

Abstract
The role of Wnt antagonists in the carcinogenesis of esophageal adenocarcinoma (EAC) remains unclear. We hypothesized that downregulation of the Wnt inhibitory factor‐1 (WIF‐1) might be involved in the neoplastic progression of Barrett's esophagus (BE). We analyzed the DNA methylation status of the WIF‐1 promoter in normal, preneoplastic, and neoplastic samples from BE patients and in EAC cell lines. We investigated the role of WIF‐1 on EAC cell growth and the chemosensitization of the cells to cisplatin. We found that silencing of WIF‐1 correlated with promoter hypermethylation. EAC tissue samples showed higher levels of WIF‐1 methylation compared to the matched normal epithelium. In addition, we found that WIF‐1 hypermethylation was more frequent in BE samples from patients with EAC than in BE samples from patients who had not progressed to EAC. Restoration of WIF‐1 in cell lines where WIF‐1 was methylation‐silenced resulted in growth suppression. Restoration of WIF‐1 could sensitize the EAC cells to the chemotherapy drug cisplatin. Our results suggest that silencing of WIF‐1 through promoter hypermethylation is an early and common event in the carcinogenesis of BE. Restoring functional WIF‐1 might be used as a new targeted therapy for the treatment of this malignancy. (Cancer Sci 2008; 99: 46–53)
Abbreviations:
- 5‐azadC
5‐aza‐2′‐deoxycytidine
- BE
Barrett's esophagus
- EAC
esophageal adenocarcinoma
- MSP
methylation‐specific polymerase chain reaction
- MS‐SSCA
methylation‐sensitive single‐strand conformation analysis
- PCR
polymerase chain reaction
- RT‐PCR
reverse transcription–polymerase chain reaction
- SFRP
secreted Frizzled‐related protein
- WIF‐1
Wnt inhibitory factor‐1.
BE is an acquired condition in which the normal squamous epithelium in the distal esophagus has been replaced by a metaplastic columnar epithelium, as a complication of chronic gastroesophageal reflux. The clinical significance of this disease is its associated predisposition to EAC, which arises ultimately after progression through a sequence of increasing degrees of dysplasia.( 1 ) Although the incidence of BE‐associated EAC in developed countries has been constantly increasing for the last two decades, the reasons for this remain unknown.( 2 ) Despite advances in treatment, EAC is still a highly lethal disease. New therapies based on better understanding of the molecular alterations occurring during the evolution from BE to EAC are therefore in great need.
Growing evidence has implicated aberrant activation of the Wnt signaling pathway in the pathogenesis of a broad range of cancers.( 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 ) In the absence of the secreted Wnt molecules, cytosolic β‐catenin is phosphorylated by glycogen synthase kinase β in a multiprotein complex including adenomatous polyposis coli, axin, and casein kinase 1. Following phosphorylation, β‐catenin is targeted for degradation through the ubiquitin proteosome pathway. Binding of Wnt proteins to their Frizzled membrane receptors triggers the phosphorylation of the cytoplasmic effector Dishevelled. Dishevelled activity alters the glycogen synthase kinase β phosphorylation of the adenomatous polyposis coli–axin complex and therefore the phosphorylation of β‐catenin. This results in an increased cytoplasmic pool of β‐catenin, which can subsequently translocate to the nucleus, where it binds to members of the TCF/LEF family of transcription factors to promote the expression of TCF‐target genes.( 11 , 12 )
Modulation of Wnt signaling on the cell surface occurs through two classes of Wnt antagonists based on different mechanisms of action. The first class, including SFRP and WIF‐1, binds directly to the Wnt proteins. The second class inhibits Wnt signaling through binding to the Frizzled co‐receptor LRP5/6 and comprises the Dickkopf family.( 13 )
Epigenetic silencing of the WIF‐1 gene has already been described in various types of human cancer,( 14 , 15 , 16 , 17 , 18 , 19 , 20 ) including the esophagus. Interestingly, one published study was exclusively based on squamous carcinomas,( 21 ) and in another histology was not specified.( 19 ) This highlights the novelty of this study. We determined to elucidate whether the WIF‐1 gene is silenced by promoter hypermethylation in EAC cell lines and tissue samples. In addition, we established when in the neoplastic progression of BE the methylation of the WIF‐1 promoter occurs. We also evaluated the role of WIF‐1 on the growth and chemosensitivity of EAC cells.
