

Abstract
Autophagy, a cellular degradation system has been demonstrated in some hematopoietic malignant cell lines, but there is much still remaining to be known about its role and the mechanisms. We observed the excessive autophagy in chronic myelogenous leukemia (CML) cell line, K562, associated with treatment of 12‐O‐tetradecanoyl‐phorbol‐13‐acetate (TPA), which can induce K562 cells to differentiate into megakaryocytic lineage. Confocal microscopic analysis demonstrated that autophagic cells did not express a megakaryocyte marker, the CD41 molecule, indicating that the autophagy was independent of megakaryocytic differentiation. After remarkable autophagic degradation, the cells finally underwent autophagic cell death (APCD). On the other hand, a block of TPA‐induced autophagy by chloroquine rapidly promoted cell death that was not APCD. This result suggested that autophagy regulated two mechanisms in K562 cells: both the cell survival system and APCD. To confirm that autophagy regulates the cell survival system in K562 cells, imatinib was used to induce cell death in K562 cells. Autophagy has not been considered during imatinib treatment; nonetheless, co‐treatment with imatinib and chloroquine markedly enhanced imatinib‐induced cell death, compared to K562 cells treated only with imatinib. Furthermore, imatinib‐resistant cell lines, BaF3/T315I and BaF3/E255K, also underwent cell death by co‐treatment with imatinib and chloroquine. From these data, we concluded that autophagy is deeply related to the cell survival system and that inhibition of autophagy accelerates TPA‐ or imatinib‐induced cell death. The block of autophagy could be a new strategy in the treatment of CML. (Cancer Sci 2008; 99: 2200–2208)
Autophagy has been demonstrated to be a self‐proteolysis system that survives under nutrient‐deprived conditions in yeast.( 1 , 2 ) First, the isolation of membrane expands and autophagic vacuoles that have double membrane are formed. Second, cytoplasmic organelle is isolated in the vacuoles. Third, autophagic vacuoles fuse with lysosomes and start degradation of contents.
Autophagic degradation system is seen not only in yeast but also in eukaryotic cells such as insects, embryonic cells and human malignant cells; therefore, the mechanisms and signal transductions have been studied. Moreover, recent studies have demonstrated the various roles of autophagy, outside the cell survival system; for example, development, differentiation, tumorigenesis and cell death. Inhibition of autophagy induces abnormal growth and disease including cancer, muscular disorders, neurodegeneration, pathogenesis and aging.( 3 , 4 ) In the human embryo, the degradation of cellular proteins proceeds through an autophagosome–lysosomal pathway, and autophagolysosomal proteolysis is important in normal growth control of cells and defense against the proliferation of tumor cells.( 5 )
On the other hand, autophagy appears to play an important role in the cell death that is independent of apoptosis. Autophagic cell death (APCD) has been investigated as type II programmed cell death in recent years.( 6 , 7 ) The appearance of APCD is differentiated from that of apoptosis by the presence of autophagosomes and autophagolysosomes in the dying cells and is characterized by the preservation of nucleus until the last phase of cell death.( 8 , 9 ) APCD is complementarily associated with caspase activation and the bcl‐2 family. Caspase plays a central role for the mitochondrial apoptosis pathway, involving bcl‐2 family members, cytochrome c release, and the formation and activation of the apoptosome.( 3 ) Caspase‐8‐inhibition inhibits apoptosis and induces APCD, whereas the inhibitions of autophagy gene ATG7 and ATG6 by RNA interference‐inhibited Z‐VAD‐induced APCD, which is a potent inhibitor of caspase‐8.( 10 ) Similarly, embryonic fibroblasts that lack the function bcl‐2 family members are resistant to apoptosis, but die by APCD requiring ATG5 and ATG6 function.( 11 ) Bcl‐2 downregulation causes autophagy in a caspase‐independent manner in a human leukemic cell line, HL60. HL60 cells transfected with the antisense of the bcl‐2 gene experienced regulated cell growth and underwent cell death by autophagy.( 12 )
K562 is a chronic myelogenous leukemia (CML) cell line and has the ability of both erythroid and megakaryocytic differentiation. Co‐treatment with hemin and anthracyclin leads to terminal erythroid maturation. The cytoplasmic organelles, such as mitochondria, endoplasmic reticulum, ribosomes and Golgi apparatus as well as proteins are digested during maturation to red blood cells in the autophagic vacuoles. It is thought that autophagic degradation system plays a role in terminal differentiation of erythroid cells.( 13 ) In this process, although the number of mitochondria decreased during differentiation, the released cytochrome c into the cytoplasm was not detected. Mitochondrias were sequestered into autophagic structures and digested in the closed compartment. The digestion of mitochondria in autolysosomes may allow the cells to escape from rapid apoptotic cell death through concomitant removal of mitochondrial death‐promoting factors such as cytochrome c.( 13 , 14 )
The roles of autophagy in hematopoietic cells have been demonstrated, but much remains to be learned. The relationship between autophagy and megakaryocytic differentiation is not yet well known, compared to that with erythroid differentiation. Additionally, the appearance of autophagy in the process of cell death of CML cells has not been well studied. Herein, we investigate some of the mechanisms of autophagy in megakaryocytic differentiation and cell death using K562 cells. This report indicates a new concept of autophagy in CML cells and begins to clarify of the mechanisms of autophagy in CML cells, which could lead to successful clinical treatment.
Materials and Methods
Cell cultures and transfection. K562 cell lines were obtained from ATCC; BaF3/wild‐type cells, BaF3/E255K cells and BaF3/T315I cells were kindly provided by Shinya Kimura (Kyoto University, Kyoto, Japan). A mitogen‐activated protein kinase kinase (MAPKK) dominant negative mutant, MKK1 K97A gene, was kindly provided by Dr Yukiko Gotoh (Tokyo University, Tokyo, Japan). To regulate the expression of the transfected gene, we used Tet‐on gene expression systems (Clontech). The pTet on/K562 cell lines developed from wild‐type K562 cells by transfection with pTet‐on plasmid using lipofection. The vectors of pTRE2hyg‐MAPKK K97A were transfected to pTet on/K562 cells and were maintained in 300 µg/mL geneticine and 100 µg/mL hygromycin. We screened the cells expressing MKK1 K97A genes with or without 1 µg/mL doxycycline.
