

Abstract
Malignant pleural mesothelioma (MPM) is closely related to exposure to asbestos, and a rapid increase in the number of MPM patients in Japan is estimated in the years 2010–2050. The purpose of the present study was to establish a clinically relevant animal model that shows human patient‐like progression of MPM. Here, we demonstrate that a human MPM cell line (EHMES‐10) inoculated orthotopically (thoracic cavity) into severe combined immunodeficiency (SCID) mice produces highly vascularized thoracic tumors with pleural dissemination and bloody pleural effusions by 5 weeks, suggesting a patient‐like progression of this cell line after orthotopic inoculation. EHMES‐10 cells overexpressed vascular endothelial growth factor (VEGF), a molecule responsible for malignant effusions, and its receptor. Treatment with cisplatin, but not gemcitabine, significantly inhibited the production of pleural effusions, but it was not effective for thoracic tumors, consistent with chemotherapy refractory characteristics of MPM in patients. Our patient‐like orthotopic model using EHMES‐10 cells overexpressing VEGF and its receptor may be useful for examining the molecular pathogenesis of MPM and may contribute to the development of novel treatment strategies for MPM. (Cancer Sci 2006; 97: 183 –191)
Abbreviations:
- CDDP
cisplatin
- CEA
carcinoembryonic antigen
- DOC
docetaxel
- EMA
epithelial membrane antigen
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- GEM
gemcitabine
- H&E
hematoxylin–eosin
- HMVEC
human microvascular endothelial cells
- MPM
malignant pleural mesothelioma
- OCT
optimal cutting temperature
- RT‐PCR
reverse transcription–polymerase chain reaction
- SCID
severe combined immunodeficiency
- VEGF
vascular endothelial growth factor
- VEGFR
VEGF receptor
- VNR
vinorelbine
Malignant pleural mesothelioma arises from the mesothelial cells that line the thoracic cavity. MPM grows aggressively with dissemination in the thoracic cavity and frequently produces malignant pleural effusions.( 1 ) MPM causes respiratory symptoms, including dyspnea, shortness of breath and chest pain, which extremely limits the quality of life of the patients with this disease.
Several etiologic factors, including asbestos,( 2 , 3 ) iron( 4 ) and simian virus 40 (SV40)( 1 ) are reported to be involved in the development of MPM. Of these factors, exposure to asbestos is most closely related to development of MPM, and individuals with a history of exposure to asbestos have a much greater risk of developing MPM. A large amount of asbestos was imported during the 1970s to 1980s and used widely in Japan.( 3 ) Considering that the latent period between first exposure to asbestos and the occurrence of MPM is 30–40 years, a rapid increase in the number of MPM patients is estimated in Japan during the 2010s to 2040s.( 3 )
Although surgical resection at an early stage is the only curative therapeutic modality, the majority of MPM patients are diagnosed at the advanced stage.( 1 ) In addition, MPM is refractory to conventional chemotherapy and radiotherapy, and has a poor prognosis (median survival time from onset is approximately 1 year). Therefore, novel effective therapies are necessary to improve the prognosis of this disease.
To develop a novel therapeutic approach, the cellular and molecular pathogenesis of MPM should be clarified. For such purposes, a suitable animal model of MPM that shows patient‐like tumor progression is required. It has become clear that the orthotopic sites of implantation or on‐plantation are critical for the development of transplanted tumors in nude or SCID mice.( 5 ) When colon tumor cell lines were implanted into the cecum and the spleen, they formed metastases at various sites, including the liver (patient‐like progression). However, when they were implanted subcutaneously they grew to form a primary tumor, but did not metastasize.
The purpose of the present study was to establish a clinically relevant animal model that shows patient‐like progression of MPM. We used a human MPM cell line, named EHMES‐10,( 6 ) and examined whether EHMES‐10 developed tumors when implanted into the thoracic cavity (orthotopic site) of SCID mice.
Materials and Methods
Cell lines
RPMI 1640 medium supplemented with 10% FBS, penicillin (100 U/mL) and streptomycin (50 µg/mL), designated as CRPMI1640, was used for cell culture in this study. Cells were cultured in a humidified CO2 incubator at 37°C.
The human MPM cell line EHMES‐10 was established from pleural effusion of a patient with MPM (biphasic type, the diagnosis was made after autopsy) at Ehime University (Ehime, Japan).( 6 ) Briefly, bloody pleural effusion was harvested aseptically by percutaneous puncture. The effusion was centrifuged at 267 g for 5 min. The supernatant was removed and the cell pellet was resuspended in the CRPMI1640. The cell solution was briefly centrifuged twice at 119 g for 5–10 s to enrich for large cells (tumor cells). To remove non‐tumor cells (such as macrophages and fibroblasts), the resultant cells were incubated in a plastic flask in CRPMI1640 for 2 h, and only non‐adherent cells were harvested. After two cycles of this process, large cells (tumor cells) were further enriched. The resultant cells were cultured in CRPMI1640. Adherent cells were repeatedly subcultured and named as EHMES‐10. The human lung adenocarcinoma cell line PC14PE6( 7 ) was kindly provided by Dr I. J. Fidler (MD Anderson Cancer Center, Houston, TX, USA).
Animals
Male SCID mice (6–8 weeks old) were obtained from CLEA Japan (Osaka, Japan) and maintained under specific pathogen‐free conditions throughout the study. Experiments were carried out in accordance with the guidelines established by the Tokushima University Committee on Animal Care and Use.
Orthotopic implantation of MPM cells into SCID mice
Human EHMES‐10 cells were harvested and washed with Ca2+‐ and Mg2+‐free PBS, and suspended in the same solution.
For orthotopic implantation, SCID mice were anesthetized with ether and had their right chest wall shaved. After sterilization of the chest wall with 70% ethanol, their right chest skin and subcutaneous tissue was cut, and their parietal pleura was exposed. They were then injected with EHMES‐10 cells (3 × 106 cells/100 µL) into the thoracic cavity through the parietal pleura using a 27G needle. Finally, they were sutured to close the wound.