Materials and Methods
Cell lines and tissue samples. The human Barrett's‐associated adenocarcinoma cell lines TE‐7, BIC‐1, and SEG‐1 were kindly provided by Dr Michael Korn (University of California, San Francisco). OE19 and OE33 cell lines were obtained from the European Collection of Cell Culture (Salisbury, UK). TE‐7, OE19, and OE33 cells were routinely cultured in RPMI‐1640 medium and BIC‐1 and SEG‐1 cells in Dulbecco's modified Eagle's medium (DMEM). Both media were supplemented with 10% fetal bovine serum, penicillin (100 units/mL) and streptomycin (100 µg/mL). All cells were cultured at 37°C in a humid incubator with 5% CO2.
Fresh esophageal adenocarcinoma tissue (18 samples) and adjacent normal squamous esophageal epithelium (17 samples) from patients undergoing esophagectomy were collected at the time of surgery (University of California, San Francisco) and immediately snap‐frozen in liquid nitrogen. All 47 formalin‐fixed and paraffin‐embedded tissues analyzed in this study were microdissected. They included 15 biopsies from BE mucosa from patients who had not progressed to dysplasia or adenocarcinoma during follow‐up from 4 to 10 years, as well as four normal squamous esophageal epithelia, 16 Barrett's mucosa, and 12 EAC from esophagectomy specimens occurring in the context of BE and selected from the files of the Institute of Pathology, Lausanne, Switzerland. In addition, normal colon mucosa was used as control for the methylation analysis. The use of the human tissues was according to the guidelines of the local ethics committee.
RNA extraction and RT‐PCR. Total RNA was isolated using an extraction kit (RNeasy Mini Kit; Qiagen, Valencia, CA). RT‐PCR was carried out using the SuperScript One‐step RT‐PCR with Platinum Taq Kit (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Primer sequences for the human WIF‐1 cDNA are: 5′‐CCGAAATGGAGGCTTTTGTA‐3′ (forward) and 5′‐GTGTCTTCCATGCCAACCTT‐3′ (reverse). The housekeeping gene glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was amplified as an internal control.
DNA extraction and sodium bisulfite conversion. Genomic DNA of the cell lines and fresh tissue samples was extracted using the Qiagen DNeasy Tissue Kit following the manufacturer's protocol. Bisulfite modification of genomic DNA was carried out using an EZ DNA Methylation‐Gold Kit (Zymo Research, Orange, CA). Genomic DNA from the formalin‐fixed paraffin‐embedded tissue sections was extracted after manual microdissection of the lesion of interest as previously described.( 22 ) The total amount of DNA extracted from microdissected tissues was modified in 40 µL of water with sodium bisulfite as previously described.( 23 )
MSP, MS‐SSCA and sequencing analysis. Methylation‐specific PCR amplifications were carried out using HotStar Taq DNA polymerase (Qiagen) for 40 cycles. Primer sequences for the amplification of the methylated DNA are: 5′‐GGGCGTTTTATTGGGCGTAT‐3′ (forward) and 5′‐AAACCAACAATCAACGAAAC‐3′ (reverse). Sequences of the unmethylation‐specific primers are: 5′‐GGGTGTTTTATTGGGTGTAT‐3′ (forward) and 5′‐AAACCAACAATCAACAAAAC‐3′ (reverse). The fragment amplified corresponds to the WIF‐1 promoter region –488 to –291 (the ATG start codon of WIF‐1 was defined as +1).( 18 ) Methylation analysis of DNA extracted from the formalin‐fixed paraffin‐embedded tissue sections was done by MS‐SSCA. The WIF‐1 promoter was amplified by seminested PCR using the following primers: 5′‐GCCCGGTAGGTTTTTTGGTATTTAGGT‐3′ (forward) and 5′‐GCGGCAACCATACTACTCAAAACCTC‐3′ (reverse) for the outer PCR and 5′‐GGGAATAGTTTTGGTTGAGGGAGTTGT‐3′ (forward) for the inner PCR. Forty and 20 cycles were carried out for the outer and inner PCR, respectively. The amplified product includes eight CpG sites between –110 and +6 of the WIF‐1 promoter sequence. Single‐strand conformation analysis was carried out as described previously.( 23 ) Bisulfite‐treated genomic DNA extracted from EAC cell lines was amplified using two different pairs of primers to amplify nucleotides –554 to +118 of the WIF‐1 promoter region, which covers 64 CpG sites. The sequences of the two pairs of primers used are: 5′‐GAGTGATGTTTTAGGGGTTT‐3′ (forward) and 5′‐CCTAAATACCAAAAAACCTAC‐3′ (reverse); and 5′‐GTAGGTTTTTTGGTATTTAGG‐3′ (forward) and 5′‐TCCATAAATACAAACTCTCCTC‐3′ (reverse). The PCR products were cloned into the pCR2.1‐TOPO Vector using the TOPO TA Cloning Kit (Invitrogen) according to manufacturer's protocol. Ten colonies were randomly chosen for each PCR reaction. Sequencing of the clones was carried out at Quintara Biosciences (Berkeley, CA).
