

Abstract
Oncogenic Ras proteins transform cells by way of multiple downstream signaling pathways that promote the genesis of human cancers. However, the exact cellular mechanisms by which downstream targets are regulated are not fully understood. Here, we show that oncogenic Ras reduced Clast1/LR8 transcript levels in mouse NIH3T3 fibroblasts and human WI38 fibroblasts. Clast1/LR8 transcript was undetectable in H460, A549, and H1299 cells showing high Ras activity, but was relatively abundant in DMS53 cells displaying low Ras activity. We also showed that K‐Ras siRNA restored Clast1/LR8 expression in H460 and A549 cells, and that inhibitors of DNA methylation and histone deacetylation reversed oncogenic H‐Ras‐mediated suppression of Clast1/LR8 transcription. Additionally, ectopic expression of Clast1/LR8 inhibited serum‐stimulated phosphorylation of ERK1/2 and Akt in H‐RasV12‐transformed NIH3T3 cells. We further showed that the expression of Clast1/LR8 interfered with oncogenic Ras‐induced NIH3T3 cell transformation and invasion. Finally, our results showed that Clast1/LR8 inhibited Ras‐induced proliferation of, and tumor formation by, oncogenic H‐RasV12‐transformed NIH3T3 cells in vivo. This study identifies the downregulation of Clast1/LR8 as a potentially important mechanism by which oncogenic Ras‐mediated neoplastic transformation occurs. (Cancer Sci 2010)
The ras proto‐oncogenes encode several 21‐kDa GTP‐binding proteins that act as pivotal mediators of cell signaling.( 1 ) They help to transfer extracellular signaling information from the cell membrane through the cytosol and finally into the nucleus. In normal cells, Ras proteins, including H‐, K‐, and N‐Ras, are transiently activated in response to extracellular signals and help to control cell proliferation, differentiation, and survival in all multicellular organisms.( 2 ) Consequently, proper regulation of Ras signaling is critically important for normal development. In tumor cells, the oncogenic activation of ras occurs as a consequence of point mutations that either impair Ras’s GTPase activity or enhance its GTP binding affinity, resulting in the generation of a highly active proliferative signal.( 3 ) Indeed, constitutively active Ras has been implicated in many processes underscoring malignant phenotypes, including proliferation, neoplastic transformation, invasion, and metastasis.( 4 ) Oncogenic Ras proteins are commonly detected in human cancers, including approximately 90% of pancreatic cancers, 70% of malignant neoplasias, and 30% of all human cancers, suggesting that Ras plays a key role in the development of cancer.( 5 , 6 )
Signaling events downstream of Ras are complex, nonlinear, and dynamic.( 7 , 8 ) Genome‐wide surveys have identified numerous putative Ras target genes that contribute to Ras transformation.( 2 , 9 , 10 ) Despite the identification of these diverse Ras targets, accurately determining which downstream signaling molecules represent the best therapeutic targets for blocking Ras‐mediated neoplastic transformation/tumorigenesis remains a challenge. Thus, the identification of Ras downstream targets in cancer cells is of considerable significance.
Here, we attempt to identify genes that are differentially expressed in control NIH3T3 cells and oncogenic H‐Ras‐expressing NIH3T3 cells through annealing control primer (ACP)‐based differential display RT‐PCR. We report the identification of a gene the expression of which was downregulated in H‐RasV12‐expressing NIH3T3 cells relative to control NIH3T3 cells. The sequence of this gene was found to be identical to that of mouse Clast1/LR8 (GenBank accession number AB031386).
We subsequently showed that Clast1/LR8 expression suppressed serum‐stimulated Ras signaling. Additionally, Clast1/LR8 prevented H‐RasV12‐induced cell transformation and invasion as well as tumor growth in nude mice. These results indicate that Clast1/LR8 may play an important role as a suppressor of oncogenic Ras‐mediated tumorigenesis.
Materials and Methods
Cell culture. NIH3T3 cells were grown in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS. DMS53 cells were maintained in Waymouth’s medium (Invitrogen) supplemented with 10% FBS. A549 cells were maintained in Ham’s F12 medium (Invitrogen) supplemented with 10% FBS. H1299 and H460 cells were maintained in RPMI‐1640 (Invitrogen) medium supplemented with 10% FBS and 5% glutamine. Cells were maintained in cell‐specific media at 37°C in a humidified atmosphere containing 5% CO2. All cell lines were obtained from the American Type Culture Collection (Rockville, MD, USA).
Preparation of constructs. Generation of the dominant‐positive H‐RasV12 constructs is described elsewhere.( 11 ) Mouse Clast1/LR8 cDNAs were amplified by RT‐PCR using the following primers: Clast1/LR8‐sense, ATGGTCCAGAGCACAGTGAC; Clast1/LR8‐antisense, TCACAGGATAGCAGGGATCT. After confirming their DNA sequences, mouse Clast/LR8 cDNAs were cloned into a pcDNA4/HisMax‐TOPO vector (Invitrogen).
RT‐PCR analysis. Inhibition of DNA methylation and histone deacetylation was achieved by culturing cells in 10 μM 5‐aza‐2′‐deoxycytidine (AZC; Sigma, St. Louis, MO, USA) for 2–4 days and 30–100 nM trichostatin A (TSA; Sigma) for 24 h. Total RNA was extracted from cells using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. The following primers were used: mouse Clast1‐sense, TCTCTAGGGGTGACCCAGATATT; mouse Clast1‐antisense, AGGATACAGACCACTGTGAGCAT; human Clast1‐sense, GTTCTTGGAGTGTGTCTCAGCTT; human Clast1‐antisense, GACACAATGACCTTCAAGACACA; mouse GAPDH‐sense, TAAAGGGCATCCTGGGCTACACT; mouse GADPH‐antisense, TTACTCCTTGGAGGCCATGTAGG; human GAPDH‐sense, GGTGAAGGTCGGTGTGAACGGATTT; human GADPH‐anti‐sense, AATGCCAAAGTTGTCATGGATGACC. The RT‐PCR analyses were carried out in duplicate and the results confirmed in two independent experiments. A description of ACP‐based differential display RT‐PCR is included in the Data S1.