After 28–35 days, the mice were anesthetized and killed to evaluate tumor development. The appearance of pleural effusions was checked and the volume was measured as reported previously.( 7 ) The tumors were removed and weighed, and the pleural disseminations were inspected macroscopically. The lungs were removed and fixed in Bouin's solution for 24 h, and the dissemination of visceral pleura (white spots on the surface) was inspected macroscopically.
Reagents
Cisplatin, a platinum drug that forms platinum–DNA adducts and induces cell death, was purchased from Bristol‐Myers Squibb (Princeton, NJ, USA). GEM, an antimetabolic drug that inhibits DNA synthesis, was from Eli Lilly (Indianapolis, IN, USA). DOC, a taxan that stabilizes microtubules against depolymerization, thereby inducing apoptosis, was from Aventis Pharma (Strasbourg, France). VNR, a vinca alkaloid that interacts with tubulin and disrupts microtubule function, was from Kyowa Hakko (Tokyo, Japan). Working solutions of CDDP, GEM, DOC and VNR were prepared in 0.9% NaCl before use.
Cell proliferation assay
Cell proliferation was measured using the MTT (3‐[4,5‐dimethylthiazol‐2‐yl]‐2, 5‐diphenyl tetrazolium) dye reduction method.( 8 ) Tumor cells (2 × 103 cells/100 µL) were plated into each well of the 96‐well plates in medium and incubated at 37°C under 5% CO2 in humidified air. After 24 h, 100 µL of various concentrations of anticancer drugs were added and incubated for an additional 72 h. After the incubation, 50 µL of stock MTT solution (2 mg/mL; Sigma, St Louis, MO, USA) was added to all of the wells, and the cells were incubated for 2 h at 37°C. The media containing MTT solution was removed, and the dark blue crystals were dissolved by adding 100 µL of dimethylsulfoxide. The absorbance was measured with a MTP‐120 microplate reader (Corona Electric, Ibaraki, Japan) at test and reference wavelengths of 550 and 630 nm, respectively.
VEGF production
Tumor cells (2 × 106/20 mL) were plated in duplicate in 10‐cm dishes and cultured in RPMI 1640 medium supplemented with 10% FBS for 48 h. The culture supernatants were harvested and stored until measurements were taken. Protein levels of VEGF in the culture supernatants were determined using enzyme‐linked immunosorbent assay kits for VEGF according to the manufacturer's instructions (R & D Systems, Minneapolis, MN, USA). The detection limit was 15.6 pg/mL.
Expression of VEGF isoforms and VEGFR Expression levels of VEGF isoforms and VEGFR were determined by RT‐PCR. Total cellular RNA was extracted from the culture of the cell lines using the acid guanidinium thiocyanate/phenol‐chloroform method (ISOGEN, Nippon Gene Co., Toyama, Japan) according to the manufacturer's protocol. One microgram of total RNA was reverse transcribed. The PCR program used to amplify VEGF, VEGFR and GAPDH consisted of a precycle of 5 min at 94°C, 45 s at 60°C, and 45 s at 72°C. After this cycle, the process was continued for 30 cycles of 1 min at 94°C, 45 s at 65°C, 2 min at 72°C, and concluding with 7 min at 72°C. The primers for VEGF, VEGFR and GAPDH were as follows: VEGF sense (5′‐TCC AGG AGT ACC CTG ATG AG‐3′) and antisense (5′‐CTT TCC TGG TGA GAG ATC TGG‐3′) immediately flanking the region of the VEGF open reading frame involved in the alternative splicing of several exons;( 9 ) VEGFR‐1 sense (5′‐ATT TGT GAT TTT GGC CTT GC‐3′) and antisense (5′‐CAG GCT CAT GAA CTT GAA AGC‐3′); VEGFR‐2 sense (5′‐GTG ACC AAC ATG GAG TCG TG‐3′) and antisense (5′‐CCA GAC ATT CCA TGC CAC TT‐3′);( 10 ) VEGFR‐3 sense (5′‐CCC ACG CAG ACA TCA AGA CG‐3′) and antisense (5′‐TGC AGA ACT CCA CGA TCA CC‐3′);( 11 ) and GAPDH sense (5′‐AGT CAT CCA CGA GCG ATT TG‐3′) and antisense (5′‐TGC TGC TTT TAC AGC CTC CT‐3′). RT‐PCR was carried out using a one‐step RNA PCR kit (TAKARA, Tokyo, Japan). The bands were visualized by staining with ethidium bromide.
Histology and immunohistochemistry
The lungs and tumors of the SCID mice were harvested at autopsy, cut into 5‐mm fragments and placed in OCT compound (Miles Laboratories, Elkhart, IN, USA) to be snap frozen in liquid nitrogen for histological and immunohistochemical analysis. Frozen tissue sections (8‐µm thick) were fixed with cold acetone. The slides were rinsed with PBS, and endogenous peroxidase was blocked by the use of 3% hydrogen peroxide in PBS for 12 min. The samples were washed three times with PBS and incubated for 10 min at room temperature with a protein‐blocking solution consisting of PBS (pH 7.5) containing 5% normal horse serum and 1% normal goat serum. The excess blocking solution was drained, and the samples were incubated overnight at 4°C with a 1:400 dilution of rabbit polyclonal antihuman VEGF antibody (Pharmigen, San Diego, CA, USA) or a 1:100 dilution of rat antimouse CD31 monoclonal antibody (Pharmigen).( 12 ) The samples were then rinsed three times with PBS and incubated for 60 min at room temperature with the appropriate dilution of peroxidase‐conjugated antirabbit IgG or antirat IgG. The slides were rinsed with PBS and incubated for 5 min with diaminobenzidine. The sections were then washed three times with distilled water, counterstained with Gill's hematoxylin, washed once with distilled water and once with PBS, and rinsed again with distilled water. The slides were mounted and examined using a brightfield microscope. A reddish‐brown precipitate in the cytoplasm indicated a positive reaction. Frozen sections were also stained with H&E for routine histological examination.
Statistical analysis
The statistical significance of difference in the in vivo data was analyzed using the Mann–Whitney U‐test.
Results
Proliferation of EHMES‐10 cells in vitro
A human MPM cell line, EHMES‐10, grew as adherent cultures (Fig. 1). The doubling time was 36 h.
