

Abstract
In the present study, we addressed the molecular mechanism of the downregulation of reversion‐inducing‐cysteine‐rich protein with Kazal motifs (RECK), a critical tumor suppressor that can potently inhibit angiogenesis and metastasis, in non‐small cell lung cancer and its clinical significance. The methylation status of the RECK gene promoter was studied by methylation‐specific polymerase chain reaction. RECK mRNA and protein levels were investigated by reverse transcription–polymerase chain reaction and western blot analysis. Downregulation of RECK was observed in 60% of the 55 tumors analyzed. Using methylation‐specific polymerase chain reaction analysis methylation of the RECK promoter was detected in 63.6% (35/55) of the tumor tissues. A strong correlation between downregulation and promoter methylation was found in these tumors (P = 0.000005). More importantly, downregulation of RECK significantly correlated with lymph node metastasis (P = 0.038). Mutation of codon 12 of the K‐ras gene was detected in 25.5% (14/55) of lung tumor tissues. Statistical analysis indicated that K‐ras mutation was linked with RECK promoter methylation (P = 0.047) and downregulation (P = 0.023). Promoter methylation was also detected in human lung cancer cell lines, and the DNA methyltransferase inhibitor 5′‐azacytidine reversed the expression of RECK and reduced the invasive ability of these cell lines. Collectively, our results suggest that downregulation of the metastasis suppressor RECK is caused by promoter methylation in non‐small cell lung cancer and is associated with K‐ras mutation and lymph node metastasis. (Cancer Sci 2007; 98: 169–173)
The reversion‐inducing‐cysteine‐rich protein with Kazal motifs (RECK) gene was isolated as a transformation suppressor gene that induced flat reversion in v‐K‐ras‐transformed NIH/3T3 cells.( 1 ) This gene encodes a membrane‐anchored glycoprotein that can negatively regulate matrix metalloproteinase‐2 (MMP‐2) and MMP‐9 activities and inhibit tumor angiogenesis and metastasis.( 2 , 3 ) Whereas RECK mRNA is highly expressed in most normal human tissues and untransformed cells, it is downregulated or undetectable in many tumor cell lines or in cells ectopically expressing active oncogenes.( 2 ) Pathological studies have demonstrated that RECK downregulation is found in human cancers including pancreatic cancer, breast cancer, non‐small cell lung cancer and colon cancer.( 4 , 5 , 6 , 7 ) In addition, reduced RECK expression correlates with poor prognosis in these cancers. However, the molecular mechanism that causes gene silencing of RECK in cancer cells is still unknown.
Mutations in the K‐ras proto‐oncogene are found frequently in non‐small cell lung cancer.( 8 ) Aberrant activation of this oncogene has been implicated in many aspects of malignant phenotypes including proliferation, transformation, invasion and metastasis. Numerous studies have shown that oncogenic ras increases the metastatic ability of transformed cells.( 9 , 10 , 11 ) However, the underlying mechanism is poorly characterized. Recently, we addressed the effect of oncogenic ras on RECK expression using an in vitro inducible system.( 12 ) Our results showed that ras activation stimulates the expression of DNA methyltransferase 3b (DNMT3b), promotes DNMT3b binding to the RECK promoter and represses RECK expression via methylation of the promoter. We also found that human lung cancer cells harboring K‐ras mutations exhibit increased promoter methylation and reduced expression of the RECK gene.( 12 ) To verify the results observed in our previous study, we examined the methylation status of the RECK promoter in 55 lung tumor tissues. Moreover, we studied whether RECK promoter methylation correlated with K‐ras mutation and other clinicopathological parameters.
Materials and Methods
Tissues. Fifty‐five paired normal and lung tumor tissues were obtained from patients who underwent resection of tumors at the Kaohsiung Veterans General Hospital. Detailed data about patient‐ and tumor‐related variables were collected by reviewing the patients’ medical charts. Before acquisition of these tissues, the investigational nature of this study was explained to the patients, and informed consent was obtained. Portions of resected tissues were quickly placed into RNAlater solution (Ambion, Austin, TX, USA) and stored at −20°C until use. Tissues were subjected to isolation of genomic DNA, total RNA and proteins using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions.