Chemicals. Phorbor ester (12‐O‐tetradecanoyl‐phorbol‐13‐acetate [TPA]) and chloroquine diphosphate salt were obtained from Sigma. U0126 and N‐acetyl‐s‐farnesyl‐L‐cysteine (AFC) were obtained from Biomol. Imatinib was obtained from LKT Laboratories.
Cell death and cell cycle assay. We performed transferase deoxytidyl uridine end labeling (TUNEL) assay with an in situ cell death detection kit (Roche) according to manufacturer's protocol. These treated cells were analyzed by FACScan (Becton Dickinson). For cell cycle assay, we used cycle Test Plus (Becton Dickinson) for cell cycle assay under the manufacturer's protocol and analyzed by FACScan.
Western blotting. After incubation with TPA with or without inhibitors, the cells were homogenized in lysis sodium dodecylsulfate (SDS) sample buffer. Amounts of 30 µg of proteins were separated in SDS polyacrylamide gel electrophoresis (PAGE). The proteins on the gel were transferred to a polyvinylidene fluoride membrane (Bio‐rad). The membranes were blocked by casein and were incubated with anti ERK1/2 antibody or anti‐active MAPK (Promega). The proteins on the membrane were detected by ECL‐Plus systems (Amersham Pharmacia Biotech). Antibody for LC3 was kindly provided by Dr Masahiro Shibata (Osaka University, Osaka, Japan).
Electron microscopy. Cultured cells treated with or without TPA were washed in phosphate‐buffered saline (PBS) three times and then centrifuged into 1.5‐mL microcentrifuge tubes. Pelleted cells were fixed in 2% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) containing 5% sucrose for 3 h at 22°C and then washed in the same buffer. They were postfixed in 2% osmium and 1.6% potassium ferrocyanide in 100 mM cacodylate buffer (pH 7.4) for 1 h at 4°C, dehydrated in ethanol, and then embedded in Epon resin. Ultrathin sections were cut, stained with lead citrate and uranyl acetate, and subsequently examined with a Hitachi H‐7500 transmission electron microscope.
Acid phosphatase cytochemistry. The cells treated with or without TPA were washed in PBS three times and then fixed in 2% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) containing 5% sucrose for 5 min at 4°C. They were then layered over culture dishes (35‐mm diameter) coated with 0.2% poly L‐lysine, and fixed for an additional 25 min at 4°C. After fixation, cells were washed in the same buffer three times. The cells were incubated in a cerium‐containing reaction medium for detection of acid phosphatase for 1 h at 37°C with constant agitation. This medium contained 100 mM sodium acetate buffer (pH 5.0), 2 mM cerium chloride, 2 mM‐glycerophosphate, 0.0001% Tx‐100 and 5% sucrose. Control cytochemical incubations consisted of omission of substrate from the reaction mixture and inclusion of NaF (10 mM) in the complete reaction mixture. Following the cytochemical reaction, the cells were postfixed, dehydrated and then embedded in Epon as described above.
Monodansylcadaverine assay. After incubation with TPA, the cells were treated with 50 µM monodansylcadaverine (MDC) (Sigma) for 30 min. After washing, the cells were fixed by 4% paraformaldehyde for 15 min and then we measured the fluorescence of MDC by a fluorescent microplate reader. After fixing and washing MDC‐treated cells, we adjusted the cells at the concentration of 1.0 × 105 cells/100 µL in each well and measured the fluorescence intensity by a 355‐nm excitation filter and a 538‐nm emission filter.
Double staining of immunofluorescence with MDC staining and anti‐CD41. For double staining of immunofluorescence for CD41a with MDC, we used a Zenon Mouse IgG Labeling Kit (Invitrogen). We diluted antibody for human CD41a (BD Pharmingen) in PBS buffer to 1:10 and added Alexa Fluor 647 (dilution, 1:5). After 5 min, we mixed with blocking reagent. Then, MDC diluted to 1:1000 was added to the mixture. The mixtures were incubated with K562 cells treated with TPA or nocodazole for 5 or 7 days. The cells were examined by laser confocal microscopy.
Double staining of immunofluorescence with MDC staining and anti‐LC3. After 72 h of incubation with TPA, we added 50 µM MDC for 30 min. Then, the cells were attached to preparations by cytospin and fixed by 4% paraformaldehyde. After permeabilization and blocking, the cytospin specimens were labeled with LC3‐Zenon IgG labeling complex (prepared as CD41a‐Zenon IgG complex) for 60 min. We observed the cells using laser confocal microscopy.
Results
Autophagy appeared during megakaryocytic differentiation in K562 cells. A phorbor ester, TPA has been known to induce megakaryocytic differentiation in K562 cells. However, few megakaryocyte appeared, and most cells exhibited vacuoles and degradation of membranes. After TPA treatment, cytoplasmic blebs appeared in K562 cells for a few minutes. These blebs disappeared from the cytoplasm at 24 h and the cells were aggregated, with many vacuoles appearing in the cytoplasm after 48 h. Then, the nuclei were localized at one side of the cytoplasm, and the volume of vacuoles in the cytoplasm was increased. Finally, the cytoplasmic membranes were degraded, and the nuclei were segmented (Fig. 1a). These phenomena could be distinguished from apoptosis, which is characterized as nuclear degradation at an early phase. Expressions of one of the autophagy markers, LC3, which is a mammalian homolog of yeast Atg8 (Aut7/Apg8) product,( 15 ) was increased by the treatment of TPA (Fig. 1b). These data suggested autophagy and APCD occurred in TPA‐treated K562 cells. Cell cycle analysis demonstrated that TPA induced cell cycle arrest in the G0/G1 phase. The cells in the G0/G1 phase were 48.7% before treatment with TPA; the cells accumulated in the G0/G1 phase reached 67.8% at 48 h after TPA‐treatment. The cells in the G2/M phase were 30.1% and 25.8% before and after TPA treatment, respectively (Fig. 1c). This data suggested that autophagy might occur in the cells accumulated in the G0/G1 phase after TPA treatment.
Figure 1.