Figure 1.
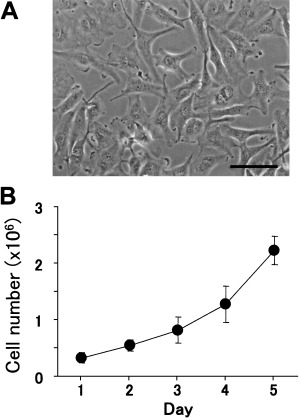
Morphology and proliferation of EHMES‐10 cells in vitro. (A) Culture of EHMES‐10 cells. Bars indicate 100 µm. (B) EHMES‐10 (closed circles) were cultured in 10‐cm dishes in MEM with 10% fetal bovine serum. The number of cells was counted daily. Values are the mean ± SD of triplicate cultures. Data shown are representative of three independent experiments with similar results.
Tumorigenicity of EHMES‐10 cells in SCID mice
We examined whether human EHMES‐10 cells could produce tumors when inoculated orthotopically (into the thoracic cavity) into SCID mice. EHMES‐10 cells inoculated orthotopically developed tumors in the thoracic cavity in seven of seven recipient mice by 5 weeks (Table 1), and showed classical patterns of spread of advanced MPM, such as lung encasement by tumor rind (Fig. 2B) and diffusely grown pleural‐based masses (Fig. 2C).( 13 ) In addition, bloody pleural effusions (up to 1 mL) were produced in seven of seven recipient mice (Table 1; Fig. 2A,D). These results suggest that EHMES‐10 cells were highly tumorigenic when inoculated into the orthotopic microenvironment (thoracic cavity). Furthermore, orthotopically inoculated EHMES‐10 cells closely resemble the clinical behavior of MPM in terms of disease progression pattern.
Table 1.
Tumorigenicity of EHMES‐10 cells in SCID mice
| Thoracic tumor | Pleural effusion | ||||
|---|---|---|---|---|---|
| Incidence | Median weight (mg) | Weight range (mg) | Incidence | Median volume (µL) | Volume range (µL) |
| 7/7 | 200 | 10–1005 | 7/7 | 300 | 10–1000 |
SCID, severe combined immunodeficiency. EHMES‐10 cells (3 × 106) were inoculated into the thoracic cavity of SCID mice on day 0. The production of thoracic tumors and pleural effusions was determined on day 35.
Figure 2.
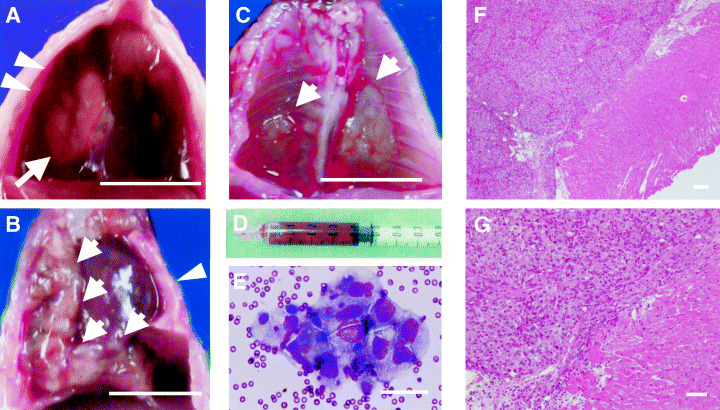
Thoracic tumors and bloody pleural effusions produced by orthotopically implanted EHMES‐10 cells in severe combined immunodeficiency (SCID) mice. EHMES‐10 cells (3 × 106) were inoculated into the thoracic cavity of SCID mice. (A) thoracic cavity on day 28. White arrow and arrow heads indicate thoracic tumor and bloody pleural effusion, respectively. (B) Thoracic cavity on day 28. White arrows indicate tumor rind encasing the right lung. White arrow heads indicate the compressed left lung. (C) Pleural‐based masses grown diffusely in the thoracic cavity (white arrow). (D) Bloody pleural effusion aspirated in the syringe. (E) Tumor cells in the bloody pleural effusion produced by EHMES‐10 cells (hematoxylin–oesin [H&E] staining, ×400). H&E staining of pleural‐based masses at low (F) and high (G) magnifications. Note: solid sheet‐like arrangement of large tumor cells was observed. Those masses diffusely invaded the parietal pleura. Bars indicate 10 mm (A–C) and 100 µm (E–G).
Hematoxylin–eosin staining of pleural‐based masses showed solid sheet‐like arrangement of large tumor cells. Those masses invaded the parietal pleura diffusely (Fig. 2F,G).
Survival of SCID mice in the orthotopic implantation model
The mice inoculated with EHMES‐10 cells into the thoracic cavity became moribund, as shown in Fig. 3. The median survival was 33 days (range 21–44 days). The major cause of the weakening of the EHMES‐10‐bearing mice was most likely respiratory failure due to oppression by tumors and pleural effusions, similar to human patients with MPM.
Figure 3.
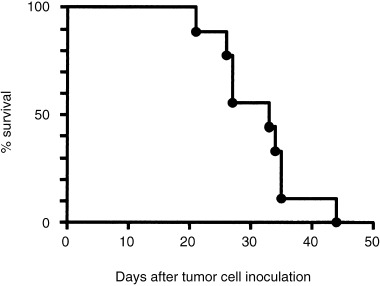
Survival of severe combined immunodeficiency (SCID) mice inoculated with EHMES‐10 cells. EHMES‐10 cells (3 × 106) were inoculated into the thoracic cavity of SCID mice. When the mice became moribund, they were killed. Survival was therefore determined up to the day the mice were killed.
Determination of optimal experimental conditions for the orthotopic implantation model with EHMES‐10 cells
To determine the optimal tumor cell number for the orthotopic MPM model, various numbers of EHMES‐10 cells were inoculated into the thoracic cavity of SCID mice. As shown in Table 2, the tumorigenicity and production of pleural effusions by EHMES‐10 cells were increased in a cell number‐dependent manner. On the basis of these results, we chose to use 3 × 106 EHMES‐10 cells for inoculation.
Table 2.