Cell lines and 5′‐azacytidine treatment. Five cell lines were used in the present study. MRC‐5 cells are human embryonic lung fibroblasts and were used as a normal control. H226 and H520 are squamous cell carcinoma cell lines and H358 and A549 are adenocarcinoma cell lines. Cells were maintained routinely in Dulbecco's modified Eagle's medium/F12 medium supplemented with 10% fetal calf serum (FCS) and antibiotics. 5′‐Azacytidine (5 µM) was added into the culture medium and incubated for 48 h. Genomic DNA, total RNA and cellular proteins were extracted using TRIzol reagent and subjected to different analyses.
RNA extraction and reverse transcription–polymerase chain reaction. Expression of RECK mRNA was investigated using the OneStep reverse transcription–polymerase chain reaction (RT‐PCR) kit according to the manufacturer's protocol (Qiagen, Valencia, CA, USA). Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as an internal control to check the efficiency of cDNA synthesis and PCR amplification. cDNA synthesis was carried out at 50°C for 30 min with the following PCR conditions: 30 cycles of denaturation (94°C/1 min), annealing (60°C/1 min) and extension (72°C/1 min), and one final cycle of extension (72°C/10 min). The predicted sizes of the RECK and GAPDH PCR products were 477 and 512 bp, respectively. The sequences of the primers were: RECK‐forward, 5′‐CCTCAGTGAGCACAG TTCAGA‐3′ RECK‐reverse, 5′‐GCAGCACACACACTGCTGTA‐3′ GAPDH‐forward, 5′‐GAGTCAACGGATTTGGTCGT‐3′ and GAPDH‐reverse, 5′‐TGTGGTCATGAGTCCTTCCA‐3′. After reaction, PCR products were separated on a 2% agarose gel, stained with ethidium bromide and visualized under ultraviolet light.
Western blot analysis. Equal amounts of cellular proteins were subjected to sodium dodecylsulfate–polyacrylamide gel electrophoresis as described previously.( 13 ) Proteins were transferred to nitrocellulose membranes and the blots were probed with various primary antibodies. Enhanced chemiluminescence reagents were used to detect the proteins on the membranes. Anti‐RECK antibody (Clone 32C10A) was purchased from MBL (Nagoya, Japan) and anti‐actin antibody (Clone C4) was obtained from Chemicon (Temecula, CA, USA).
Methylation‐specific PCR analysis. Genomic DNA was modified with sodium bisulfite and analyzed according to the procedures of the CpGenome DNA Modification Kit (Chemicon). We used the following primers for the detection of human RECK promoter methylation: M‐sense, 5′‐AATAAAGAGTTTTGGTACGGGGTAC‐3′ and M‐antisense, 5′‐AAAACCGCGAAATACTCGAA‐3′ for the methylated sequence of the human RECK promoter; U‐sense, 5′‐TAAAGAGTTTTGGTATGGGGTATGT‐3′ and U‐antisense, 5′‐CTCCAAACCACAAAATACTCAAA‐3′ for the unmethylated sequence of the human RECK promoter. The predicted products for methylated and unmethylated DNA were 195 and 199 bp, respectively. Modified DNA was amplified in 50‐µL reaction mixtures containing 5 µL of 10× PCR buffer, 14 µL of 25 mM MgCl2, 2.5 µL of 25 mM dNTP, 1 µL of each primer (300 ng/µL), and 0.5 units of AmpliTaq Gold DNA polymerase (Roche, Basel, Switzerland). PCR was carried out in a thermal cycler for 35 cycles (denaturation at 95°C for 1 min, annealing at 56°C for 2 min and extension at 72°C for 1 min), followed by a final 5‐min extension at 72°C. PCR products were separated on 2% agarose gels, stained with ethidium bromide and visualized under ultraviolet illumination.
In vitro invasion assay. The in vitro invasion assay was carried out using 24‐well transwell units with polycarbonate filters (pore size 8 µM) coated on the upper side with Matrigel (Becton Dickinson Labware, Bedford, MA, USA). Cells were collected, and 5 × 103 cells in 100 µL of medium containing 5′‐azacytidine (5 µM) or antibodies was placed in the upper part of the transwell unit and allowed to invade for 24 h. The lower part of the transwell unit was filled with medium containing 10% FCS. After incubation, non‐invaded cells on the upper part of the membrane were removed with a cotton swab. Invaded cells on the bottom surface of the membrane were fixed in formaldehyde, stained with Giemsa solution and counted under a microscope.