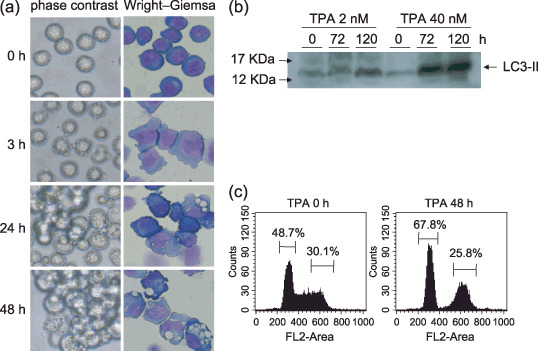
Induction of autophagy by 12‐O‐tetradecanoyl‐phorbol‐13‐acetate (TPA). (a) K562 cells were incubated with TPA (2–40 nM) at 37°C, and morphological change was observed at the indicated times with contrasted phases by microscopy (left columns). The cytospin specimens were prepared and Wright–Giemsa staining was performed (right columns). (Original magnification, ×400.) (b) After treatment of TPA for indicated times, western blotting was performed with anti‐LC3 antibody. (c) The K562 cells seeded at 2 × 105 cells/mL and TPA (40 nM) was added. After 48 h, the cell cycle analysis was performed. The data represents average of triplicate experiments.
Electron microscopy exhibited autophagy in TPA‐treated K562 cells. Electron microscopy demonstrated that cells untreated with TPA showed few, if any, autophagosome‐like structure (Fig. 2a,b). On the other hand, the TPA‐treated cells had many autophagosomes with a double membrane structure that contained degradative organelles and cytoplasmic components (Fig. 2c,d). Acid phosphatase is one of the marker enzymes for lysosomes; cytochemistry for acid phosphatase revealed that some autophagosomes fused with lysosomes and subsequently became autophagolysosomes (Fig. 2e,f). Hydrolytic enzymes (e.g. acid phosphatase) were supplied to the autophagosome, and the membrane organelles and cytoplasm‐derived contents were degraded. The ultrastructure shown by the electron microscopy demonstrated that TPA induced autophagic degradation in K562 cells.
Figure 2.
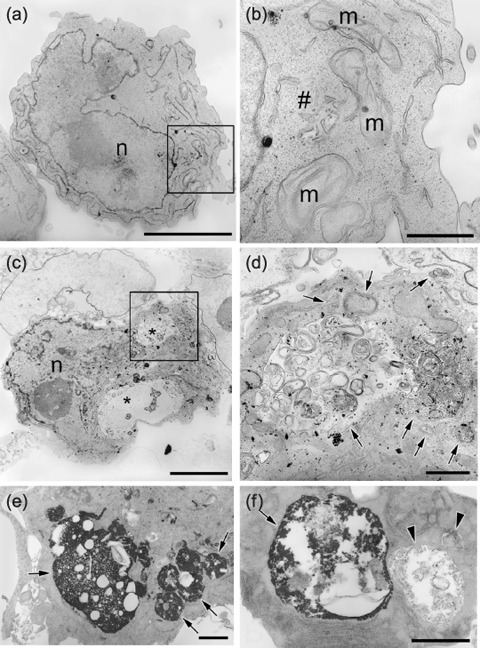
Ultrastructural feature of 12‐O‐tetradecanoyl‐phorbol‐13‐acetate (TPA)‐treated K562 cells. K562 cells treated with or without TPA (4 nM) for 48 h were harvest and fixed; electron microscopic observation was performed. (a) TPA‐untreated cells lack autophagosome‐like structure. The nucleus is evident (n). (b) Higher magnification of a portion of the section shown in panel (a) square. Mitochondria (m) and Golgi (#) are evident. (c) Cells treated with TPA have many autophagosomes (*). The nucleus is evident (n). (d) Higher magnification of a portion of the section shown in panel (c) square. In autophagosomes, degradative organelles and cytoplasmic components are shown (arrows). Scale lines: (a,c) = 5 µm; (b,d) = 1 µm. (e,f) Acid phosphatase cytochemistry. The cells treated with TPA were fixed and incubated in a cerium‐containing reaction medium for detection of acid phosphatase for 1 h at 37°C with constant agitation. (e) Electron‐dense deposits of cerium phosphate indicating acid phosphatase are present in autophagolysosomes (arrows). In autophagolysosomes, degradative organelles are evident (arrows). (f) Two autophagosomes prior (the right) or after (the left) to fusion with lysosomes. The autophagosome before fused with lysosomes lacks electron dense deposits. Scale lines = 1 µm.
Autophagy did not occur in the CD41‐expressed cells. The autofluorescent agent MDC is considered to accumulate in the acidic compartment that is constructed by fusion of lysosomes with autophagosomes,( 16 , 17 ) although MDC is not specific marker of autophagy. TPA‐treated K562 cells showed much accumulation of MDC in the cytoplasmic vacuoles (Fig. 3a, left middle) and MDC‐positive cells were very few in the untreated cells (data not shown). LC3 is known to be associated with the completed autophagosomes, and it can be recognized as a marker specific for autophagy.( 18 , 19 ) We performed double staining of LC3 antibody with MDC. The confocal microscopy demonstrated that LC3 expressed around the staining of MDC in the TPA treated K562 cells (Fig. 3a). Neither LC3 nor MDC did not express in the untreated K562 cells (data not shown). These data indicated that MDC accumulated acidic compartments of autophagosomes during TPA treatment. To quantify autophagosome formation, the fluorescent intensity of intracellular MDC was measured by fluorescent microplate reader at 355 and 538 nm wavelengths. The fluorescent intensity of MDC in the TPA‐treated K562 cells increased dependent on time (Fig. 3b, left). The data obtained from the microplate reader were consistent with percentages of MDC‐positive cells under fluorescent microscopy (Fig. 3b. middle and right). The peak of MDC‐positive cells matched that of LC3 as detected by western blotting in these cells (Fig. 1b). From these data, we concluded that the accumulation of MDC was consistent with the appearance of autophagy in TPA treatment. To investigate whether it is necessary for autophagy to appear during megakaryocytic differentiation, we performed double staining with MDC and anti‐CD41 antibody, one of the specific markers for megakaryocyte. The TPA‐treated cells showed accumulation of MDC at 3 days, and the expression of CD41 was observed at 5 or 7 days. Interestingly, there were few cells that were double‐positive of both MDC and CD41 (Fig. 3c, left). Next, K562 cells were cultured with nocodazole (50 ng/mL), which is known to be another inducer of polyploidization and megakaryocytic differentiation, for 5–7 days. Double staining was performed in these treated cells. In the treatment with nocodazole, the cells exhibited only huge shapes and expressed the intensity of neither MDC nor CD41 (Fig. 3c, middle). On the other hand, after treatment with nocodazole for 2 days, we replaced the medium containing TPA and cultured for an additional 3–5 days. Confocal microscopic analysis demonstrated that the cells expressed CD41 after 7 days; nonetheless, the MDC‐positive cells decreased (Fig. 3c, right). Autophagy did not appear during TPA‐induced terminal maturation for megakaryocyte after polyploidization by nocodazole. These data suggested that degradation of cytoplasmic component by autophagy is not necessary for megakaryocytic differentiation.