Effect of tumor cell number on the production of thoracic tumors and pleural effusions by EHMES‐10 cells in SCID mice
| No. cells inoculated | Thoracic tumor | Pleural effusion | ||||
|---|---|---|---|---|---|---|
| Incidence | Median weight (mg) | Weight range (mg) | Incidence | Median volume (µL) | Volume range (µL) | |
| 3 × 105 | 6/8 (75%) | 50 | 0–300 | 4/8 (50%) | 50 | 0–900 |
| 1 × 106 | 5/6 (83%) | 119 | 0–474 | 3/6 (50%) | 400 | 0–1000 |
| 3 × 106 | 6/7 (86%) | 255 | 0–1730 | 6/7 (86%) | 400 | 0–600 |
SCID, severe combined immunodeficiency. Indicated numbers of EHMES‐10 cells were inoculated into the thoracic cavity of SCID mice on day 0. SCID mice were killed on day 28, and the production of thoracic tumors and pleural effusions was evaluated.
We further determined the time kinetics of the production of thoracic tumors and pleural effusions by EHMES‐10 cells. Both thoracic tumors and pleural effusions were detected as early as 14 days after tumor cell inoculation, and the incidence was increased in a time‐dependent manner (Table 3). As some mice became moribund by day 35, we chose to use 3 × 106 EHMES‐10 cells for inoculation and to determine the production of thoracic tumors and pleural effusions 28 days after tumor cell inoculation in the following experiments.
Table 3.
Time kinetics of the production of thoracic tumors and pleural effusions due to EHMES‐10 cells in SCID mice
| Day after inoculation | Thoracic tumor | Pleural effusion | ||||
|---|---|---|---|---|---|---|
| Incidence | Median weight (mg) | Weight range (mg) | Incidence | Median volume (µL) | Volume range (µL) | |
| 7 | 0/5 | All | 0 | 0/5 | All | 0 |
| 14 | 3/5 | 12 | 0–80 | 1/5 | 0 | 0–40 |
| 21 | 3/5 | 27 | 0–104 | 1/5 | 0 | 0–150 |
| 28 | 5/5 | 44 | 14–254 | 4/5 | 630 | 0–860 |
| 35 | 5/5 | 136 | 48–400 | 4/5 | 480 | 0–900 |
SCID, severe combined immunodeficiency. EHMES‐10 cells (3 × 106) were inoculated into the thoracic cavity of SCID mice on day 0. The mice were killed at the indicated times, and production of thoracic tumors and pleural effusions was evaluated.
Expression of VEGF‐related molecules
Using the lung cancer cell line PC14PE6, we demonstrated recently that overexpression of VEGF in the thoracic cavity induces vascular hyperpermeability and hence causes bloody pleural effusion.( 7 ) Therefore, we explored VEGF production in EHMES‐10 cells compared with PC14PE6 cells. As shown in Fig. 4A, the VEGF level of EHMES‐10 cells was as high as that of PC14PE6 cells, suggesting the involvement of VEGF overexpression by EHMES‐10 cells in the production of bloody pleural effusions.
Figure 4.
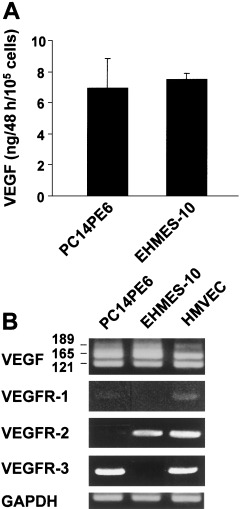
Expression of vascular endothelial growth factor (VEGF) and VEGF receptors (VEGFR) by EHMES‐10 cells. (A) Tumor cells (2 × 105) were cultured for 48 h in 2 mL MEM with 10% fetal bovine serum. The culture supernatants were harvested and their VEGF level was measured by enzyme‐linked immunosorbent assay. Bars are the SD of duplicate cultures. EHMES‐10 cells produced VEGF at levels as high as in lung adenocarcinoma PC14PE6 cells. (B) The levels of VEGF isoforms and VEGFR were determined by reverse transcription–polymerase chain reaction. HMVEC were used as a positive control VEGFR expression. Data shown are representative of three independent experiments with similar results.
Vascular endothelial growth factor expresses several isoforms (121, 165, 189 and 206 amino acids) resulting from alternative mRNA splicing of a single gene.( 14 ) Recent studies suggest that the expression patterns of certain VEGF isoforms are tissue specific, implying that these isoforms have defined roles in vasculogenesis and tumor angiogenesis.( 15 ) Therefore, we next examined the expression of VEGF isoforms by RT‐PCR and found that both EHMES‐10 and PC14PE6 cells predominantly expressed VEGF165 (Fig. 4B), which is biologically the most potent isoform.( 14 )
Vascular endothelial growth factor receptors are thought to play pivotal roles in angiogenesis and lymphangiogenesis.( 15 ) VEGF binds to specific receptors, VEGFR‐1 (also referred to as fms‐like tyrosine kinase‐1 [Flt‐1])( 16 ) and VEGFR‐2 (also referred to as KDR, and the murine homolog, Flk‐1).( 17 , 18 Recently it was reported that VEGFR‐1 and VEGFR‐2 are expressed not only on endothelial cells, but also on other types of cells, including hematopoietic stem cells and tumor cells.( 15 ) Thus, we next examined the expression of VEGFR on MPM cells by means of RT‐PCR (Fig. 4B). EHMES‐10 cells only expressed VEGFR‐2, which is considered to be the most important mediator of VEGF‐induced signaling responses.( 15 )
We further evaluated VEGF production in EHMES‐10 cells in vivo. Immunohistochemical analyses revealed that the thoracic tumors produced by EHMES‐10 expressed the VEGF protein (Fig. 5). In addition, the tumors were highly angiogenic when examined by staining for CD31 (a pan‐endothelial marker).
Figure 5.