Statistical analysis. The associations between RECK and clinicopathological parameters were assessed using the χ2‐test and Fisher's exact test. Statistical significance was defined as P < 0.05.
Results
RECK expression is downregulated in lung cancers. Tissues of 55 patients were included in the present study and the patients’ characteristics are described in Table 1. We first examined the expression of RECK in paired normal and lung tumor tissues by RT‐PCR and western blot analysis. The signal intensities on the blots were calculated and compared. The RECK protein level of most of the tumor tissues investigated in the present study was very low. However, three cases showed a marginal reduction (7–12%) of RECK protein in tumor tissues. Therefore, we decided to use 20% reduction as a cut‐off value in our study. More than 20% reduction of RECK protein levels in tumor tissues was defined as downregulation. Our results indicated that RECK was downregulated in 60% (33/55) of tumor tissues. Representative data are shown in Fig. 1 and the distributions of the RECK/actin ratio of normal and tumor groups is demonstrated in Fig. 1C. We next tested the correlation between RECK expression and clinicopathological parameters. Our data indicated that RECK downregulation was not associated with sex, age, T‐stage, stage or histology (Table 1). On the contrary, RECK downregulation was significantly associated with increased lymph node metastasis (Table 1).
Table 1.
Correlation between reversion‐inducing‐cysteine‐rich protein with Kazal motifs (RECK) and clinicopathological parameters
| Parameter | Patient no. (n = 55) | RECK | P‐value | |
|---|---|---|---|---|
| No change | Downregulation | |||
| Sex | ||||
| Male | 40 | 14 | 26 | 0.216 |
| Female | 15 | 8 | 7 | |
| Age (years) | ||||
| ≥70 | 27 | 13 | 14 | 0.226 |
| ≤70 | 28 | 9 | 19 | |
| T‐stage | ||||
| T0, T1 | 18 | 9 | 9 | 0.291 |
| T3, T4 | 37 | 13 | 24 | |
| Stage | ||||
| I, II | 40 | 18 | 22 | 0.216 |
| III, IV | 15 | 4 | 11 | |
| Histology | ||||
| Adenocarcinoma | 36 | 15 | 21 | 0.728a |
| Squamous cell carcinoma | 16 | 6 | 10 | |
| Large cell carcinoma | 3 | 1 | 2 | |
| Lymph node metastasis | ||||
| + | 13 | 2 | 11 | 0.038* |
| − | 42 | 20 | 22 | |
P < 0.05. a: Due to the small case number of large cell carcinoma, the statistical significance was tested between adenocarcinoma and squamous cell carcinoma groups.
Figure 1.
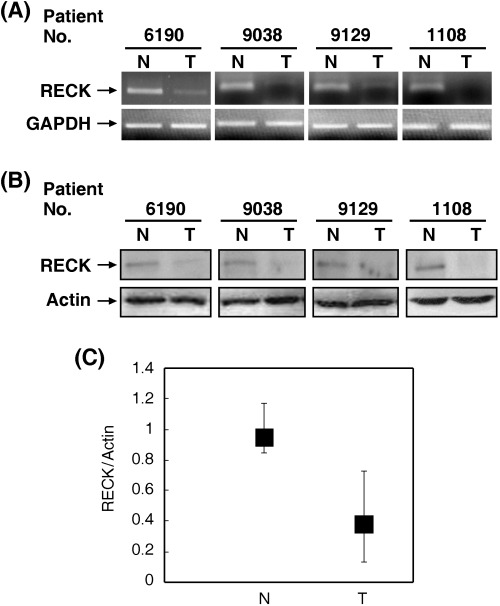
Expression of reversion‐inducing‐cysteine‐rich protein with Kazal motifs (RECK) in human lung tumor tissues (T) and their normal counterparts (N). Resected tissues placed in the RNAlater solution were subjected to isolation of mRNA and proteins using TRIzol reagent. (A) Reverse transcriptase–polymerase chain reaction assays were carried out to investigate RECK mRNA levels. Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as an internal control. (B) The protein level of RECK in N or T tissues was investigated by western blot analysis and actin was used as an internal control. (C) The distribution of the RECK/actin ratio of N and T tissues.