Figure 3.
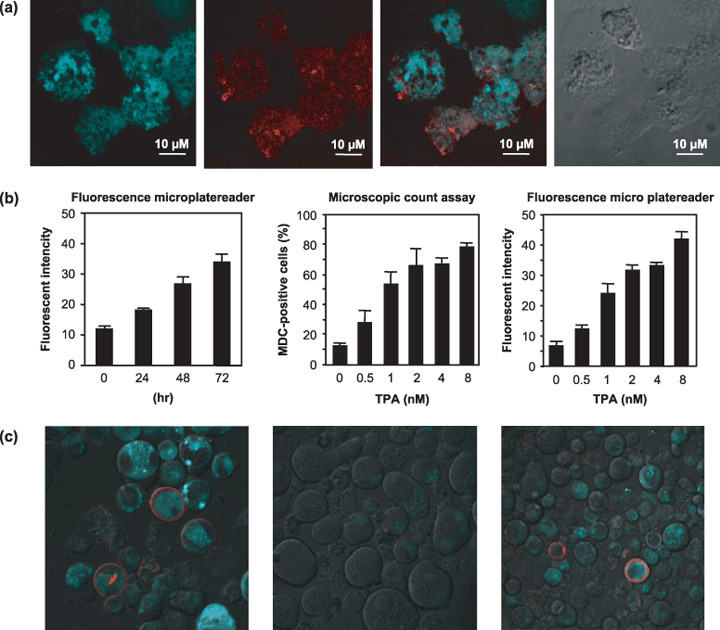
(a) K562 cells were treated with 12‐O‐tetradecanoyl‐phorbol‐13‐acetate (TPA) and double staining of LC3 antibody‐Zenon IgG complex and monodansylcadaverine (MDC) was performed using a Zenon‐IgG labeling kit. The cells examined by confocal microscopy. The MDC staining (left), LC3 staining (middle left), Marge (middle right), Nomarski Interference Contrast (right). (b) The percentage of MDC‐positive cells was determined by counting 200 cells under the fluorescent microscope. The fluorescent intensity of MDC‐positive cells were measured through a 355‐nm excitation filter and a 538‐nm emission filter with microplate reader. The cells were adjusted at the concentration of 1.0 × 105 cells/100 µL in each well before measuring the fluorescent intensity. We cultured K562 cells with TPA (40 µM) or nocodazole (50 ng/mL) for 5 or 7 days, and with for TPA 5 or 7 days after 2 days of treatment with nocodazole. The cells collected and treated with MDC staining and anti‐CD41 as described in Material and Methods. The data represents means of triplicate experiments and are shown as means ± standard deviation (n = 3). (c) Then, we analyzed by confocal microscopy. Left, the K562 cells treated with TPA for 7 days. Middle, the cells treated for 7 days with nocodazole. Right, the cells treated for 5 days with nocodazole and an additional 2 days with TPA.
Autophagy induced by TPA is dependent on activation of MAPK. TPA induces activation of Ras/MAPK that is required for megakaryocytic differentiation in K562 cells. Most CML cells, such as K562 cells have fusion proteins, BCR‐ABL. BCR‐ABL‐tyrosine kinase is continuously activated, and the signals from tyrosine kinase activate Ras and downstream signaling proteins belonging to the MAPK pathways.( 20 ) In our assay, western blotting analysis revealed that MAPK was strongly activated 1 h after treatment with TPA and continued for 24 h or more (Fig. 4a). K562 cells were treated with TPA in the presence or absence of a Ras inhibitor, N‐acetyl‐S‐farnesyl‐L‐cysteine (AFC; 50 µM). MDC assay showed that AFC inhibited TPA‐induced autophagy (Fig. 4b) and MAPK inhibitor, U0126, significantly inhibited autophagy in K562 cells (Fig. 4c). Furthermore, dominant negative MAPKK mutant K97A (K562/K97A) was transfected to K562 cells under a Tet‐on system. In this system, addition of doxycycline (DOX) led to dominant negative MAPKK expression, and then phosphorylation of endogenous MAPK was suppressed. The cells were incubated in the presence or absence of DOX for 24 h, and TPA was added to the cell culture. TPA‐induced activation of MAPK was inhibited in the K562/K97A cells compared to that in wild‐type K562 cells (Fig. 4d). TPA‐induced autophagy was decreased in K562/K97A cells in the presence of DOX compared to those in the absence of DOX (Fig. 4e). These results strongly suggested that TPA‐induced autophagy was mediated by the Ras/MAPK pathway.
Figure 4.
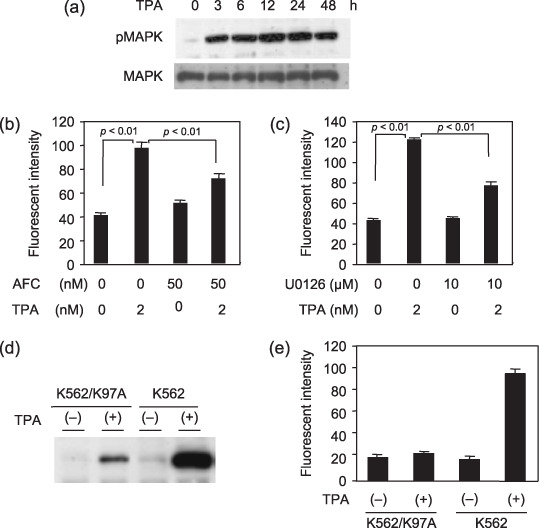
12‐O‐tetradecanoyl‐phorbol‐13‐acetate (TPA)‐induced autophagy is dependent on activation of mitogen‐activated protein kinase (MAPK). (a) The expressions of total MAPK and phosphorylated MAPK (pMAPK)in K562 cells during treatment with TPA. (b,c) K562 cells were treated with TPA for 48 h with each inhibitor, and monodansylcadaverine (MDC) assay was performed. The data represents means of triplicate experiments and are shown as means ± standard deviation (n = 3). (b) Ras inhibitor N‐acetyl‐s‐farnesyl‐L‐cysteine (AFC) (50 nM) (c) MAPK inhibitor U0126 (10 µM) Two‐sided Student's t‐test was performed to determine statistical significance. (d) The immunoblotting of pMAPK in transfectant cell, K562/K97A and K562 cells after treatment with TPA. The K562/K97A cells was transfectant under Tet on system. The pTet on/K562 cells were derived from wild‐type K562 cells by transfection with pTet vector. The vectors of pTRE2hyg‐MAPKK‐K97A were transfected to pTet on/K562 cells and were maintained with 300 µg/mL G418 and 100 µg/mL hygromycin. After 24 h incubation in the presence of DOX, the cells were treated with TPA for 48 h (e) MDC assay were performed on the K562/K97A cells and K562 cells, as described above. The data represents means of triplicate experiments and are shown as means ± standard deviation (n = 3).