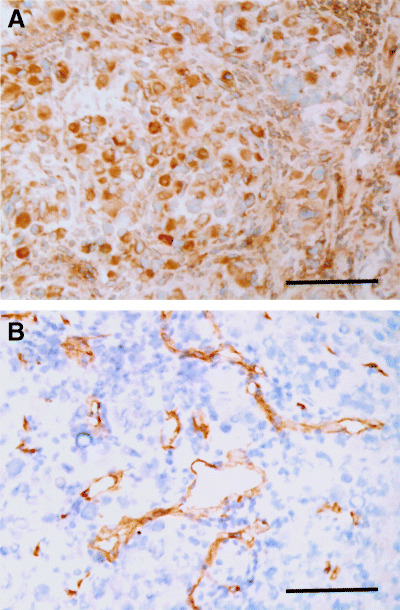
Histological examinations of thoracic tumors produced by EHMES‐10 cells. (A) vascular endothelial growth factor (VEGF); and (B) CD31 (panendothelial marker). Note: thoracic tumors produced by EHMES‐10 cells expressed VEGF protein. The tumors were highly vascularized when determined by staining for CD31 (×200). Bars indicate 100 µm.
Sensitivity of human MPM cell lines to chemotherapeutic agents in vitro
We next examined the effect of anticancer drugs against MPM cells. The in vitro cytotoxicity of various anticancer drugs was evaluated using an MTT assay. A human lung adenocarcinoma cell line, PC14PE6, was used as a control. EHMES‐10 cells were less sensitive to CDDP, compared with PC14PE6 cells. However, EHMES‐10 cells were more sensitive to GEM compared with PC14PE6 cells. In addition, EHMES‐10 was more sensitive to VNR, and both lines showed a similar sensitivity to DOC (Fig. 6).
Figure 6.
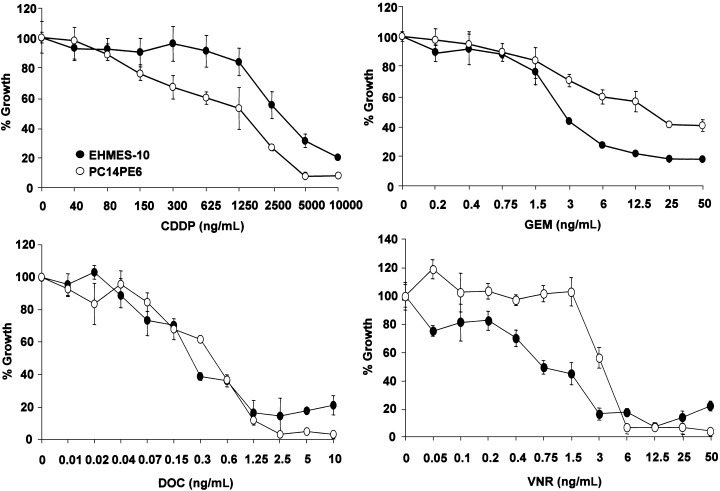
Sensitivity of EHMES‐10 cells to chemotherapeutic agents in vitro. EHMES‐10 (○) and PC14PE6 (•) (2 × 103) were cultured in 96‐well plates with or without various concentrations of cisplatin (CDDP), gemcitabine (GEM), docetaxel (DOC) or vinorelbine (VNR) for 3 days. Growth of tumor cells was determined by MTT assay. Bars are the SD of triplicate cultures. Data shown are representative of four independent experiments with similar results.
Therapeutic effects of CDDP and GEM in the orthotopic implantation model with EHMES‐10 cells
We examined the therapeutic effect of chemotherapeutic agents on tumors and pleural effusion caused by EHMES‐10 cells. We chose CDDP and GEM for the in vivo experiments because these two agents are commonly used for MPM patients.( 19 ) In set 1 (Table 4), EHMES‐10 cells were inoculated into the thoracic cavity of SCID mice, and the mice were treated with intravenous administration of CDDP (3 mg/kg or 6 mg/kg) on day 14, or by intraperitoneal administration of GEM (175 mg/kg or 350 mg/kg) on days 14, 18, 21 and 25. Treatment with 3 mg/kg CDDP had no effect either on tumor weight or pleural effusions. Treatment with 6 mg/kg CDDP significantly inhibited the production of pleural effusions, although it was less effective for tumor weight.
Table 4.
Therapeutic effects of anticancer drugs in the orthotopic implantation model with EHMES‐10 cells in SCID mice
| Treatment | Thoracic tumor | Pleural effusion | ||||
|---|---|---|---|---|---|---|
| Incidence | Median weight (mg) | Weight range (mg) | Incidence | Median volume (µL) | Volume range (µL) | |
| Set 1 | ||||||
| PBS | 5/5 | 190 | 50–350 | 4/5 | 500 | <20–600 |
| CDDP 3 mg/kg | 3/4 | 85 | 0–320 | 3/4 | 225 | <20–800 |
| CDDP 6 mg/kg | 5/5 | 220 | 90–380 | 2/5 | 0* | <20–50 |
| Set 2 | ||||||
| PBS | 5/5 | 197 | 23–222 | 4/5 | 450 | <20–800 |
| GEM 60 mg/kg | 5/5 | 113 | 101–131 | 3/5 | 700 | <20–900 |
| GEM 300 mg/kg | 5/5 | 100 | 60–155 | 3/5 | 300 | <20–680 |
SCID, severe combined immunodeficiency. EHMES‐10 cells (3 × 106) were inoculated into the thoracic cavity of SCID mice on day 0. Cisplatin (CDDP) or gemcitabine (GEM) were administered intravenously on day 14 or intraperitoneally on days 14 and 21, respectively. The production of thoracic tumors and pleural effusions was evaluated on day 28. *P < 0.05, Mann–Whitney U‐test.
In contrast, treatment with 175 mg/kg and 350 mg/kg GEM on days 14, 18, 21 and 25 completely inhibited tumors and pleural effusions (data not shown); however, these treatments caused severe bodyweight loss (7 g/2 weeks), suggesting that treatment with this schedule is not feasible. Therefore, we chose 60 mg/kg and 300 mg/kg on days 14 and 21 in set 2. GEM in this treatment protocol did not cause loss of bodyweight; however, it did not inhibit production of thoracic tumors or pleural effusions. These results are consistent with chemotherapy refractory characteristics of MPM in human patients.