RECK downregulation is associated with promoter methylation. We next addressed whether downregulation of RECK in tumor tissues was caused by promoter methylation, an epigenetic alteration that frequently leads to gene silencing. Methylation‐specific PCR (MSP) analysis showed that RECK promoter methylation was found in 63.6% (35/55) of lung tumor tissues. Figure 2 demonstrates the methylation status of the RECK promoter of four paired normal and lung tumor tissues. We tested the correlation between RECK expression and promoter methylation and our data indicated that downregulation of RECK was strongly correlated with promoter methylation (P = 0.000005; Table 2).
Figure 2.

Methylation of the reversion‐inducing‐cysteine‐rich protein with Kazal motifs (RECK) promoter in human lung tumor tissues. Genomic DNA was isolated from lung tumors or their normal counterparts and subjected to sodium bisulfite modification. Methylation‐specific polymerase chain reaction was used to detect the methylation status of the human RECK promoter. The predicted products for methylated (M) and unmethylated (U) DNA were 195 and 199 bp, respectively.
Table 2. Reversion‐inducing‐cysteine‐rich protein with Kazal motifs (.
RECK) downregulation was associated with promoter methylation
| RECK promoter | RECK | P‐value | |
|---|---|---|---|
| No change | Downregulation | ||
| Methylated | 6 | 29 | 0.000005 |
| Unmethylated | 16 | 4 | |
K‐ras mutation is correlated with RECK promoter methylation and downregulation. Our previous study demonstrated that oncogenic ras increases the expression of DNMT3b and induces promoter methylation of the RECK gene.( 12 ) However, these findings have not been verified in primary tumor tissues. Therefore, we investigated the correlation between K‐ras mutation and RECK promoter methylation in lung tumors. Our results showed that K‐ras mutation was detected in 25.5% (14/55) of the 55 tumors tested in the present study and was significantly correlated with RECK promoter methylation (P = 0.047; Table 3). In addition, K‐ras mutation also correlated with RECK downregulation (P = 0.023).
Table 3.
K‐ras mutation was associated with reversion‐inducing‐cysteine‐rich protein with Kazal motifs (RECK) downregulation and promoter methylation
| RECK | K‐ras | P‐value | |
|---|---|---|---|
| Wild type | Mutant | ||
| Promoter | |||
| Methylated | 23 | 12 | 0.047 |
| Unmethylated | 18 | 2 | |
| Expression | |||
| No change | 20 | 2 | 0.023 |
| Downregulation | 21 | 12 | |
DNMT inhibitor 5′‐azacytidine restores RECK expression and reduces the invasive ability of human lung cancer cell lines. To examine the role of promoter methylation in the silencing of the RECK gene in lung cancer cells, we first investigated the methylation status of the RECK promoter and its expression. As demonstrated in Fig. 3A, MRC‐5 human lung fibroblasts expressed high levels of RECK mRNA and protein. RECK was also highly expressed in H226 human lung cancer cells. In contrast, the expression of RECK in H520, H358 and A549 lung cancer cells was very low or undetectable. MSP analysis indicated that significant RECK promoter methylation was found in these three lung cancer cells but not in MRC‐5 and H226 cells (Fig. 3B). We tested the effect of the DNMT inhibitor 5′‐azacytidine on H520, H358, A549 and H226 cells. Our results demonstrated that this drug restored the expression of RECK and reduced the invasive ability of the H520, H358 and A549 cell lines (Fig. 3C,D). Conversely, 5′‐azacytidine did not obviously increase the RECK protein level in H226 cells (Fig. 3C). Only 20% inhibition of cell invasion was observed in this cell line after drug treatment (Fig. 3D).
Figure 3.

Downregulation of reversion‐inducing‐cysteine‐rich protein with Kazal motifs (RECK) in human lung cancer cell lines and its restoration by 5′‐azacytidine. (A) Human lung fibroblast (1, MRC‐5) and lung cancer cell lines (2, H226; 3, H520; 4, H358; 5, A549) were harvested and subjected to isolation of genomic DNA, RNA and protein by TRIzol reagent. Reverse transcription–polymerase chain reaction (RT‐PCR) and western blot analysis were carried out to investigate RECK expression. (B) The methylation status of the RECK promoter was also addressed by methylation‐specific polymerase chain reaction. M, methylated; U, unmethylated. (C) H520 (3), H358 (4), A549 (5) and H226 (2) cells were treated with 5′‐azacytidine for 48 h and RECK expression was examined by RT‐PCR and western blot analysis. (D) In vitro cell invasion assays were carried out and the data are shown as mean ± SD. A, 5′‐azacytidine; C, control (0.1% dimethylsulfoxide). GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase.