Inhibition of autophagy accelerated non‐autophagic cell death in K562 cells. Chloroquine blocks the fusion of autophagosome and lysosome, and then inhibits autophagosomal degradation.( 19 , 21 ) Boya et al. described that hydoroxychloroquine induced an increase in autophagic vacuoles, it inhibited the progression of the autophagic process, on the other hand, hydroxychloroquine reduced turnover of long‐lived proteins.( 22 ) To investigate the role of autophagy during megakaryocytic differentiation, K562 cells were treated with TPA and chloroquine diphosphate salt. Interestingly, most cells co‐treated cells with TPA and chloroquine diphosphate salt underwent cell death in the early phase within 48 h which is non‐autophagic cell death. There was few viable cells at 5–7 days (data not shown). From this result, we concluded that autophagy might be a cell survival system in K562 cells. Then, to investigate whether the autophagic cell survival system was activated during cell death of K562 cells, we focused on imatinib treatment. Imatinib mesilate is a tyrosine kinase inhibitor and clinically effective drug for CML. We treated K562 cells by imatinib with or without chloroquine diphosphate salt. After 48 h of treatment, although no remarkable change was seen in only chloroquine‐ or imatinib‐treated cells under a Trypan blue exclusion test, co‐treatment with imatinib and chloroquine diphosphate salt remarkably inhibited the proliferation of K562 cells, even at a low dose of imatinib (Fig. 5a). Equally, TUNEL assay demonstrated the acceleration of apoptotic cell death under co‐treatment with imatinib and chloroquine diphosphate salt compared to only imatinib‐treated cells after 48 h of treatment (Fig. 5b). These results indicated that the inhibition of autophagy induced acceleration of imatinib‐induced cell death.
Figure 5.
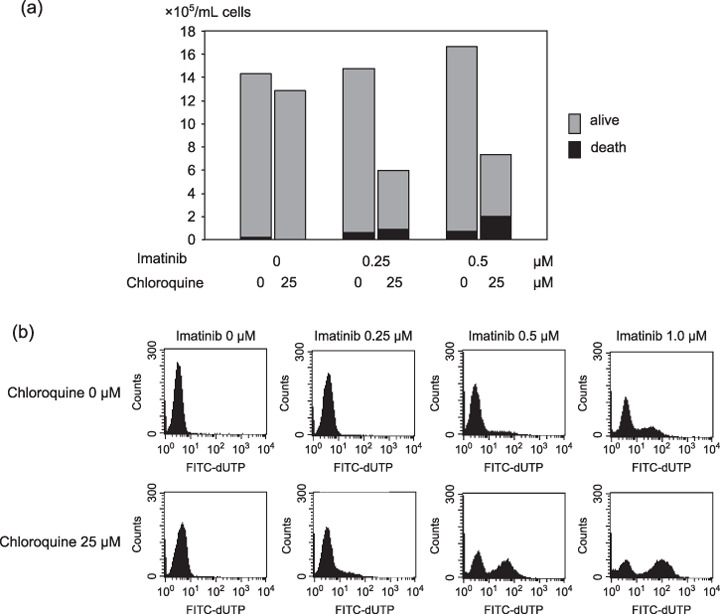
(a) Trypan blue count of K562 cells treated by imatinib (0–0.5 µM) with or without chloroquine diphosphate salt (25 µM) for 48 h. Gray bar indicates total count and black bar is dead cell count. The data represents average of triplicate experiments. (b) Transferase deoxytidyl uridine end labeling (TUNEL) assay of K562 cells after treatment of imatinib (0–5 µM) with or without chloroquine diphosphate salt (25 µM) for 48 h. The data represents average of triplicate experiments.
Inhibition of autophagy accelerates imatinib‐induced cell death of imatinib‐resistant cells. To further analyze the relationship between the cell survival system during imatinib treatment and autophagy, we used and imatinib‐sensitive cell line, BaF3/wild type, that was transfected with the wild type of the BCR‐ABL gene to a mouse lymphoid cell line, BaF3, and imatinib‐resistant cell lines, BaF3/E255K and BaF3/T315I, that were transfected with mutants of the BCR‐ABL gene. E255K and T315I are popular point mutations of the BCR‐ABL gene after imatinib treatment, and both types show remarkable resistance to imatinib.( 23 ) Especially, T315I also resists other novel agents such as dasatinib and nilotinib.( 24 , 25 ) We performed a Trypan blue exclusion test and TUNEL assay after 3 days of treatment with imatinib with or without chloroquine diphosphate salt. In BaF3/wild‐type cells, chloroquine diphosphate salt inhibited cell growth and accelerated cell death in imatinib treatment (Fig. 6a). Furthermore, although a lower dose of imatinib (0.5 µM) did not induce cell death, a higher concentration of imatinib (5–10 µM) inhibited cell growth in both BaF3/E255K and BaF3/T315I cells under chloroquine diphosphate salt treatment (Fig. 6b,c). TUNEL analysis demonstrated the acceleration of cell death by co‐treatment with imatinib and chloroquine diphosphate salt even in BaF3/T315I cells (Fig. 6d). These results suggested that autophagy is related to resistance to imatinib and inhibition of autophagy increased the effect of imatinib in imatinib‐resistant cells. To clarify the role of autophagy during treatment of imatinib with chloroquine diphosphate salt, we investigated LC3 expression by immunoblotting. LC3 protein was expressed in normal status of K562 cells and highly expressed in chloroquine‐treated cells. This result indicated that autophagy continuously occurred in normal status of K562 cells. Chloroquine blocks autophagosomal degradation and caused accumulation of autophagic vacuole marker, LC3‐II.( 22 ) Only imatinib treatment decreased the level of LC3, but co‐treatment with imatinib and chloroquine diphosphate salt induced higher expression of LC3, suggesting that autophagosomes were constructed under imatinib treatment and that chloroquine blocks the digestion of autophagosomes (Fig. 7a). Equally, BaF3/wild‐type and BaF3/E255K cells demonstrated the increase of LC3 under treatment of chloroquine diphosphate salt, but treatment with imatinib only did not change the level of LC3 in imatinib‐resistant cell lines compared to K562 cells (Fig. 7b,c). Especially, BaF3/T315I exhibited marked LC3 expression with or without chloroquine diphosphate salt (Fig. 7d). From these data, we concluded that autophagy also occurred in imatinib‐resistant cell lines and was related to resistance to imatinib.