Discussion
Experimental animal models are valuable for clarifying the cellular and molecular pathogenesis of malignant diseases and the preclinical evaluation of anticancer agents. Tumor progression is dependent on the properties both of tumor cells and host factors,( 20 ) and the orthotopic environment is suggested to be the most appropriate for disease progression. In fact, orthotopic implantation models have been reported for various cancers, including lung,( 21 ) breast,( 22 ) stomach,( 23 ) colon,( 24 ) renal,( 25 ) pancreas,( 26 ) prostate( 27 ) and bladder( 28 ) cancers, and patient‐like disease progression or metastasis was observed in these models. Thus, the sites of tumor inoculation should be chosen carefully when animal models are established. For MPM, whereas several models with ectopic (peritoneal or subcutaneous) transplantation have been reported previously,( 29 , 30 ) only subcutaneous tumors were available and they did not seem to represent the clinical behavior of MPM. In contrast, Colt et al. reported an orthotopic model by thoracically implanted intact human pleural tumors into athymic nude mice.( 31 ) This model mimicked the clinical behavior of human MPM in terms of pattern of disease progression, but surgical resection of clinical specimens is necessary for experiments and thoracic tumors require a long period (up to 6 months) to develop. In the present study, we established an orthotopic implantation model of MPM in SCID mice using a human MPM cell line, EHMES‐10. EHMES‐10 cells orthotopically inoculated into the thoracic cavity reproducibly develop thoracic tumors and bloody pleural effusions, showing a human patient‐like progression of MPM. This model is technically easy to perform, highly reproducible, and requires only 4 weeks to develop disease. Therefore, it seems to be useful not only for mechanism analyses of disease progression, but also for the screening of anticancer agents.
Malignant pleural mesothelioma needs to be distinguished from metastatic carcinoma, particularly lung adenocarcinoma. This is a very difficult or even impossible task at the H&E level in a biopsy specimen, and sometimes even in a surgical specimen, because of the capacity of some lung adenocarcinoma to grow in a mesothelioma‐like fashion.( 13 ) Recently, immunohistochemical staining for several markers has been reported to be useful for distinguishing epithelial‐type MPM from lung adenocarcinoma. Of these markers, calretinin and cytokeratin are frequently positive and CEA is negative for MPM,( 32 ) although the specificity of diagnosis using these stains is not 100%.( 13 ) Immunohistochemical staining revealed that EHMES‐10 tumors are calretinin negative, cytokeratin negative, mesothelin negative, vimentin positive, EMA negative and CEA negative, and that tumors from MPM patients are calretinin negative, cytokeratin positive, mesothelin positive, vimentin positive, EMA positive and CEA negative (data not shown). There are two ways in which EHMES‐10 cells could have lost markers for MPM: (i) the tumor cells of the MPM patient were heterogeneous, and cells from this population that were negative for MPM markers were established as this cell line; or (ii) the MPM markers were lost from the EHMES‐10 cells during in vitro culture. On the other hand, orthotopically inoculated EHMES‐10 cells (established from a MPM patient) demonstrated classical patterns of spread of advanced MPM, such as lung encasement by tumor rind, pleural‐based masses and bloody pleural effusions.( 13 ) Therefore, EHMES‐10 cells seem to retain the characteristics of MPM in terms of disease progression patterns.
Recent studies suggest that some growth factors play a role in the growth and progression of MPM, such as VEGF, VEGF‐C, epidermal growth factor, platelet‐derived growth factor‐B, insulin‐like growth factor‐1, transforming growth factor‐α and interleukin‐8.( 33 , 34 , 35 , 36 , 37 ) Of these factors, VEGF, a proangiogenic factor, is the best‐studied molecule for MPM. VEGF was originally discovered because of its ability to render venules and small veins hyperpermeable to circulating macromolecules and was therefore initially termed vascular permeability factor.( 38 ) Later, VEGF was isolated and cloned as an endothelial‐specific mitogen. There are two specific receptors, VEGFR‐1 (Flt‐1) and VEGFR‐2 (Flk‐1/KDR), which were reported to be predominantly expressed on endothelial cells.( 15 ) VEGF is thought to play at least three critical roles in MPM. First, VEGF is overexpressed in MPM cells and induces angiogenesis.( 14 , 15 ) In fact, VEGF expression in MPM correlates with intratumoral microvessel density and VEGFR‐2 expression( 39 ) and serum VEGF levels are inversely correlated with survival of MPM patients.( 40 ) Consistent with these findings, EHMES‐10 cells secreted a high level of VEGF protein both in vitro and in vivo (4, 5), and thoracic tumors produced by EHMES‐10 cells were highly vascularized (Fig. 5). Second, VEGF acts as an autocrine growth factor of MPM cells. Recent reports have demonstrated that VEGFR (VEGFR‐1, VEGFR‐2 and VEGFR‐3)( 37 , 41 ) are also expressed in MPM cells (including EHMES‐1 and EHMES‐10 cells [Fig. 4]) and blockade of the VEGFR‐mediated signal suppresses the proliferation of the MPM cells. Third, VEGF facilitates the production of pleural effusions.( 7 ) Malignant pleural effusion frequently develops in patients with MPM. We reported recently that the VEGF levels in pleural effusions were elevated in patients with malignant diseases, including MPM,( 42 ) compared with heart failure or inflammatory lung diseases. We further reported that the VEGF that was overexpressed in the thoracic cavity, but not in the lung parenchyma, induced hyperpermeability of the thoracic cavity and caused malignant pleural effusions in an experimental nude mouse model with PC14PE6 cells overexpressing VEGF.( 7 ) Moreover, we have shown that treatment with an inhibitor of VEGFR‐2 (PTK787) suppresses vascular permeability and reduces the production of pleural effusions developed by PC14PE6 cells.( 43 ) In keeping with this, VEGF‐overexpressing EHMES‐10 cells produced bloody pleural effusions when they were inoculated directly into the thoracic cavity, strongly suggesting that VEGF may be a cause of the pleural effusions produced by EHMES‐10 cells. Collectively, VEGF and its receptors critically regulate the progression of MPM, and therefore inhibition of the VEGF–VEGFR signal is considered to be a promising therapeutic target. Experiments evaluating the therapeutic efficiency of VEGF inhibitors in an orthotopic model with EHMES‐10 cells are ongoing in our department.