Small interfering RNAs and transfection. To knock down Clast1/LR8 expression, two siRNA target sites in the mouse Clast1/LR8 mRNA sequence (GenBank accession number AB031386), obtained from the NCBI Entrez Nucleotide database (http://www.ncbi.nlm.nih.gov/nuccore/5804793), were chosen. The sequences of the Clast1/LR8 siRNAs were: sense, UUCUAGCUGGAGUUGGUACdTdT; and antisense, GUACCAACUCCAGCUAGAAdTdT. K‐Ras siRNA#1 (sc‐35731), K‐Ras siRNA#2 (sc‐43874) and control siRNA (sc‐37007) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Cells were transiently transfected with siRNA duplexes using RNAiMAX (Invitrogen), according to the manufacturer’s instructions.
Western blot analysis. Whole‐cell lysates were prepared in lysis buffer (50 mM Tris–HCl [pH 7.5], 150 mM NaCl, 1% NP‐40, 5 mM EDTA, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, and 1× protease inhibitor cocktail) (Roche Diagnostics, Indianapolis, IN, USA). Each sample’s protein concentration was determined using the Bradford assay. Equal amounts of protein were loaded to 10% SDS‐polyacrylamide gels, separated by electrophoresis, and transferred to PVDF membranes (Millipore, Bedford, MA, USA). Following transfer, membranes were blocked with 5% skim milk for 1 h, then incubated for 16 h with anti‐Xpress (Invitrogen), anti‐AKT, anti‐phospho‐AKT, anti‐ERK1/2, and anti‐phospho‐ERK1/2 antibodies (Cell Signaling Technology, Beverly, MA, USA). They were then washed, incubated with the appropriate secondary antibody (diluted 1:4000 in blocking buffer) for 2 h, and washed again. Blotted proteins were detected using an enhanced chemiluminescence detection system (iNtRON Biotechnology, Seoul, Korea).
Anchorage‐independent growth assay. Cells (2 × 104) were suspended in cell–specific medium containing 0.3% agar, plated on a 0.5% agar base, then covered with medium supplemented with 5% FBS. Cultures were maintained at 37°C for 20 days (the growth medium being replaced every 3 days). Numbers of colonies in five randomly‐selected fields were counted.
In vitro invasion assays. An in vitro invasion assay was carried out using 24‐well Transwell units with polycarbonate filters (pore size, 8 μm; Corning, Corning, NY, USA) coated on their upper sides with Matrigel (Becton Dickinson Labware, Bedford, MA, USA). Cells (5 × 104) were transferred to the upper part of each Transwell unit and were allowed 48 h to invade. The lower part of the Transwell unit was filled with 10% FBS‐supplemented medium. After incubation, non‐invaded cells on the upper surface of the membrane were removed from the chamber, and the invaded cells on the lower surface of the membrane were stained with Diff‐Quick (Kokusai Shiyaku, Kobe, Japan). Finally, numbers of cells in five randomly‐selected microscopic fields (200×) per membrane were counted.
Pull‐down experiment. The Ras‐binding domain (RBD) of Raf was coupled to GST, and a pull‐down experiment was carried out using an EZ‐Detect Ras Activation Kit (Pierce, Rockford, IL, USA) according to the manufacturer’s protocol. Briefly, cell lysates, prepared by lysing cells in commercial buffer, were incubated with pre‐coupled GST‐RBD for 30 min at 4°C. Beads were then collected by centrifugation, washed three times in lysis buffer, and re‐suspended in SDS loading buffer. Samples were then analyzed by Western blot using an anti‐pan‐Ras antibody.
In vivo tumor formation assays. Six‐week‐old nude female mice, purchased from Charles River Korea (Seoul, Korea), were used in in vivo tumor formation assays. Tumor cells (1 × 106) were re‐suspended in 150 μL sterile PBS and injected s.c. into the flanks of the nude mice. Tumor sizes were measured every 2–5 days using calipers. The lengths (l) and widths (w) of the developing tumors were used to calculate tumor volumes according to the following equation: volume (v) = (w2 × l)/2. Mice were killed when tumor diameters reached 2 cm.
Data analysis. All data are presented as the mean ± SD. Statistical comparisons were carried out using the unpaired Student’s t‐test. P‐values <0.01 were deemed to indicate statistical significance.
Results
Oncogenic H‐Ras‐mediated downregulation of Clast1. In an attempt to discover novel downstream effectors of Ras‐mediated cellular transformation, we carried out ACP‐based RT‐PCR analysis and compared the expression profiles of mouse NIH3T3 fibroblasts stably transfected with H‐RasV12 (H‐RasV12 NIH3T3) or empty pcDNA3 vector (NIH3T3/vector). We identified a partial cDNA sequence that was significantly downregulated in H‐RasV12‐expressing cells. The 343‐bp amplicon displayed complete homology with Clast1/LRr8 (GenBank accession number AB031386; Fig. S1). The Clast1/LR8 gene is ubiquitously expressed in various organs of the adult mouse. In splenic B cells, its expression is induced following activation with CD40 ligand.( 12 ) However, its physiological function remains largely unknown.
To confirm the results of our ACP‐based RT‐PCR analysis, we carried out semiquantitative RT‐PCR analyses of the H‐RasV12 NIH3T3 and NIH3T3/vector cells. Using the Clast1/LR8‐specific primers, we showed the expression of the Clast1/LR8 gene to be dramatically decreased in cells transfected with H‐RasV12 expression vector, compared with the parental NIH3T3 cells and vector control cells (Fig. 1a). We also investigated the effect of oncogenic Ras on human Clast1/LR8 transcription in human lung WI38 fibroblasts. We found that oncogenic H‐RasV12 downregulated Clast1/LR8 mRNA expression in WI38 cells (Fig. 1b).
Figure 1.