Figure 6.
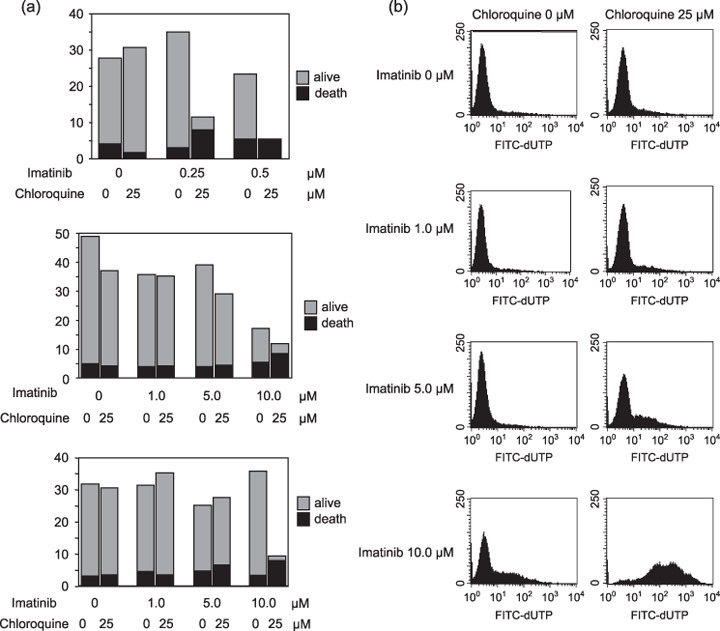
(a) Trypan blue count of BaF3/wild‐type cells (upper), BaF3/E255K cells (middle), BaF3/T315I cells (lower) after treatment imatinib (indicated dose) with or without chloroquine diphosphate salt (25 µM) for 48 h. Gray bar indicates total count and black bar indicates dead cell count. The data represents average of triplicate experiments. (b) TUNEL assay of BaF3/T315I cells after treatment with imatinib (0–10 µM) with or without chloroquine diphosphate salt (25 µM) for 48 h. The data represents average of triplicate experiments.
Figure 7.
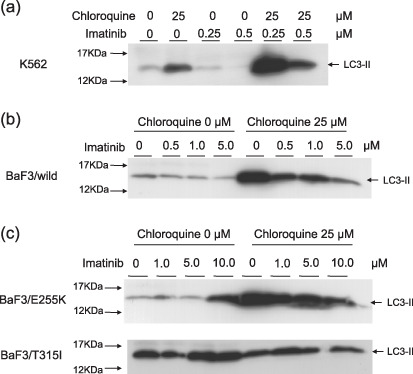
Immunoblotting of LC3 after treatment with imatinib (indicated dose) with or without chloroquine diphosphate salt (25 µM) for 48 h (a) K562 (b) BaF3/wild‐type and (c) BaF3/E255K (upper) BaF3/T315I (lower).
Discussion
At first, we thought that autophagy occurred as one process of megakaryocytic differentiation in K562 cells, but our data indicated that autophagy was an independent system on megakaryocytic differentiation, and autophagy was a cell survival system from cell stress induced by TPA. TPA induces expression of the cyclin‐dependent kinase inhibitor p21 and the G0/G1 arrest in K562 cells.( 26 ) P21 inhibits the activity of CDC2 kinase that is essential for G2/M transition, thereby allowing cells to skip the M phase.( 27 ) Kikuchi et al. have reported that megakaryocytic differentiation consists of two processes: polyploidization and functional maturation.( 28 ) Skipping the M phase induces polyploidization of cells and functional maturations for megakaryocyte likely to occur after polyploidation in the G1 phase. TPA and microtubule‐depolymerizing agent nocodazole‐induced polyploidation and differentiation into megakaryocyte in the megakaryocytic leukemia cell line, UT‐7 cells. TPA mainly influences the late phase of megakaryocytic differentiation, which is functional maturation.( 28 ) Similarly, Cavalloni et al. described no significant increase of CD41 expression in nocodazole‐treated K562 cells, whereas CD41 expression was significantly induced by TPA after 7 days of treatment.( 22 ) In our experiments, TPA induced high expression of CD41, and many autophagic vacuoles were induced. The appearance of autophagy was independent of the CD41 molecule. Additionally, nocodazole is known to inhibit autophagy by blockade of fusion of lysozomes and autophagosomes.( 29 ) These data indicated that megakaryocytic differentiation is independent of autophagy. When we treated K562 cells with TPA after nocodazole‐induced hyperploidy, CD41 expressions occurred, but the autophagic cells were decreased compared to those treated with TPA only. Kikuchi et al. have described that hyperploid cells are already on the way to differentiation and can express more mature phenotypes in response to TPA than other non‐hyperploid cells.( 28 ) From these data, we thought that after TPA‐induced cell cycle arrest, in a few cells in the G2/M phase, DNA polyploidy would occur and cytoplasmic maturation reached megakaryocytic differentiation. But then, most cells in the G0/G1 phase showed no cytoplasmic maturation and increase of autophagy‐induced cytoplasmic degradation. Finally, the cells underwent APCD after over‐digestion of important components for cell survival, such as mitochondria.