Whereas MPM is known to be one of the tumors most refractory to conventional chemotherapy( 1 ) and recent clinical trials show that GEM in combination with CDDP has some benefit in the treatment of MPM patients( 44 ), no single agent has consistently induced a response rate greater than 20%.( 1 ) Although EHMES‐10 cells showed higher sensitivity to GEM compared with a lung adenocarcinoma cell line (PC14PE6) in vitro, in vivo treatment with CDDP or GEM at feasible doses did not significantly reduce production of thoracic tumors of EHMES‐10 cells, and only CDDP inhibited production of pleural effusions, resembling chemotherapy refractory characteristics of MPM in patients. More recently, Alimta (pemetrexed), a structurally novel antifolate, has been reported to be clinically beneficial for MPM patients in terms of survival prolongation.( 45 ) Thus, we are going to evaluate effect of Alimta in combination with conventional chemotherapy or molecular targeted drugs, such as VEGFR inhibitors.
In conclusion, we established an orthotopic implantation SCID mouse model of human MPM using EHMES‐10 cells overexpressing VEGF and its receptor. This model shows patient‐like disease progression, such as tumors growing locally in the thoracic cavity, pleural dissemination and malignant pleural effusions. Therefore, this model may be useful for examining the molecular pathogenesis of MPM and may contribute to the development of novel treatment strategies.
Acknowledgments
This work was supported by Grants‐in‐Aid for Cancer Research from the Ministry of Education, Science, Sports and Culture of Japan.
References
- 1. Light RW. Pleural Diseases, 4th edn. Philadelphia: Lippincott, Williams and Wilkins, 2001. [Google Scholar]
- 2. Broaddus VC. Asbestos, the mesothelial cell and malignancy: a matter of life or death. Am J Respir Cell Mol Biol 1997; 17: 657–9. [DOI] [PubMed] [Google Scholar]
- 3. Morinaga K, Kishimoto T, Sakatani M, Akira M, Yokoyama K, Sera Y. Asbestos‐related lung cancer and mesothelioma in Japan. Industrial Health 2001; 39: 65–74. [DOI] [PubMed] [Google Scholar]
- 4. Mineral fiber content of lungs in patients with mesothelioma seeking compensation in Quebec. Am J Respir Crit Care Med 1996; 153: 711–18. [DOI] [PubMed] [Google Scholar]
- 5. Hoffman RM. Patient‐like models of human cancer in mice. A review and critique of their development. Curr Perspect Mol Cell Oncol 1992; 1: 311–29. [Google Scholar]
- 6. Yokoyama A, Kohno N, Fujino S et al. Origin of heterogeneity of interleukin‐6 (IL‐6) levels in malignant pleural effusions. Oncol Rep 1994; 1: 507–11. [DOI] [PubMed] [Google Scholar]
- 7. Yano S, Shinohara H, Herbst RS et al. Production of experimental malignant pleural effusions is dependent on invasion of the pleura and expression of vascular endothelial growth factor/vascular permeability factor by human lung cancer cells. Am J Pathol 2000; 157: 1893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Green LM, Reade JL, Ware CF. Rapid colometric assay for cell viability: application to the quantitation of cytotoxic and growth inhibitory lymphokines. J Immunol Meth 1984; 70: 257–68. [DOI] [PubMed] [Google Scholar]
- 9. Yano S, Shinohara H, Herbst RS et al. Expression of vascular endothelial growth factor is necessary but not sufficient for production and growth of brain metastasis. Cancer Res 2000; 60: 4959–67. [PubMed] [Google Scholar]
- 10. Dias S, Hattori K, Zhu Z et al. Autocrine stimulation of VEGFR‐2 activates human leukemic cell growth and migration. J Clin Invest 2000; 106: 511–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shushanov S, Bronstein M, Adelaide J et al. VEGFc and VEGFR3 expression in human thyroid pathologies. Int J Cancer 2000; 86: 47–52. [DOI] [PubMed] [Google Scholar]
- 12. Yano S, Shinohara H, Herbst RS et al. Expression of vascular endothelial growth factor is necessary but not sufficient for production and growth of brain metastasis. Cancer Res 2000; 60: 4959–67. [PubMed] [Google Scholar]
- 13. Rosai J. Rosao and Ackerman's Surgical Pathology, 9th edn. New York: Mosby, 2004. [Google Scholar]
- 14. Ferrara N. The role of vascular endothelial growth factor in the regulation of blood vessel growth. In: Bicknell R, Lewis CE, Ferrara N, eds. Tumor Angiogenesis. New York: Oxford University Press, Inc., 1997; 185–99. [Google Scholar]
- 15. Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol 2005; 23: 1011–27. [DOI] [PubMed] [Google Scholar]
- 16. Shibuya M, Yamaguchi S, Yamane A et al. Nucleotide sequence and expression of a novel human receptor‐type tyrosine kinase gene (flt) closely related to the fms family. Oncogene 1990; 5: 519–24. [PubMed] [Google Scholar]
- 17. Terman BI, Dougher‐Vermazen M, Carrion ME et al. Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem Biophys Res Commun 1992; 187: 1579–86. [DOI] [PubMed] [Google Scholar]
- 18. Matthews W, Jordan CT, Gavin M, Jenkins NA, Copeland NG, Lemischka IR. A receptor tyrosine kinase cDNA isolated from a population of enriched primitive hematopoietic cells and exhibiting close genetic linkage to c‐kit. Proc Natl Acad Sci USA 1991; 88: 9026–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Byrne MJ, Davidson JA, Musk AW et al. Cisplatin and gemcitabine treatment for malignant mesothelioma: a phase II study. J Clin Oncol 1999; 17: 25–30. [DOI] [PubMed] [Google Scholar]
- 20. Fidler IJ. Critical determinants of metastasis. Semin Cancer Biol 2002; 12: 89–96. [DOI] [PubMed] [Google Scholar]