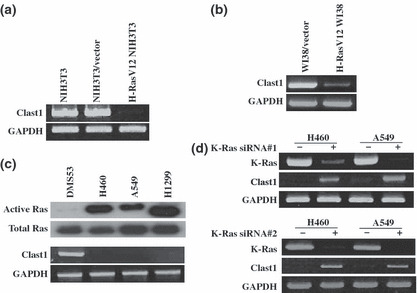
Clast1/LR8 expression is downregulated as a result of oncogenic Ras activation. (a) Total RNA extracts prepared from parental NIH3T3 cells, NIH3T3/vector cells, and H‐RasV12 NIH3T3 cells were analyzed by semiquantitative RT‐PCR using mouse Clast1/LR8‐specific primers. (b) WI38 cells were transiently transfected with an H‐RasV12 expression vector or control vector. Total RNA was isolated and human Clast1/LR8 mRNA expression was then examined by RT‐PCR using human Clast1/LR8‐specific primers. (c) Ras activity (upper panel) and Clast1/LR8 mRNA expression (determined by RT‐PCR) (lower panel) in four lung cancer cell lines. (d) Clast1/LR8 expression in H460 and A549 cells transfected with control siRNA, K‐Ras siRNA#1, or K‐Ras siRNA #2.
Because Clast1/LR8 mRNA was originally detected in human lung fibroblasts,( 13 ) and active mutations in Kras gene have been frequently noted in lung cancer tissues,( 14 , 15 ) we next investigated Clast1/LR8 expression in human lung cancer cell lines including DMS53, H460 A549, and H1299. H460 and A549 cells harbor constitutively active K‐Ras proteins (as a result of mutations in codons 61 and 12 respectively), and H1299 cells have oncogenic N‐Ras (mutations in codon 61). We initially confirmed and compared Ras activity in lung cancer cell lines through pull‐down experiments using GST‐Raf‐RBD. Consistent with previous reports, we found that the above lung cancer cell lines, with the exception of DMS53 cells, expressed constitutively active forms of Ras, (Fig. 1c, upper panel). We next examined the levels of Clast1/LR8 mRNA in these cell lines. We found Clast1/LR8 mRNA expression to be undetectable in H460, A549, and H1299 cells, which show increased Ras activity, but was detectable in DMS53 cells, which display only low Ras activity (Fig. 1c, lower panel). To test whether non‐expression of Clast1/LR8 in H460 and A549 cells could be attributed to the persistent activation of K‐Ras, we assessed the effect of K‐Ras knockdown on Clast1/LR8 expression. We found that transfection with K‐Ras siRNA restored Clast1/LR8 expression in H460 and A549 cells (Fig. 1d). Thus, an inverse correlation exists between Clast1/LR8 levels and Ras activation.
DNA methylation and histone deacetylation contribute to oncogenic Ras‐mediated downregulation of Clast1/LR8 expression. Oncogenic Ras has been shown to suppress the expression of several tumor suppressor genes.( 16 , 17 , 18 , 19 ) Although the mechanism by which oncogenic Ras suppresses gene transcription remains largely unknown, recent studies suggest that altered activation of histone deacetylase and hypermethylation of CpG islands may contribute to the downregulation of tumor suppressor genes by oncogenic Ras.( 18 , 19 , 20 , 21 ) Thus, we investigated whether the downregulation of Clast1/LR8 by H‐RasV12 involves a similar mechanism. NIH3T3/vector and H‐RasV12 NIH3T3 cells were treated with or without AZC, an inhibitor of DNA methylation, and the histone deacetylase inhibitor TSA. Clast1/LR8 mRNA levels were then determined by RT‐PCR analysis. AZC and TSA effectively reversed the suppression of Clast1/LR8 mRNA expression by H‐RasV12 (Fig. 2a).
Figure 2.
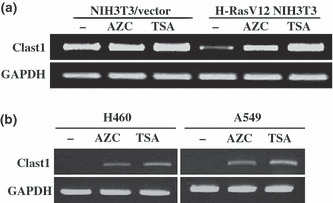
Effects of inhibitors of DNA methylation and histone deacetylation on Clast1/LR8 expression. (a) NIH3T3/vector and H‐RasV12 NIH3T3 cells were treated with or without (–) 10 μM 5‐aza‐2′‐deoxycytidine (AZC) for 48 h, or 30 nM trichostatin A (TSA) for 24 h. Clast1/LR8 expression was then analyzed by RT‐PCR. (b) The human lung cancer cell lines H460 and A549 were treated with or without 10 μM AZC for 4 days, or 100 nM TSA for 24 h. Clast1/LR8 expression was then analyzed by RT‐PCR using human Clast1/LR8‐specific primers.
We next determined whether the above regulatory mechanism was replicated in human lung cancer cells harboring the constitutively active K‐Ras. We chose three human lung cancer cell lines (H460, H1299, and A549) to address this issue. We found that treatment with AZC and TSA increased Clast1/LR8 mRNA expression in all three cell lines (Fig. 2b). These results suggest that hypermethylation and histone deacetylation may contribute to the downregulation of Clast1/LR8 transcription by oncogenic Ras in mouse fibroblasts and human lung cancer cells.
Clast1/LR8 blocks serum‐stimulated Ras signaling. We next determined the effects of ectopic Clast1/LR8 expression on two major signal transduction pathways, Raf/MEK/ERK and the PI3K/Akt. To this end, we stably transfected H‐RasV12 NIH3T3 cells with a Clast/LR8 expression vector (Fig. 3a,b). The resulting cells were serum‐starved for 24 h then incubated with or without 20% FBS for 15 and 30 min. Cellular proteins were analyzed by Western blotting using anti‐phospho‐Akt and anti‐phospho‐ERK1/2 antibodies. We found that serum stimulation increased the levels of phospho‐Akt and phospho‐ERK1/2 in control H‐RasV12 NIH3T3 cells (Fig. 3c). Clast1/LR8 expression significantly suppressed serum‐induced phosphorylation of Akt and ERK1/2 in H‐RasV12 NIH3T3 cells. We also analyzed the effects of Clast1/LR8 overexpression on ERK1/2 and Akt phosphorylation in NIH3T3 cells. As seen in Figure 3(c), the overexpression of Clast1/LR8 did not affect the phosphorylation of ERK1/2 or Akt in response to serum stimulation. These results suggest that Clast1/LR8 suppresses oncogenic H‐Ras‐mediated activation of Raf/MEK/ERK and PI3K/Akt signaling.