Discussion
RECK is an important MMP inhibitor that is involved in the regulation of various physiological and pathological processes. Mice lacking RECK die in utero with developmental defects in blood vessels, the neural tube and mesenchymal tissues.( 3 ) RECK is also a target of myogenic regulatory factors and participates in the control of myogenesis.( 14 ) These results suggest that RECK is a key player in embryonic development. Under pathological conditions, RECK has been shown to be downregulated in human cancers.( 4 , 5 , 6 , 7 ) Two recent studies demonstrated that the 5‐year survival rate of lung cancer patients with tumors with strong RECK expression was significantly higher than that of patients with weakly expressing tumors.( 6 , 15 ) Therefore, RECK is an important prognostic factor for lung cancer patients. However, the molecular mechanism by which RECK was downregulated in tumors has never been characterized. In the present study, we provide the first evidence that silencing of the RECK gene in lung cancer cells is caused by promoter methylation. We also found that RECK downregulation is closely linked with increased lymph node metastasis. In addition, we showed that 5′‐azacytidine (also named Vidaza), a DNMT inhibitor that has recently been approved by the US Food and Drug Administration (FDA) for treatment of myelodysplastic syndrome,( 16 ) may restore RECK expression and inhibit invasion of cultured lung cancer cells.( 12 ) Our results strongly support the notion that RECK is a metastasis suppressor and a better prognostic factor for lung cancer.
Another important finding of the present study is that RECK promoter methylation and downregulation in lung cancer is associated with K‐ras mutation. We and others have demonstrated that the ras oncogene represses RECK expression.( 17 , 18 ) Our recent study extended these works and revealed that oncogenic ras acts through DNMT3b‐mediated promoter methylation to repress RECK expression.( 12 ) Our hypothesis is that oncogenic ras upregulates the expression of DNMT3b and increases the binding of this protein to the RECK gene promoter, which results in promoter methylation and gene silencing. To verify this hypothesis, two issues should be proved. First, whether ras mutation is indeed linked with RECK gene silencing in primary tumors. We investigated this correlation in lung tumor tissues and verified that K‐ras mutation and RECK downregulation are strongly correlated. Similar findings were observed in cultured lung cancer cell lines. H358 and A549 cells have been shown to harbor K‐ras mutation.( 19 ) In the present study, the expression of RECK was low and the RECK promoter was highly methylated. In contrast, H226 cells that harbor wild‐type K‐ras exhibited high RECK expression and low promoter methylation. The genetic alterations of H520 have been little reported. H520 cells harbor the wild‐type K‐ras gene.( 19 ) However, a very recent study demonstrated RASSF8 (RAS association RalGDS/AF‐6 domain family 8), a candidate tumor suppressor gene involved in the RAS signaling pathway, is downregulated in H520 cells.( 20 ) Therefore, our results suggest that hyperactivation of the ras signaling pathway suppresses RECK expression in vivo. The second issue is whether DNMT3b is upregulated in human lung tumor tissues and whether its overexpression is correlated with ras mutation. Two previous studies demonstrated that DNMT3b promoter polymorphisms are linked with an increased risk of lung cancer.( 21 , 22 ) Recently, two independent studies have shown that DNMT3b is overexpressed in human lung cancer.( 23 , 24 ) However, the status of ras mutation in tumor tissues was not addressed in these two studies. Therefore, the correlation between ras mutation and DNMT3b overexpression is still unclear and needs to be investigated in future works.
Previous studies on the oncogenic activity of ras focused mainly on its effect on cell proliferation. However, the effect of ras on tumor metastasis is less clear. Recently, several potential mediators have been identified for ras‐induced metastasis, including cytoskeletal proteins,( 25 ) integrins,( 26 ) cadherins,( 27 ) angiogenic factors( 28 , 29 ) and MMP. A genome‐wide screening of ras targets demonstrated that ras suppresses the expression of thrombospondin 1 and tissue inhibitor of metalloproteinase 1, two important antimetastatic genes, to enhance cell invasion.( 30 ) Pathological studies have also shown that inactivation of RASSF1A, a tumor suppressor that exhibits antimetastatic activity, is closely linked with ras mutation in cancer cells.( 31 , 32 ) We now identify RECK as a molecular target for the ras oncogene and show that downregulation of RECK is associated with K‐ras mutation and lymph node metastasis in non‐small cell lung tumor tissues
Acknowledgments
This study was supported by grants VGHNSU94‐003 and VGHNSU 95‐001 to Huang‐Chou Chang and a grant from National Sun Yat‐Sen University–Kaohsiung Medical University Joint Research Center to Wen‐Chun Hung.