5‐azadC treatment. EAC cells were seeded in 6‐well plates and treated after 24 h with 2 µM 5‐azadC (Sigma, St. Louis, MO) in cultured medium for 6 days. Medium was changed after 3 days and fresh 5‐azadC was added. Then cells were harvested and genomic DNA and total RNA were extracted. The level of demethylation was checked by MSP and the expression of WIF‐1 was analyzed by RT‐PCR.
TOPflash assay, cell proliferation and cisplatin treatment. OE19 and BIC‐1 cells were plated in a 12‐well plate 24 h before transfection. Lipofectamine 2000 (Invitrogen) was used to mediate co‐transfection of TOPflash (0.5 µg) or FOPflash (0.5 µg) (vectors were kindly provided by H. Clevers) with a WIF‐1 cDNA construct in pcDNA3.1 vector (1.5 µg)( 24 ) or an empty pcDNA3.1 vector (1.5 µg; Invitrogen). The Renilla luciferase reporter vector (0.05 µg) (Promega, Madison, WI) was simultaneously transfected as the control for transfection efficiency. The expression level of introduced WIF‐1 was analyzed by RT‐PCR as described above. As a control for the assay, an SFRP4 cDNA construct in pcDNA3 vector( 25 ) was co‐transfected with the TOPflash or FOPflash in OE19 cells. Cells were harvested 48 h after transfection. Luciferase assays were carried out using the Dual‐Luciferase Reporter Assay System (Promega). The experiments were done in triplicate and repeated independently at least four times.
Cell proliferation was determined using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega). Briefly, OE19 cells were plated in a 6‐well plate 24 h before transfection. Transient transfection was carried out using 4 µg of the WIF‐1 cDNA construct or the empty pcDNA3.1 vector. Twenty‐four hours after transfection, cells were seeded in a 96‐well plate and cultured for a further 24 h before addition of the CellTiter 96 AQueous One Solution (day 0). Cisplatin (Sicor; SICOR Pharmaceuticals, Irvine, CA) was diluted in medium culture at a concentration of 0.1 µg/mL (0.33 µM) at day 0 and added fresh every day. Absorbance was reported at 490 nm. Each experiment was done in sixplicate and repeated at least three times.
Colony formation assay. Transfection was carried out with OE19 cells in 6‐well plates with 4 µg of WIF‐1 cDNA construct or empty pcDNA3.1 vector as described above. Transfected cells were collected and plated on 10‐cm cell culture dishes 24 h after transfection. The cells were then selected by G418 (400 µg/mL). After 10 days, cells were counted and 2000 stable transfected cells were plated on 10‐cm cell culture dishes in triplicate. The selection by G418 was maintained for four additional weeks. Colonies were stained by using 0.5% crystal violet and counted.
Statistical analysis. Data shown represent mean values (± standard deviation). The statistical significance of differences in levels of methylation between the two groups of BE (BE without EAC vs BE with EAC) was established using Fisher's exact test. Student's t‐test was used for comparing activities of different constructs and treatments.