Figure 3.
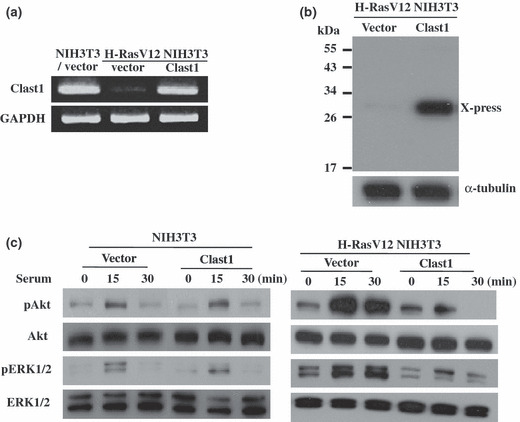
Effect of Clast1/LR8 on the Ras signaling pathway. (a) RT‐PCR analysis of Clast1/LR8 expression in NIH3T3/vector cells, H‐RasV12 NIH3T3 cells, and H‐RasV12 NIH3T3 cells transfected with a Clast1/LR8 expression vector. (b) Western blot analysis for Clast1/LR8 (X‐press) in H‐RasV12 NIH3T3 cells transfected with a Clast1/LR8 expression vector or empty vector. (c) The indicated cells were plated in DMEM supplemented with 5% FBS, washed twice in PBS 48 h later, then incubated in serum‐free medium for 24 h. Cells were then stimulated with or without 20% FBS for 15 and 30 min. Phosphorylated ERK1/2 (pERK1/2) and Akt (pAkt) levels, as well as total ERK1/2 and Akt protein levels, were analyzed by Western blotting.
Clast1/LR8 inhibits oncogenic Ras‐mediated transforma‐tion. Downregulation of Clast1/LR8 expression by oncogenic Ras may be an important event in oncogenic Ras‐induced cellular transformation. To gain further insights into this issue, we transiently or stably expressed Clast1/LR8 in H‐RasV12 NIH3T3 cells to determine whether exogenous Clast1/LR8 could prevent oncogene‐induced cellular transformation. We reasoned that if decreased levels of Clast1/LR8 are indeed necessary for oncogenic Ras to successfully transform cells, then exogenously expressed Clast1/LR8 would be expected to modulate oncogenic Ras‐induced cellular transformation. We assessed the effect of Clast1/LR8 expression on anchorage‐independent growth of mock H‐RasV12 NIH3T3 cells, empty vector‐transfected H‐RasV12 NIH3T3 cells, and Clast1/LR8‐transfected H‐RasV12 NIH3T3 cells on soft agar. We found colony formation to be significantly inhibited in Clast1/LR8‐expressing H‐RasV12 NIH3T3 cells, compared with vector‐transfected H‐RasV12 NIH3T3 cells (Figs 4,S2). However, knockdown of Clast1/LR8 did not affect the anchorage‐independent growth of NIH3T3 cells, suggesting that Clast1/LR8 limits the oncogenic potential of oncogenic Ras‐transformed NIH3T3 cells.
Figure 4.
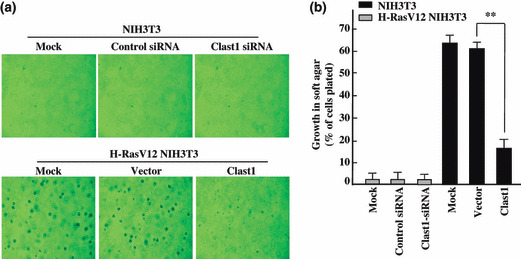
Clast1/LR8 expression inhibits an H‐RasV12‐mediated increase in colony formation in soft agar. (a) The indicated stable transfectants of NIH3T3 and H‐RasV12 NIH3T3 cells (2 × 104) were plated in medium containing 0.3% agar and supplemented with 10% FBS on a 0.5% agar base. Representative images of colony formation were taken after 20 days of growth. (b) Percentages of cells present in colonies >50 μm in diameter. Values represent the mean ± SD of three separate experiments. **P < 0.01.
Clast1/LR8 overexpression interferes with cell invasion. To investigate the potential inhibitory effects of Clast1/LR8 on oncogenic Ras‐induced NIH3T3 cell invasion, we carried out an in vitro invasion assay using 24‐well Transwell units with polycarbonate filters coated on their upper sides with Matrigel. Numbers of invasive cells on the lower surfaces of these inserts were quantified 48 h after the application of a cell suspension to the Transwell units. We found that the overexpression of Clast1/LR8 significantly reduced the invasive potential of H‐RasV12 NIH3T3 cells when compared with mock and empty vector‐transfected cells (Figs 5,S3). Approximately 2.7‐fold fewer Clast1/LR8‐transfected H‐RasV12 NIH3T3 cells crossed the Matrigel barrier than did empty vector‐transfected cells.
Figure 5.
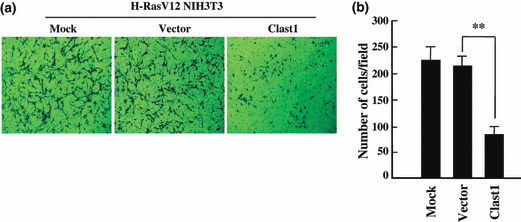
Clast1/LR8 overexpression suppresses H‐RasV12‐induced cell invasion. (a) An in vitro invasion assay was carried out using 24‐well Transwell units with polycarbonate filters coated on their upper sides with Matrigel. Cells were placed in the upper part of each Transwell unit and allowed 48 h to invade. Cells that penetrated to the lower surface of each membrane were fixed, stained, and counted under a microscope. Images are representative of invasion by each cell type. (b) Mean number of invading cells. Values represent the mean ± SD of three separate experiments. **P < 0.01.