Observing that the mechanisms of TPA‐induced remarkable autophagy and APCD, we investigated the TPA‐induced activation of MAPK. In CML cells, BCR‐ABL have activities of ABL‐kinase and autophosphorylation, and, furthermore, transmits signals to lower levels such as Ras/MAPK. From our assay, we concluded that activation of MAPK plays an important role in autophagy. In other reports, externally regulated kinase (ERK) activation was also required for starvation‐induced autophagic proteolysis. Amino acids impaired starvation‐induced ERK activation and inhibited autophagy.( 30 ) Corcelle et al. have reported that Sertoli cells induced sustained MAPK and autophagic vacuolation by the carcinogen, lindane.( 31 ) In their experiments, the inhibition of the ERK pathway did not affect the formation of autophagosomes in MEK1‐transfected cells, but elicited the disappearance of giant autolysosomes and concomitantly induced abnormal accumulation of electron‐dense materials. Thus, they concluded that MAPK/ERK activation controlled the maturation of autophagosomes into autolysosomes rather than the formation of autophagosomes. In our assay, a low level of LC3 protein was expressed in K562 cells before stimulation. More activation of MAPK by TPA induced remarkably increased expression of LC3. These data suggested that Ras/MAPK signaling by BCR‐ABL induced a continuous activation of MAPK, then small amount of autophagy were occurred in normal status. Furthermore, TPA‐induced, greater activation of MAPK accelerated remarkable autophagic degradation and finally underwent APCD.
On the other hand, our data suggested that autophagy would regulate the cell survival system. To clarify the mechanisms of autophagy during cell death, we used imatinib to induce cell death for K562 cells. Imatinib mesylate is an ABL‐kinase inhibitor and very effective clinical drug for CML. Imatinib induces apoptosis for CML cells, but the appearance of autophagy has not been considered during imatinib‐induced cell death. Imatinib inhibits autophosphorylation of BCR‐ABL and the lower signals such as Ras/MAPK. However, Ras/MAPK is stimulated by signals from various levels, so Ras/MAPK signals are not inhibited perfectly by imatinib. The remaining of stimulation of Ras/MAPK can lead to resistance to imatinib, so imatinib plus MAPK inhibitors increase cell death in CML cells.( 32 ) Our experiments indicated that the inhibition of autophagy induced accelerated imatinib‐induced cell death, so it could be considered that autophagy induced by the Ras/MAPK signal from BCR‐ABL plays a role as a resistant mechanisms in CML cells and the inhibition of autophagy increased the antitumor effect of imatinib in K562 cells. Furthermore, imatinib‐resistant BaF3/E255K cells and BaF3/T315I cells had remarkable LC3 expression in Western blotting, and a high concentration of imatinib was effective in BaF3/E255K and BaF3/T315I cells under inhibition of autophagy. These results indicated that autophagy related to cell‐resistant mechanisms in imatinib‐resistant cell lines. But imatinib mainly bind to ABL‐kinase in CML cells, and imatinib cannot bind to ABL‐kinase in imatinib‐resistant cells having a point mutation of BCR‐ABL.( 33 ) Then, as the reason for the effect of imatinib on imatinib‐resistant cells, we have to consider another target point of imatinib on CML cells. It is well known that c‐kit and platelet‐derived growth factor receptor in other cells are other target points of imatinib. Rix et al. demonstrated that an oxidoreductase, NQO2, which is a non‐kinase enzymatic target, is the other target point of imatinib in CML cells.( 34 ) Inhibition of autophagy might increase the effect of imatinib against other target points in imatinib‐resistant cells.
Interestingly, BaF3/T315I exhibited the higher expression of LC3 with or without chloroquine diphosphate salt. BaF3/T315I cells have most resistance to CML treatment. Our data suggested that autophagy plays an important role in resistance of BaF3/T315I to imatinib.
Denis and Codogno have described that autophagy can be also considered a housekeeping mechanism involved in cytoplasmic homeostasis because it controls the turnover of peroxisomes, mitochondria and the size of the endoplasmic reticulum. In addition, in cancer cells, autophagy is also a self‐defense mechanism against stress induced by drugs or ionizing radiation.( 4 ) Our data suggested that this autophagic self‐defense system would be applied to imatinib‐resistant cells. Autophagic cell survival mechanisms might be usually promoted in imatinib‐resistant cell lines, that it is proved by high expression of LC3 in BaF3/T315I cells before treatment. Then, the inhibitions of autophagy might disrupt the defense against imatinib and increase drug sensitivity. Carew et al. described that the histone deacetylase inhibitor, SAHA, induces CML apoptosis and autophagy, and that combined treatment using SAHA with an inhibitor of autophagy, chloroquine, was very effective in imatinib‐resistant cell lines, blocking autophagy promoted SAHA‐induced apoptosis.( 35 ) The mechanisms of autophagy deeply related to cell survival and cell death in CML cells. On the other hand, recent reports have indicated that chloroquine‐induced lysosomal changes can lead to p53‐dependent cell death in the absence of apoptosis.( 36 ) The effect of chloroquine might not be the only pathway in co‐treatment with imatinib.
These data could lead to a new strategy in treatment of CML. New drugs for imatinib‐resistant CML, such as dasatinib and nilotinib, have been applied in the clinical treatment. These agents are very effective but some problems still remain, such as side‐effects and high costs. Furthermore, these drugs have no effect on T315I. If the effect of imatinib with chloroquine could be applied to clinical treatment, cases of imatinib‐resistant CML could continue imatinib treatment. It will be possible to reduce the concentration of imatinib for patients with severe side‐effects, such as myelosuppression, edema and skin rash. More study on the relationship between autophagy and cell death could be a very interesting theme for new CML treatment.