Results
Silencing of WIF‐1 correlates with promoter hypermethylation in EAC cell lines. We examined WIF‐1 expression in five EAC cell lines (Fig. 1a). WIF‐1 expression was detectable in the normal esophagus control sample, but the transcript was missing in all EAC cell lines. MSP analysis revealed methylation of the WIF‐1 promoter in all the cell lines (Fig. 1b). However, for OE19, BIC‐1, and SEG‐1 cells both methylated and unmethylated bands were detected. Bisulfite sequencing showed that the CpG islands of TE‐7, OE19, and OE33 cell lines were densely methylated (Fig. 1c). Consistent with MSP results, we observed partial methylation of the CpG sites in BIC‐1 and SEG‐1 cells. We treated the EAC cells with a demethylating agent 5‐azadC and found that WIF‐1 expression was restored after the treatment (Fig. 1d). Taken together, these results suggest that the expression of WIF‐1 in EAC is regulated by hypermethylation of its promoter.
Figure 1.
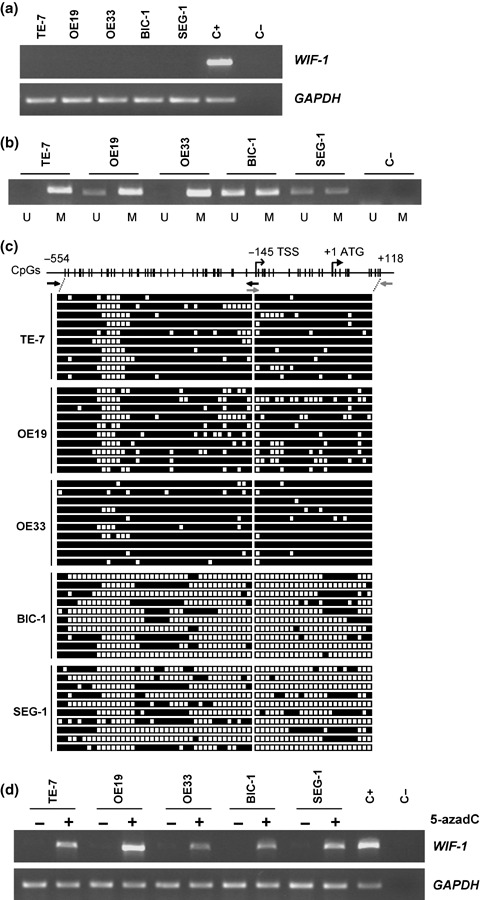
Correlation of methylation in the promoter region with silencing of the Wnt inhibitory factor‐1 (WIF‐1) gene in esophageal adenocarcinoma (EAC) cell lines. (a) Reverse transcription–polymerase chain reaction (RT‐PCR) analysis of the WIF‐1 gene in five EAC cell lines. The amplified fragment was 451 bp. A 180‐bp fragment of the glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) gene was used as a positive control for RNA quality and loading. C+ and C– corresponded to normal esophagus RNA (BD Biosciences Clontech, Palo Alto, CA) and H2O, respectively, used as a control during the RT‐PCR. (b) Methylation‐specific polymerase chain reaction analysis of WIF‐1 methylation in EAC cell lines. Bands (198 bp) in lanes labeled “U” are unmethylated DNA product amplified with unmethylation‐specific primers. Bands in lanes labeled “M” are methylated DNA product amplified with methylation‐specific primers. (c) Bisulfite‐sequencing analysis of 64 CpG sites in the region –554 to +118 of the WIF‐1 promoter in EAC cell lines. White squares represent unmethylated CpG sites and black squares indicate methylated CpG sites. Black and gray arrows represent the primers used to amplify the promoter regions –554 to –141 and –161 to +118, respectively. Ten individual clones were analyzed for each cell line. Transcription (TSS) and translation (ATG) start sites are represented (RefSeq NM_007191). (d) Reactivation of WIF‐1 expression by 5‐aza‐2′‐deoxycytidine (5‐azadC) treatment in cell lines. RT‐PCR analysis of the WIF‐1 gene in EAC cell lines after treatment (2 µM for 6 days) or without treatment with the demethylating agent 5‐azadC. The GAPDH gene was used as a positive control for RNA quality and loading. C+ and C– corresponded to normal esophagus RNA and H2O, respectively, used as a control during the RT‐PCR.