Clast1/LR8 suppresses H‐Ras oncogene‐mediated tumorigenesis in vivo. We finally examined whether Clast1/LR8 participated in oncogenic Ras‐mediated tumorigenesis in vivo. To investigate whether Clast1/LR8 expression inhibits H‐Ras‐mediated tumor formation in nude mice, parental NIH3T3 cells and H‐RasV12 NIH3T3 cells transfected with either empty vector or Clast1/LR8 expression vector were implanted s.c. into nude mice and subsequent tumor formation was monitored over a 20‐day period. We found that empty vector‐transfected H‐RasV12 NIH3T3 cells gave rise to significantly larger tumors than Clast1/LR8‐overexpressing H‐RasV12 NIH3T3 (Figs 6,S4). These results indicate that Clast1/LR8 expression markedly reduced in vivo tumor formation resulting from the injection of oncogenic H‐RasV12‐transformed NIH3T3 cells.
Figure 6.
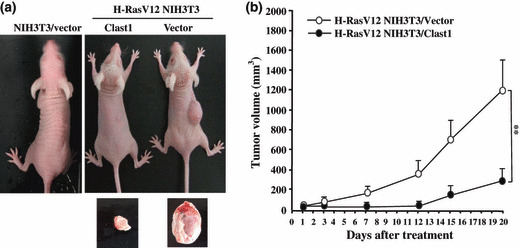
Ectopic expression of Clast1/LR8 inhibits H‐RasV12‐induced tumor formation in nude mice. (a) One million cells of each type indicated were injected s.c. into the right flanks of 5‐week‐old nude mice and tumor volumes were assessed every 2–5 days for 20 days. Images show representative mice from each injection group. (b) Tumor growth rates in mice injected with H‐RasV12 NIH3T3 cells transfected with either empty vector or Clast1/LR8 expression vector. Values represent the mean ± SD of three separate experiments. **P < 0.01.
Discussion
In this study, we identified, through differential display RT‐PCR analysis, that Clast1/LR8 is one of several genes downregulated in oncogenic H‐Ras‐expressing NIH3T3 cells. Clast1/LR8 was originally cloned in two human lung fibroblast subpopulations by mRNA differential display analysis.( 13 ) Mouse Clast1/LR8 shares 62% homology with human LR8 and contains four predicted transmembrane domains. In adult mice, Clast1 mRNA is highly expressed in various organs, notably lung, liver, kidney, and colon.( 12 ) Clast1 mRNA expression was shown to be induced by the CD40 ligand in B lymphocytes. Although Clast1/LR8 knockout mice did not show functional B cell abnormalities, they did show severe ataxia, indicating that Clast1/LR8 may be involved in the development of cerebellar granule cells. However, its physiological function is largely unknown.
A genome‐wide survey of oncogenic Ras transformation targets suggested that both gene activation and repression are critical for transformation by oncogenic Ras.( 10 ) Although the molecular mechanisms controlling the silencing of tumor suppressor genes remain unknown, recent studies suggest that DNA methyltransferases (DNMT) may be involved. A DNMT enzyme was, for example, shown to be in involved in Ras‐mediated suppression of the tumor suppressor genes FHIT, TLSC1, and RASSFIA.( 22 ) Separately, increased DNMT activity was found to be essential for transformation and gene repression by oncogenic Ras.( 23 ) Additionally, Ras‐induced repression of the pro‐apoptotic response protein PAR‐4 in epithelial cells resulted from DNA hypermethylation, realized through Raf‐dependent and ‐independent signaling.( 24 ) More recently, Chang et al. ( 19 ) reported that oncogenic Ras suppressed the metastatic suppressor RECK by upregulating DNMT expression. Ras oncogenes also suppress tumor suppressor gene expression through histone deacetylation.( 21 ) Thus, we investigated whether these mechanisms contributed to reduced Clast1/LR8 expression in cells with strong oncogenic Ras activity. We found that the suppression of Clast1/LR8 transcription in oncogenic H‐Ras‐expressing NIH3T3 cells was reversed by treatment with the DNA methylation inhibitor AZC and the histone deacetylase inhibitor TSA. These observations suggest that DNA methylation and histone deacetylation may contribute to the suppression of Clast1/LR8 expression in Ras‐activated cancer cells.
Cellular transformation is a complex process that involves a series of cellular and molecular changes. Ras proteins exert their tumorigenic effects through the activation of a complex signaling network comprising multiple downstream effectors.( 2 , 25 ) Two downstream effector pathways believed to play critical roles in Ras‐mediated transformation and tumorigenesis are the Raf/MEK/ERK pathway( 26 ) and the PI3K/Akt pathway.( 27 ) Activation of the Raf/MEK/ERK pathway is sufficient to transform NIH3T3 mouse fibroblasts.( 28 ) Injection of Ras‐transformed NIH3T3 cells into the tail veins of mice, meanwhile, induced metastatic growth, a response realized through a Raf‐dependent mechanism.( 29 ) It has been also shown that oncogenic Ras downregulated RhoB expression through a PI3K/Akt pathway, leading to Ras‐induced transformation, invasion, and metastasis in NIH3T3 fibroblasts and various cancer cell lines.( 30 ) The observed inverse correlation between Clast1/LR8 expression and Ras activation in lung cancer cells, and the downregulation of Clast1/LR8 expression in NIH3T3 cells by H‐RasV12, suggest that the disruption of Clast1/LR8 expression/function may contribute to oncogenic Ras‐induced neoplastic transformation. Thus, we next investigated whether Clast1/LR8 was involved in Ras‐mediated cellular transformation. Recent evidence indicates that Ras downregulates the expression of several molecules, including PAR‐4, Bak, and GADD153.( 31 , 32 , 33 ) Exogenous expression of the transcriptional repressors PAR‐4 and GADD153 has been reported to suppress oncogenic Ras‐induced transformation.( 32 , 33 ) Similarly, the ectopic expression of Bak in Ras‐transformed cells inhibited Ras‐induced tumorigenicity.( 31 ) Here, we report that the downregulation of Clast1/LR8 appears to be a key event during Ras‐induced cellular transformation. Transfection of Clast1/LR8 cDNA resulted in dramatic inhibition of anchorage‐independent growth and cellular invasion of oncogenic H‐Ras expressing NIH3T3 cells. Moreover, the expression of Clast1/LR8 in H‐RasV12 NIH3T3 cells significantly suppressed tumor formation in mice. However, knockdown of Clast1/LR8 using siRNA did not trigger cellular transformation of NIH3T3 cells, indicating that lack of Clast1/LR8 can only activate the transformation process when Ras is oncogenically activated. Clast1/LR8 therefore appears to be an important molecule, the downregulation of which may be a prerequisite for cellular transformation induced by oncogenic Ras. Although the mechanism by which downregulation of Clast1/LR8 facilitates cellular transformation needs to be clarified, we found that ectopic expression of Clast1/LR8 in H‐RasV12‐transformed NIH3T3 cells inhibited serum‐stimulated phosphorylation of ERK1/2 and Akt. The Raf/MEK/ERK and PI3K/Akt signaling pathways are responsible for transducing oncogenic Ras‐mediated tumorigenic signals. It is possible that the downregulation of Clast1/LR8 expression by oncogenic Ras contributes to their activation, thus facilitating cellular transformation and invasion. Precisely how downregulation of Clast1/LR8 facilitates cellular transformation requires further investigation. Because Clast1/LR8 negatively modulates oncogenic Ras‐induced cell growth and invasion, reductions in its expression and activity are likely to have profound consequences. It is possible that the oncogenic activation of growth stimulatory signals, such as those transduced by Raf/MEK/ERK and PI3K/Akt, and suppression of growth inhibitory factors, such as Par‐4, Bak, GADD153, and Clast1/LR8, may occur simultaneously and together contribute to oncogenic transformation. In this context, reduced expression of growth inhibitory molecules would allow Ras to initiate and maintain oncogenic transformation.