References
- 1. Noda M, Kitayama H, Matsuzaki T et al. Detection of genes with a potential for suppressing the transformed phenotype associated with activated ras genes. Proc Natl Acad Sci USA 1989; 86: 162 – 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Takahashi C, Sheng Z, Horan TP et al. Regulation of matrix metalloproteinase‐9 and inhibition of tumor invasion by the membrane‐anchored glycoprotein RECK. Proc Natl Acad Sci USA 1998; 95: 13 221 – 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oh J, Takahashi R, Kondo S et al. The membrane‐anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell 2001; 107: 789 – 800. [DOI] [PubMed] [Google Scholar]
- 4. Masui T, Doi R, Koshiba T et al. RECK expression in pancreatic cancer: its correlation with lower invasiveness and better prognosis. Clin Cancer Res 2003; 9: 1779 – 84. [PubMed] [Google Scholar]
- 5. Span PN, Sweep CG, Manders P, Beex LV, Leppert D, Lindberg RL. Matrix metalloproteinase inhibitor reversion‐inducing cysteine‐rich protein with Kazal motifs: a prognostic marker for good clinical outcome in human breast carcinoma. Cancer 2003; 97: 2710 – 15. [DOI] [PubMed] [Google Scholar]
- 6. Takenaka K, Ishikawa S, Kawano Y et al. Expression of a novel matrix metalloproteinase regulator, RECK, and its clinical significance in resected non‐small cell lung cancer. Eur J Cancer 2004; 40: 1617 – 23. [DOI] [PubMed] [Google Scholar]
- 7. Takeuchi T, Hisanaga M, Nagano M et al. The membrane‐anchored matrix metalloproteinase (MMP) regulator RECK in combination with MMP‐9 serves as an informative prognostic indicator for colorectal cancer. Clin Cancer Res 2004; 10: 5572 – 9. [DOI] [PubMed] [Google Scholar]
- 8. Bos JL. Ras oncogenes in human cancer: a review. Cancer Res 1989; 49: 4682 – 9. [PubMed] [Google Scholar]
- 9. Bondy GP, Wilson S, Chambers AF. Experimental metastatic ability of H‐ras‐transformed NIH3T3 cells. Cancer Res 1985; 45: 6005 – 9. [PubMed] [Google Scholar]
- 10. Muschel RJ, Williams JE, Lowy DR, Liotta LA. Harvey ras induction of metastatic potential depends upon oncogene activation and the type of recipient cell. Am J Pathol 1985; 121: 1 – 8. [PMC free article] [PubMed] [Google Scholar]
- 11. Ichikawa T, Kiprianou N, Isaacs JT. Genetic instability and the acquisition of metastatic ability by rat mammary cancer cells following v‐H‐ras oncogene transfection. Cancer Res 1990; 50: 6349 – 57. [PubMed] [Google Scholar]
- 12. Chang HC, Cho CY, Hung WC. Silencing of the metastasis suppressor RECK by RAS oncogene is mediated by DNA methyltransferase 3b‐induced promoter methylation. Cancer Res 2006; 66: 8413 – 20. [DOI] [PubMed] [Google Scholar]
- 13. Liu LT, Chang HC, Chiang LC, Hung WC. Histone deacetylase inhibitor up‐regulates RECK to inhibit MMP‐2 activation and cancer cell invasion. Cancer Res 2003; 63: 3069 – 72. [PubMed] [Google Scholar]
- 14. Echizenya M, Kondo S, Takahashi R et al. The membrane‐anchored MMP‐regulator RECK is a target of myogenic regulatory factors. Oncogene 2005; 24: 5850 – 7. [DOI] [PubMed] [Google Scholar]
- 15. Takenaka K, Ishikawa S, Yanagihara K et al. Prognostic significance of reversion‐inducing cysteine‐rich protein with kazal motifs expression in resected pathological stage IIIA N2 non‐small cell lung cancer. Ann Surg Oncol 2005; 12: 817 – 24. [DOI] [PubMed] [Google Scholar]
