dis 2008; 40: 141–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Buonamici S, Li D, Chi Y et al . EVI1 induces myelodysplastic syndrome in mice. J Clin Invest 2004; 114: 713–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Laricchia‐Robbio L, Fazzina R, Li D et al . Point mutations in two EVI1 Zn fingers abolish EVI1–GATA1 interaction and allow erythroid differentiation of murine bone marrow cells. Mol Cell Biol 2006; 26: 7658–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sitailo S, Sood R, Barton K, Nucifora G. Forced expression of the leukemia‐associated gene EVI1 in ES cells: a model for myeloid leukemia with 3q26 rearrangements. Leukemia 1999; 13: 1639–45. [DOI] [PubMed] [Google Scholar]
- 38. Shimizu S, Nagasawa T, Katoh O, Komatsu N, Yokota J, Morishita K. EVI1 is expressed in megakaryocyte cell lineage and enforced expression of EVI1 in UT‐7/GM cells induces megakaryocyte differentiation. Biochem Biophys Res Commun 2002; 292: 609–16. [DOI] [PubMed] [Google Scholar]
- 39. Metais JY, Dunbar CE. The MDS1–EVI1 gene complex as a retrovirus integration site: impact on behavior of hematopoietic cells and implications for gene therapy. Mol Ther 2008; 16: 439–49. [DOI] [PubMed] [Google Scholar]
- 40. Ott MG, Schmidt M, Schwarzwaelder K et al . Correction of X‐linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1–EVI1, PRDM16 or SETBP1. Nat Med 2006; 12: 401–9. [DOI] [PubMed] [Google Scholar]
- 41. Calmels B, Ferguson C, Laukkanen MO et al . Recurrent retroviral vector integration at the Mds1/Evi1 locus in nonhuman primate hematopoietic cells. Blood 2005; 106: 2530–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kustikova O, Fehse B, Modlich U et al . Clonal dominance of hematopoietic stem cells triggered by retroviral gene marking. Science 2005; 308: 1171–4. [DOI] [PubMed] [Google Scholar]
- 43. Louz D, Van Den Broek M, Verbakel S et al . Erythroid defects and increased retrovirally‐induced tumor formation in Evi1 transgenic mice. Leukemia 2000; 14: 1876–84. [DOI] [PubMed] [Google Scholar]
- 44. Cuenco GM, Ren R. Both AML1 and EVI1 oncogenic components are required for the cooperation of AML1/MDS1/EVI1 with BCR/ABL in the induction of acute myelogenous leukemia in mice. Oncogene 2004; 23: 569–79. [DOI] [PubMed] [Google Scholar]
- 45. Jin G, Yamazaki Y, Takuwa M et al . Trib1 and Evi1 cooperate with Hoxa and Meis1 in myeloid leukemogenesis. Blood 2007; 109: 3998–4005. [DOI] [PubMed] [Google Scholar]
- 46. Watanabe‐Okochi N, Kitaura J, Ono R et al . AML1 mutations induced MDS and MDS/AML in a mouse BMT model. Blood 2008; 111: 4297–308. [DOI] [PubMed] [Google Scholar]
- 47. Peeters P, Wlodarska I, Baens M et al . Fusion of ETV6 to MDS1/EVI1 as a result of t(3;12)(q26;p13) in myeloproliferative disorders. Cancer Res 1997; 57: 564–9. [PubMed] [Google Scholar]
- 48. Nucifora G, Begy CR, Kobayashi H et al . Consistent intergenic splicing and production of multiple transcripts between AML1 at 21q22 and unrelated genes at 3q26 in (3;21)(q26;q22) translocations. Proc Natl Acad Sci USA 1994; 91: 4004–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Morishita K, Parganas E, William CL et al . Activation of EVI1 gene expression in human acute myelogenous leukemias by translocations spanning 300–400 kilobases on chromosome band 3q26. Proc Natl Acad Sci USA 1992; 89: 3937–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Suzukawa K, Parganas E, Gajjar A et al . Identification of a breakpoint cluster region 3′ of the ribophorin I gene at 3q21 associated with the transcriptional activation of the EVI1 gene in acute myelogenous leukemias with inv(3)(q21q26). Blood 1994; 84: 2681–8. [PubMed] [Google Scholar]
- 51. Pintado T, Ferro MT, San Roman C, Mayayo M, Larana JG. Clinical correlations of the 3q21;q26 cytogenetic anomaly. A leukemic or myelodysplastic syndrome with preserved or increased platelet production and lack of response to cytotoxic drug therapy. Cancer 1985; 55: 535–41. [DOI] [PubMed] [Google Scholar]
- 52. Valk PJ, Verhaak RG, Beijen MA et al . Prognostically useful gene‐expression profiles in acute myeloid leukemia. N Engl J Med 2004; 350: 1617–28. [DOI] [PubMed] [Google Scholar]
- 53. Barjesteh van Waalwijk van Doorn‐Khosrovani S, Erpelinck C, Van Putten WL et al . High EVI1 expression predicts poor survival in acute myeloid leukemia: a study of 319 de novo AML patients. Blood 2003; 101: 837–45. [DOI] [PubMed] [Google Scholar]
- 54. Lugthart S, Van Drunen E, Van Norden Y et al . High EVI1 levels predict adverse outcome in acute myeloid leukemia: prevalence of EVI1 overexpression and chromosome 3q26 abnormalities underestimated. Blood 2008; 111: 4329–37. [DOI] [PubMed] [Google Scholar]
- 55. Haas K, Kundi M, Sperr WR et al . Expression and prognostic significance of different mRNA 5′‐end variants of the oncogene EVI1 in 266 patients with de novo AML. EVI1 and MDS1/EVI1 overexpression both predict short remission duration. Genes Chromosomes Cancer 2008; 47: 288–98. [DOI] [PubMed] [Google Scholar]
- 56. Shackelford D, Kenific C, Blusztajn A, Waxman S, Ren R. Targeted degradation of the AML1/MDS1/EVI1 oncoprotein by arsenic trioxide. Cancer Res 2006; 66: 11 360–9. [DOI] [PubMed] [Google Scholar]
- 57. Raza A, Buonamici S, Lisak L et al . Arsenic trioxide and thalidomide combination produces multi‐lineage hematological responses in myelodysplastic syndromes patients, particularly in those with high pre‐therapy EVI1 expression. Leuk Res 2004; 28: 791–803. [DOI] [PubMed] [Google Scholar]