In summary, the data generated by this study provide information on a novel downstream target molecule, Clast1/LR8, which may play an important role in tumor suppression in Ras‐activated cancer cells. We have shown that Clast1/LR8 is a suppressor of cellular transformation induced by oncogenic Ras. Thus, Clast1/LR8 may represent a new potential candidate for pharmacological re‐expression in Ras‐activated cancer cells, in what would be a novel form of cancer treatment.
Supporting information
Fig. S1. Clast1/LR8 expression is downregulated as a result of oncogenic Ras activation (a) Differential display RT‐PCR analysis of H‐RasV12‐transfected and empty vector‐transfected NIH3T3 cells. NIH3T3 cells were stably transfected with empty vector (NIH3T3/vector) or an H‐RasV12 expression vector (H‑RasV12 NIH3T3). mRNA extracted from these cells was analyzed by annealing control primer (ACP)‐based differential display RT‐PCR. It was first used to synthesize first‐strand cDNA using the primer dT‐ACP1 (cDNA synthesis primer). Second‐strand cDNA was then amplified by PCR using ACP34 (arbitrary; forward primer) and dT‐ACP2 (reverse primer), yielding a 343‐bp product. Arrows indicate differential expression between H‐RasV12‐transfected and empty vector‐transfected cells. (b) Sequence of the cloned cDNA. The sequence between 937 and 1211 (highlighted in red) was completely homologous to that of mouse Clast1/LR8 (GenBank accession number AB031386).
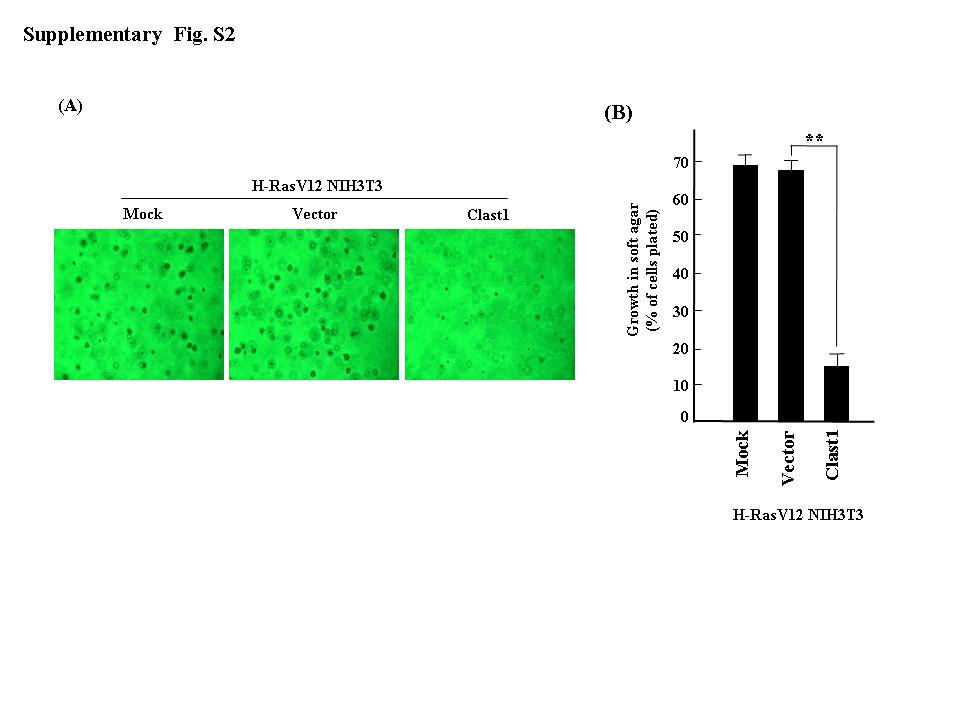
Fig. S2. Transient expression of Clast1/LR8 inhibits an H‐RasV12‐mediated increase in colony formation in soft agar. (a) The indicated plasmids were transiently transfected into H‐RasV12 NIH3T3 cells, and colony formation assay was carried out in soft agar. (b) Percentages of cells present in colonies >50 μm in diameter. Values represent the mean ± SD of three separate experiments. **P ≤ 0.01.