References
- 1. Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. Autophagy in yeast demonstrated with proteinase‐deficient mutants and conditions for its induction. J Cell Biol 1992; 119: 301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baba M, Takeshige K, Baba N, Ohsumi Y. Ultrastructural analysis of the autophagic process in yeast: detection of autophagosomes and their characterization. J Cell Biol 1994; 124: 903–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol 2005; 6: 505–10. [DOI] [PubMed] [Google Scholar]
- 4. Ogier‐Denis E, Codogno P. Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta 2003; 1603: 113–28. [DOI] [PubMed] [Google Scholar]
- 5. Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990; 181: 195–213. [DOI] [PubMed] [Google Scholar]
- 6. Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology 1973; 7: 253–66. [DOI] [PubMed] [Google Scholar]
- 7. Bursch W, Ellinger A. Autophagy – a basic mechanism and a potential role for neurodegeneration. Folia Neuropathol 2005; 43: 297–310. [PubMed] [Google Scholar]
- 8. Bursch W, Ellinger A, Gerner C, Frohwein U, Schulte‐Hermann R. Programmed cell death (PCD). Apoptosis, autophagic PCD, or others? Ann NY Acad Sci 2000; 926: 1–12. [DOI] [PubMed] [Google Scholar]
- 9. Bursch W, Hochegger K, Torok L, Marian B, Ellinger A, Hermann RS. Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. J Cell Sci 2000; 113 (7): 1189–98. [DOI] [PubMed] [Google Scholar]
- 10. Yu L, Lenardo MJ, Baehrecke EH. Autophagy and caspases: a new cell death program. Cell Cycle 2004; 3: 1124–6. [PubMed] [Google Scholar]
- 11. Shimizu S, Kanaseki T, Mizushima N et al . Role of Bcl‐2 family proteins in a non‐apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 2004; 6: 1221–8. [DOI] [PubMed] [Google Scholar]
- 12. Saeki K, Yuo A, Okuma E et al . Bcl‐2 down‐regulation causes autophagy in a caspase‐independent manner in human leukemic HL60 cells. Cell Death Differ 2000; 7: 1263–9. [DOI] [PubMed] [Google Scholar]
- 13. Fader CM, Savina A, Sanchez D, Colombo MI. Exosome secretion and red cell maturation. Exploring molecular components involved in the docking and fusion of multivesicular bodies in K562 cells. Blood Cells Mol Dis 2005; 35: 153–7. [DOI] [PubMed] [Google Scholar]
- 14. Takano‐Ohmuro H, Mukaida M, Kominami E, Morioka K. Autophagy in embryonic erythroid cells: its role in maturation. Eur J Cell Biol 2000; 79: 759–64. [DOI] [PubMed] [Google Scholar]
- 15. Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15: 1101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Biederbick A, Kern HF, Elsasser HP. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol 1995; 66: 3–14. [PubMed] [Google Scholar]
- 17. Munafo DB, Colombo MI. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci 2001; 114: 3619–29. [DOI] [PubMed] [Google Scholar]
- 18. Kabeya Y, Mizushima N, Ueno T et al . LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19: 5720–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov 2007; 6: 304–12. [DOI] [PubMed] [Google Scholar]
- 20. Cortez D, Stoica G, Pierce JH, Pendergast AM. The BCR‐ABL tyrosine kinase inhibits apoptosis by activating a Ras‐dependent signaling pathway. Oncogene 1996; 13: 2589–94. [PubMed] [Google Scholar]
- 21. Shacka JJ, Klocke BJ, Shibata M et al . Bafilomycin A1 inhibits chloroquine‐induced death of cerebellar granule neurons. Mol Pharmacol 2006; 69: 1125–36. [DOI] [PubMed] [Google Scholar]
- 22. Boya P, González‐Polo RA, Casares N et al . Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25: 1025–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Branford S, Rudzki Z, Walsh S et al . High frequency of point mutations clustered within the adenosine triphosphate‐binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph‐positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood 2002; 99: 3472–5. [DOI] [PubMed] [Google Scholar]
- 24. Quintas‐Cardama A, Kantarjian H, Cortes J. Flying under the radar: the new wave of BCR‐ABL inhibitors. Nat Rev Drug Discov 2007; 6: 834–48. [DOI] [PubMed] [Google Scholar]
- 25. Kantarjian HM, Giles F, Quintas‐Cardama A, Cortes J. Important therapeutic targets in chronic myelogenous leukemia. Clin Cancer Res 2007; 13: 1089–97. [DOI] [PubMed] [Google Scholar]
- 26. Cavalloni G, Dane A, Piacibello W et al . The involvement of human‐nuc gene in polyploidization of K562 cell line. Exp Hematol 2000; 28: 1432–40. [DOI] [PubMed] [Google Scholar]
- 27. Baccini V, Roy L, Vitrat N et al . Role of p21 (Cip1/Waf1) in cell‐cycle exit of endomitotic megakaryocytes. Blood 2001; 98: 3274–82. [DOI] [PubMed] [Google Scholar]
- 28. Kikuchi J, Furukawa Y, Iwase S et al . Polyploidization and functional maturation are two distinct processes during megakaryocytic differentiation: involvement of cyclin‐dependent kinase inhibitor p21 in polyploidization. Blood 1997; 89: 3980–90. [PubMed] [Google Scholar]
- 29. Webb JL, Ravikumar B, Rubinsztein DC. Microtubule disruption inhibits autophagosome‐lysosome fusion: implications for studying the roles of aggresomes in polyglutamine diseases. Int J Biochem Cell Biol 2004; 36: 2541–50. [DOI] [PubMed] [Google Scholar]
- 30. Pattingre S, Bauvy C, Codogno P. Amino acids interfere with the ERK1/2‐dependent control of macroautophagy by controlling the activation of Raf‐1 in human colon cancer HT‐29 cells. J Biol Chem 2003; 278: 16 667–74. [DOI] [PubMed] [Google Scholar]
- 31. Corcelle E, Nebout M, Bekri S et al . Disruption of autophagy at the maturation step by the carcinogen lindane is associated with the sustained mitogen‐activated protein kinase/extracellular signal‐regulated kinase activity. Cancer Res 2006; 66: 6861–70. [DOI] [PubMed] [Google Scholar]
- 32. Yu C, Krystal G, Varticovksi L et al . Pharmacologic mitogen‐activated protein/extracellular signal‐regulated kinase kinase/mitogen‐activated protein kinase inhibitors interact synergistically with STI571 to induce apoptosis in Bcr/Abl‐expressing human leukemia cells. Cancer Res 2002; 62: 188–99. [PubMed] [Google Scholar]
- 33. O’Hare T, Eide CA, Deininger MW. Bcr‐Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007; 110: 2242–9. [DOI] [PubMed] [Google Scholar]
- 34. Rix U, Hantschel O, Durnberger G et al . Chemical proteomic profiles of the BCR‐ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood 2007; 110: 4055–63. [DOI] [PubMed] [Google Scholar]
- 35. Carew JS, Nawrocki ST, Kahue CN et al . Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr‐Abl‐mediated drug resistance. Blood 2007; 110: 313–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maclean KH, Dorsey FC, Cleveland JL, Kastan MB. Targeting lysosomal degradation induces p53‐dependent cell death and prevents cancer in mouse models of lymphomagenesis. J Clin Invest 2008; 118: 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]