- 21. Miyoshi T, Kondo K, Ishikura H, Kinoshita H, Matsumori Y, Monden Y. SCID mouse lymphogenous metastatic model of human lung cancer constructed using orthotopic inoculation of cancer cells. Anticancer Res 2000; 20: 161–3. [PubMed] [Google Scholar]
- 22. Fu X, Le P, Hoffman RM. A metastatic orthotopic‐transplant nude‐mouse model of human patient breast cancer. Anticancer Res 1993; 13: 901–4. [PubMed] [Google Scholar]
- 23. Furukawa T, Fu X, Kubota T, Watanabe M, Kitajima M, Hoffman RM. Nude mouse metastatic models of human stomach cancer constructed using orthotopic implantation of histologically intact tissue. Cancer Res 1993; 53: 1204–8. [PubMed] [Google Scholar]
- 24. Morikawa K, Walker SM, Nakajima M, Pathak S, Jessup JM, Fidler IJ. Influence of organ environment on the growth, selection, and metastasis of human colon carcinoma cells in nude mice. Cancer Res 1988; 48: 6863–71. [PubMed] [Google Scholar]
- 25. Naito S, von Eschenbach AC, Giavazzi R, Fidler IJ. Growth and metastasis of tumor cells isolated from a human renal cell carcinoma implanted into different organs of nude mice. Cancer Res 1986; 46: 4109–15. [PubMed] [Google Scholar]
- 26. Fu X, Guadagni F, Hoffman RM. A metastatic nude‐mouse model of human pancreatic cancer constructed orthotopically with histologically intact patient specimens. Proc Natl Acad Sci USA 1992; 89: 5645–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stephenson RA, Dinney CP, Gohji K, Ordonez NG, Killion JJ, Fidler IJ. Metastatic model for human prostate cancer using orthotopic implantation in nude mice. J Natl Cancer Inst 1992; 84: 951–7. [DOI] [PubMed] [Google Scholar]
- 28. Slaton JW, Perrotte P, Inoue K, Dinney CP, Fidler IJ. Interferon‐alpha‐mediated down‐regulation of angiogenesis‐related genes and therapy of bladder cancer are dependent on optimization of biological dose and schedule. Clin Cancer Res 1999; 5: 2726–34. [PubMed] [Google Scholar]
- 29. Lee JM, Bruckner HW, Szrajer L, Brenne U, Schindelheim G, Andreotti PE. Taxol inhibits growth of mesothelioma xenografts. Anticancer Res 1995; 15: 693–6. [PubMed] [Google Scholar]
- 30. Smythe WR, Hwang HC, Elshami AA et al. Treatment of experimental human mesothelioma using adenovirus transfer of the herpes simplex thymidine kinase gene. Ann Surg 1995; 222: 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Colt HG, Astoul P, Wang X, Yi ES, Boutin C, Hoffman RM. Clinical course of human epithelial‐type malignant pleural mesothelioma replicated in an orthotopic‐transplant nude mouse model. Anticancer Res 1996; 16: 633–9. [PubMed] [Google Scholar]
- 32. Ordonez NG. The immunohistochemical diagnosis of mesothelioma: a comparative study of epithelioid mesothelioma and lung adenocarcinoma. Am J Surg Pathol 2003; 27: 1031–51. [DOI] [PubMed] [Google Scholar]
- 33. Versnel MA, Claesson‐Welsh L, Hammacher A et al. Human malignant mesothelioma cell lines express PDGF beta‐receptors whereas cultured normal mesothelial cells express predominantly PDGF alpha‐receptors. Oncogene 1991; 6: 2005–11. [PubMed] [Google Scholar]
- 34. Lee TC, Zhang Y, Aston C et al. Normal human mesothelial cells and mesothelioma cell lines express insulin‐like growth factor I and associated molecules. Cancer Res 1993; 53: 2858–64. [PubMed] [Google Scholar]
- 35. Morocz IA, Schmitter D, Lauber B, Stahel RA. Autocrine stimulation of a human lung mesothelioma cell line is mediated through the transforming growth factor alpha/epidermal growth factor receptor mitogenic pathway. Br J Cancer 1994; 70: 850–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Galffy G, Mohammed KA, Dowling PA, Nasreen N, Ward MJ, Antony VB. Interleukin 8: an autocrine growth factor for malignant mesothelioma. Cancer Res 1999; 59: 367–71. [PubMed] [Google Scholar]
- 37. Ohta Y, Shridhar V, Bright RK et al. VEGF and VEGF type C play an important role in angiogenesis and lymphangiogenesis in human malignant mesothelioma tumours. Br J Cancer 1999; 81: 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983; 219: 983–5. [DOI] [PubMed] [Google Scholar]
- 39. Konig JE, Tolnay E, Wiethege T, Muller KM. Thrombospondin‐1 expression and clinical implications in malignant pleural mesothelioma. Cancer 1999; 85: 2570–6. [PubMed] [Google Scholar]
- 40. Strizzi L, Catalano A, Vianale G et al. Vascular endothelial growth factor is an autocrine growth factor in human malignant mesothelioma. J Pathol 2001; 193: 468–75. [DOI] [PubMed] [Google Scholar]
- 41. Konig J, Tolnay E, Wiethege T, Muller K. Co‐expression of vascular endothelial growth factor and its receptor flt‐1 in malignant pleural mesothelioma. Respiration 2000; 67: 36–40. [DOI] [PubMed] [Google Scholar]
- 42. Zebrowski BK, Yano S, Liu W et al. Vascular endothelial growth factor levels and induction of permeability in malignant pleural effusions. Clin Cancer Res 1999; 5: 3364–8. [PubMed] [Google Scholar]
- 43. Yano S, Herbst RS, Shinohara H et al. Treatment for malignant pleural effusion of human lung adenocarcinoma by inhibition of vascular endothelial growth factor receptor tyrosine kinase phosphorylation. Clin Cancer Res 2000; 6: 957–65. [PubMed] [Google Scholar]
- 44. Byrne MJ, Davidson JA, Musk AW et al. Cisplatin and gemcitabine treatment for malignant mesothelioma: a phase II study. J Clin Oncol 1999; 17: 25–30. [DOI] [PubMed] [Google Scholar]
- 45. Hazarika M, White RM Jr, Booth BP et al. Pemetrexed in malignant pleural mesothelioma. Clin Cancer Res 2005, 2005; 11: 982–92. [PubMed] [Google Scholar]