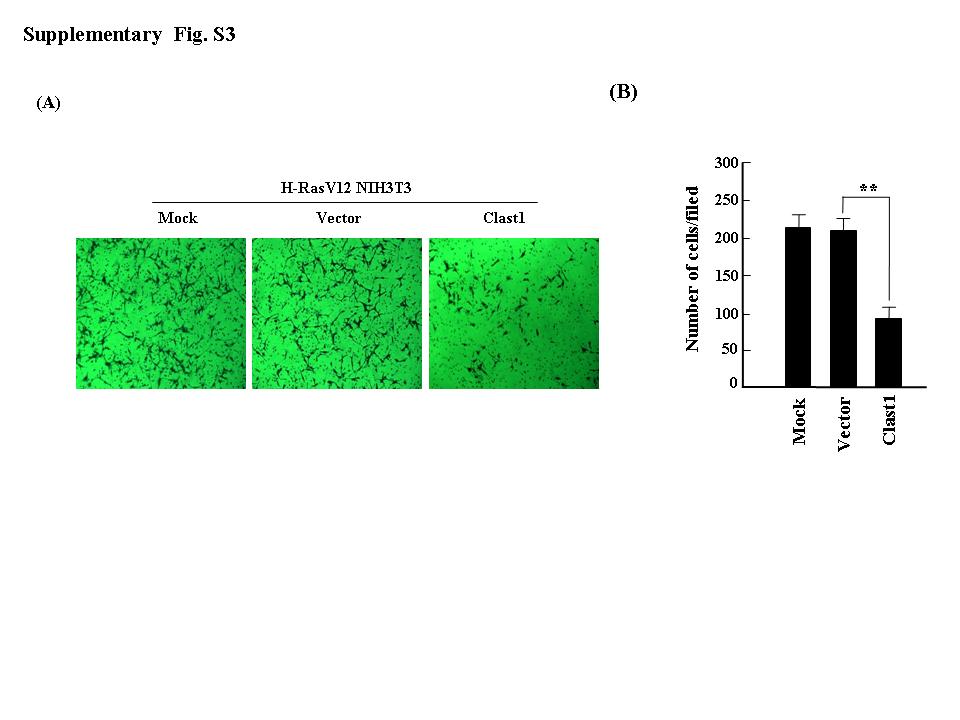
Fig. S3. Transient expression of Clast1/LR8 suppresses H‐RasV12‐induced cell invasion. (a) The indicated plasmids were transiently transfected into H‐RasV12 NIH3T3 cells, and an in vitro invasion assay was carried out. (b) Mean number of invading cells. Values represent the mean ± SD of three separate experiments. **P ≤ 0.01.
Fig. S4. Transient expression of Clast1/LR8 inhibits H‐RasV12‐induced tumor formation in nude mice. One million cells of each type indicated were injected s.c. into the right flanks of 5‐week‐old nude mice and tumor volumes were assessed every 2–5 days for 20 days. Images show representative mice from each injection group.
Data S1. Annealing control primer (ACP)‐based differential display RT‐PCR.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Acknowledgment
This research was supported by Chosun University, 2003.
References
- 1. Lowy DR, Willumsen BM. Function and regulation of ras. Annu Rev Biochem 1993; 62: 851–91. [DOI] [PubMed] [Google Scholar]
- 2. Campbell SL, Khosravi‐Far R, Rossman KL, Clark GJ, Der CJ. Increasing complexity of Ras signaling. Oncogene 1998; 17: 1395–413. [DOI] [PubMed] [Google Scholar]
- 3. White MA, Nicolette C, Minden A et al. Multiple Ras functions can contribute to mammalian cell transformation. Cell 1995; 80: 533–41. [DOI] [PubMed] [Google Scholar]
- 4. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 2007; 7: 295–308. [DOI] [PubMed] [Google Scholar]
- 5. Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer 2002; 2: 897–909. [DOI] [PubMed] [Google Scholar]
- 6. Bos JL. ras oncogenes in human cancer: a review. Cancer Res 1989; 49: 4682–9. [PubMed] [Google Scholar]
- 7. Huang E, Ishida S, Pittman J et al. Gene expression phenotypic models that predict the activity of oncogenic pathways. Nat Genet 2003; 34: 226–30. [DOI] [PubMed] [Google Scholar]
- 8. Bild AH, Yao G, Chang JT et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 2006; 439: 353–7. [DOI] [PubMed] [Google Scholar]
- 9. Ulku AS, Der CJ. Ras signaling, deregulation of gene expression and oncogenesis. Cancer Treat Res 2003; 115: 189–208. [PubMed] [Google Scholar]
- 10. Zuber J, Tchernitsa OI, Hinzmann B et al. A genome‐wide survey of RAS transformation targets. Nat Genet 2000; 24: 1441–52. [DOI] [PubMed] [Google Scholar]
- 11. Chang IY, Youn CK, Kim HB et al. Oncogenic H‐Ras up‐regulates expression of Ku80 to protect cells from gamma‐ray irradiation in NIH3T3 cells. Cancer Res 2005; 65: 6811–9. [DOI] [PubMed] [Google Scholar]
- 12. Maeda Y, Fujimura L, O‐Wang J et al. Role of Clast1 in development of cerebellar granule cells. Brain Res 2006; 1104: 18–26. [DOI] [PubMed] [Google Scholar]
- 13. Lurton J, Rose TM, Raghu G, Narayanan AS. Isolation of a gene product expressed by a subpopulation of human lung fibroblasts by differential display. Am J Respir Cell Mol Biol 1999; 20: 327–31. [DOI] [PubMed] [Google Scholar]
- 14. Rodenhuis S, Slebos RJ, Boot AJ et al. Incidence and possible clinical significance of K‐ras oncogene activation in adenocarcinoma of the human lung. Cancer Res 1988; 48: 5738–41. [PubMed] [Google Scholar]
- 15. You M, Candrian U, Maronpot RR, Stoner GD, Anderson MW. Activation of the Ki‐ras protooncogene in spontaneously occurring and chemically induced lung tumors of the strain A mouse. Proc Natl Acad Sci USA 1989; 86: 3070–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Contente S, Kenyon K, Sriraman P, Subramanyan S, Friedman RM. Epigenetic inhibition of lysyl oxidase transcription after transformation by ras oncogene. Mol Cell Biochem 1999; 194: 79–91. [DOI] [PubMed] [Google Scholar]
- 17. Lorentz O, Cadoret A, Duluc I et al. Downregulation of the colon tumour‐suppressor homeobox gene Cdx‐2 by oncogenic ras. Oncogene 1999; 18: 87–92. [DOI] [PubMed] [Google Scholar]
- 18. Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat Genet 2000; 25: 315–9. [DOI] [PubMed] [Google Scholar]
- 19. Chang HC, Cho CY, Hung WC. Silencing of the metastasis suppressor RECK by RAS oncogene is mediated by DNA methyltransferase 3b‐induced promoter methylation. Cancer Res 2006; 66: 8413–20. [DOI] [PubMed] [Google Scholar]
- 20. Akino K, Toyota M, Suzuki H et al. The Ras effector RASSF2 is a novel tumor‐suppressor gene in human colorectal cancer. Gastroenterology 2005; 129: 156–69. [DOI] [PubMed] [Google Scholar]
- 21. Chang HC, Liu LT, Hung WC. Involvement of histone deacetylation in ras‐induced down‐regulation of the metastasis suppressor RECK. Cell Signal 2004; 16: 675–9. [DOI] [PubMed] [Google Scholar]