- 16. Das PM, Singal R. DNA methylation and cancer. J Clin Oncol 2004; 22: 4632 – 42. [DOI] [PubMed] [Google Scholar]
- 17. Sasahara RM, Takahashi C, Noda M. Involvement of the Sp1 site in ras‐mediated downregulation of the RECK metastasis suppressor gene. Biochem Biophys Res Commun 1999; 264: 668 – 75. [DOI] [PubMed] [Google Scholar]
- 18. Chang HC, Liu LT, Hung WC. Involvement of histone deacetylation in ras‐induced down‐regulation of the metastasis suppressor RECK. Cell Signal 2004; 16: 675 – 9. [DOI] [PubMed] [Google Scholar]
- 19. Mitsudomi T, Steinberg SM, Nau MM et al. p53 gene mutations in non‐small‐cell lung cancer cell lines and their correlation with the presence of ras mutations and clinical features. Oncogene 1992; 7: 171 – 80. [PubMed] [Google Scholar]
- 20. Falvella FS, Manenti G, Spinola M et al. Identification of RASSF8 as a candidate lung tumor suppressor gene. Oncogene 2006; 25: 3934 – 8. [DOI] [PubMed] [Google Scholar]
- 21. Shen H, Wang L, Spitz MR, Hong WK, Mao L, Wei Q. A novel polymorphism in human cytosine DNA‐methyltransferase‐3B promoter is associated with an increased risk of lung cancer. Cancer Res 2002; 62: 4992 – 5. [PubMed] [Google Scholar]
- 22. Lee SJ, Jeon HS, Jang JS et al. DNMT3B polymorphisms and risk of primary lung cancer. Carcinogenesis 2005; 26: 403 – 9. [DOI] [PubMed] [Google Scholar]
- 23. Vallbohmer D, Brabender J, Yang D et al. DNA methyltransferase messenger RNA expression and aberrant methylation of CpG islands in non‐small‐cell lung cancer: association and prognostic value. Clin Lung Cancer 2006; 8: 39 – 44. [DOI] [PubMed] [Google Scholar]
- 24. Wang J, Walsh G, Liu DD, Lee JJ, Mao L. Expression of ΔDNMT3B variants and its association with promoter methylation of p16 and RASSF1A in primary non‐small cell lung cancer. Cancer Res 2006; 66: 8361 – 6. [DOI] [PubMed] [Google Scholar]
- 25. Hall A. Ras‐related GTPases and cytoskeleton. Mol Biol Cell 1992; 3: 475 – 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang Z, Vuori K, Wang H, Reed JC, Ruoslahti E. Integrin activation by R‐ras. Cell 1996; 85: 61 – 9. [DOI] [PubMed] [Google Scholar]
- 27. Schmidt CR, Gi YJ, Coffey RJ, Beauchamp RD, Pearson AS. Oncogenic Ras dominates overexpression of E‐cadherin in malignant transformation of intestinal epithelial cells. Surgery 2004; 136: 303 – 9. [DOI] [PubMed] [Google Scholar]
- 28. Rak J, Mitsuhashi Y, Bayko L et al. Mutant ras oncogenes upregulate VEGF/VPF expression: implications for induction and inhibition of tumor angiogenesis. Cancer Res 1995; 55: 4575 – 80. [PubMed] [Google Scholar]
- 29. Pironin M, Clement G, Benzakour O, Lawrence D, Vigier P. Growth in serum‐free medium of NIH3T3 cells transformed by the EJ‐H‐ras oncogene: evidence for multiple autocrine growth factors. Int J Cancer 1992; 51: 980 – 8. [DOI] [PubMed] [Google Scholar]
- 30. Zuber J, Tchernitsa OI, Hinzmann B et al. A genome‐wide survey of RAS transformation targets. Nat Genet 2002; 24: 144 – 52. [DOI] [PubMed] [Google Scholar]
- 31. Dammann R, Schagdarsurengin U, Liu L et al. Frequent RASSF1A promoter hypermethylation and K‐ras mutations in pancreatic carcinoma. Oncogene 2003; 22: 3806 – 12. [DOI] [PubMed] [Google Scholar]
- 32. Kim DH, Kim JS, Park JH et al. Relationship of Ras association domain family 1 methylation and K‐ras mutation in primary non‐small cell lung cancer. Cancer Res 2003; 63: 6206 – 11. [PubMed] [Google Scholar]