WIF‐1 promoter is methylated in EAC tissue samples. We investigated the methylation status of the WIF‐1 promoter in 18 frozen EAC tissue samples (17 were matched pairs of normal and tumor tissue). We found aberrant methylation of the WIF‐1 promoter in 11/18 tumor samples (61%), whereas methylation was not detected in seven matched pairs of normal and tumor samples (Fig. 2a). Cases 6, 7, 10 and 14 showed some methylation in both tumor and their matched normal samples. This might be due to contamination of tumor cells in the normal specimen or to premalignant changes of the squamous epithelium adjacent to EAC. Similarly, MSP revealed unmethylated bands in every methylated tumor sample. This could be interpreted as contamination of normal cells in the tumor specimens or partial methylation of the WIF‐1 promoter (mixture of methylated and unmethylated alleles).
Figure 2.
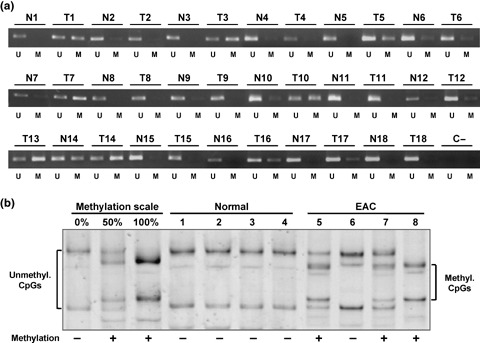
Methylation analysis of the Wnt inhibitory factor‐1 (WIF‐1) promoter in esophageal adenocarcinoma (EAC) tissue samples. (a) Methylation‐specific polymerase chain reaction analysis of 17 matched pairs of frozen normal (N) and EAC (T) tissue samples and of one unpaired EAC sample (T13). Bands (198 bp) in lanes labeled “U” are unmethylated DNA product amplified with unmethylation‐specific primers. Bands in lanes labeled “M” are methylated DNA product amplified with methylation‐specific primers. (b) WIF‐1 promoter methylation by methylation‐sensitive single‐strand conformation analysis in four microdissected formalin‐fixed paraffin‐embedded normal squamous epithelia and four EAC samples. Square brackets indicate the migration of either the umethylated DNA (unmethyl. CpGs) or the methylated DNA (methyl. CpGs). + and – indicate the methylation status of each sample.
In addition, we examined the methylation status of the WIF‐1 promoter in 12 microdissected formalin‐fixed paraffin‐embedded EAC specimens and four adjacent normal squamous epithelia using MS‐SSCA (Fig. 2b). The methylation scale was carried out as described previously.( 26 ) None of the normal squamous epithelia were methylated. Ten of 12 (83%) microdissected tumor samples were methylated for the WIF‐1 promoter. Among those, three showed full methylation, and seven had unmethylated and methylated WIF‐1 alleles at varying ratios. A mixture of methylated and unmethylated alleles can be explained by either monoallelic methylation of the WIF‐1 promoter or by the existence of subpopulations of cells, some with methylation of the WIF‐1 promoter and some without.
Hypermethylation of the WIF‐1 promoter occurs early in the carcinogenesis of EAC. We further analyzed the methylation status of the WIF‐1 promoter in the premalignant lesions of EAC. Fifteen microdissected formalin‐fixed paraffin‐embedded BE samples from patients who had not progressed to dysplasia or EAC, as well as 16 non‐dysplastic microdissected fixed BE samples adjacent to EAC were subjected to MS‐SSCA (Fig. 3a,b). Methylation was detected in 3/15 (20%) BE samples from patients without EAC. Among those three samples, one showed complete CpG island methylation of the WIF‐1 promoter and the other two a mixture of methylated and unmethylated alleles. In contrast, WIF‐1 promoter hypermethylation was found in 13/16 (81%) BE samples from patients with EAC. Among those samples, four showed complete methylation of the WIF‐1 promoter, and nine had a mixture of methylated and unmethylated WIF‐1 alleles. Statistical analysis of the difference between the methylated and unmethylated samples in the two groups of BE (BE without EAC versus BE with EAC) was highly significant (P < 0.001).