- 22. Soejima K, Fang W, Rollins BJ. DNA methyltransferase 3b contributes to oncogenic transformation induced by SV40T antigen and activated Ras. Oncogene 2003; 22: 4723–33. [DOI] [PubMed] [Google Scholar]
- 23. Ordway JM, Williams K, Curran T. Transcription repression in oncogenic transformation: common targets of epigenetic repression in cells transformed by Fos, Ras or Dnmt1. Oncogene 2004; 23: 3737–48. [DOI] [PubMed] [Google Scholar]
- 24. Pruitt K, Ulku AS, Frantz K et al. Ras‐mediated loss of the pro‐apoptotic response protein Par‐4 is mediated by DNA hypermethylation through Raf‐independent and Raf‐dependent signaling cascades in epithelial cells. J Biol Chem 2005; 280: 23363–70. [DOI] [PubMed] [Google Scholar]
- 25. Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer 2003; 3: 459–65. [DOI] [PubMed] [Google Scholar]
- 26. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 2003; 3: 11–22. [DOI] [PubMed] [Google Scholar]
- 27. Rodriguez‐Viciana P, Warne PH, Khwaja A et al. Role of phosphoinositide 3‐OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell 1997; 89: 457–67. [DOI] [PubMed] [Google Scholar]
- 28. Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell 1994; 77: 841–52. [DOI] [PubMed] [Google Scholar]
- 29. Bradley MO, Kraynak AR, Storer RD, Gibbs JB. Experimental metastasis in nude mice of NIH 3T3 cells containing various ras genes. Proc Natl Acad Sci USA 1986; 83: 5277–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jiang K, Sun J, Cheng J, Djeu JY, Wei S, Sebti S. Akt mediates Ras downregulation of RhoB, a suppressor of transformation, invasion, and metastasis. Mol Cell Biol 2004; 24: 5565–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rosen K, Rak J, Jin J, Kerbel RS, Newman MJ, Filmus J. Downregulation of the pro‐apoptotic protein Bak is required for the ras‐induced transformation of intestinal epithelial cells. Curr Biol 1998; 8: 1331–4. [DOI] [PubMed] [Google Scholar]
- 32. Barradas M, Monjas A, Diaz‐Meco MT, Serrano M, Moscat J. The downregulation of the pro‐apoptotic protein Par‐4 is critical for Ras‐induced survival and tumor progression. EMBO J 1999; 18: 6362–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rong R, Montalbano J, Jin W et al. Oncogenic Ras‐mediated downregulation of Gadd153/CHOP is required for Ras‐induced cellular transformation. Oncogene 2005; 24: 4867–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Clast1/LR8 expression is downregulated as a result of oncogenic Ras activation (a) Differential display RT‐PCR analysis of H‐RasV12‐transfected and empty vector‐transfected NIH3T3 cells. NIH3T3 cells were stably transfected with empty vector (NIH3T3/vector) or an H‐RasV12 expression vector (H‑RasV12 NIH3T3). mRNA extracted from these cells was analyzed by annealing control primer (ACP)‐based differential display RT‐PCR. It was first used to synthesize first‐strand cDNA using the primer dT‐ACP1 (cDNA synthesis primer). Second‐strand cDNA was then amplified by PCR using ACP34 (arbitrary; forward primer) and dT‐ACP2 (reverse primer), yielding a 343‐bp product. Arrows indicate differential expression between H‐RasV12‐transfected and empty vector‐transfected cells. (b) Sequence of the cloned cDNA. The sequence between 937 and 1211 (highlighted in red) was completely homologous to that of mouse Clast1/LR8 (GenBank accession number AB031386).
Fig. S2. Transient expression of Clast1/LR8 inhibits an H‐RasV12‐mediated increase in colony formation in soft agar. (a) The indicated plasmids were transiently transfected into H‐RasV12 NIH3T3 cells, and colony formation assay was carried out in soft agar. (b) Percentages of cells present in colonies >50 μm in diameter. Values represent the mean ± SD of three separate experiments. **P ≤ 0.01.
Fig. S3. Transient expression of Clast1/LR8 suppresses H‐RasV12‐induced cell invasion. (a) The indicated plasmids were transiently transfected into H‐RasV12 NIH3T3 cells, and an in vitro invasion assay was carried out. (b) Mean number of invading cells. Values represent the mean ± SD of three separate experiments. **P ≤ 0.01.
Fig. S4. Transient expression of Clast1/LR8 inhibits H‐RasV12‐induced tumor formation in nude mice. One million cells of each type indicated were injected s.c. into the right flanks of 5‐week‐old nude mice and tumor volumes were assessed every 2–5 days for 20 days. Images show representative mice from each injection group.
Data S1. Annealing control primer (ACP)‐based differential display RT‐PCR.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item