Figure 3.
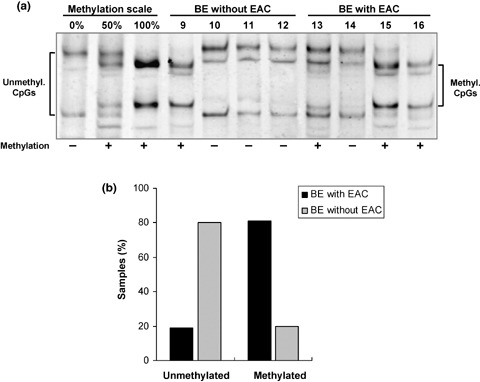
Methylation analysis of the Wnt inhibitory factor‐1 (WIF‐1) promoter in Barrett's esophagus (BE) tissue samples. (a) WIF‐1 promoter methylation by methylation‐sensitive single‐strand conformation analysis in four BE patients without esophageal adenocarcinoma (EAC) and in four BE patients with EAC. Square brackets indicate the migration of either the umethylated DNA (unmethyl. CpGs) or the methylated DNA (methyl. CpGs). + and – indicate the methylation status of each sample. (b) Graphical representation of the methylated and unmethylated samples of BE without and BE with EAC.
WIF‐1 expression modulates the Wnt signaling pathway in EAC cell lines. We subsequently restored WIF‐1 expression in the OE19 and BIC‐1 cell lines by transiently transfecting a WIF‐1 expression vector and analyzed the Wnt signaling pathway activity by simultaneously co‐transfecting the TOPflash vector. RT‐PCR analysis confirmed that WIF‐1 was overexpressed in the cells transiently transfected with the WIF‐1 expression vector compared to the cells transfected with the empty vector pcDNA3.1 (Fig. 4). Restoration of WIF‐1 was able to decrease the TOPflash activity by approximately 39% and 36% in OE19 and BIC‐1 cells (P < 0.05), respectively, compared to the TOPflash activity after co‐transfection with the empty expression vector pcDNA3.1 (Fig. 4). To validate the accuracy of this assay in our cell lines, the TOPflash vector was co‐transfected with an SFRP4 expression vector in OE19 cells that lacked SFRP4 expression (data not shown), because restoration of SFRP4 has been shown to modulate the Wnt signaling pathway.( 27 ) As expected, restoration of SFRP4 decreased the TOPflash activity by approximately 20%. As a negative control for this assay, we confirmed that forced expression of SOCS‐3, a Wnt signaling independent factor,( 28 ) did not modulate the Wnt signaling pathway (data not shown).
Figure 4.
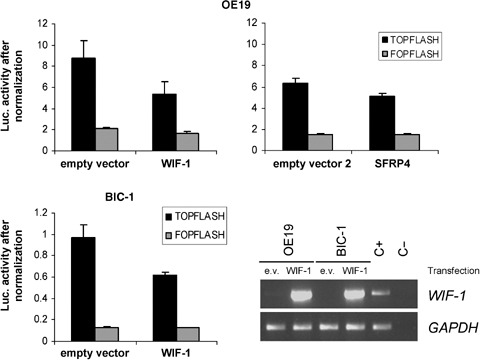
Effect of the restoration of Wnt inhibitory factor‐1 (WIF‐1) expression on the Wnt‐dependent transcription activity. OE19 and BIC‐1 cells were co‐transfected with 1.5 µg of WIF‐1 expression vector or empty vector (pcDNA3.1) and 0.5 µg of TOPflash or FOPflash reporter. As a positive control, OE19 cells were cotransfected with 1.5 µg of SFPR4 expression vector or empty vector 2 (pcDNA3) and 0.5 µg of TOPflash or FOPflash reporter. Firefly luciferase activity of the reporters is represented after normalization of each sample with the Renilla luciferase activity. The experiment was carried out in triplicate. The expression level of WIF‐1 in OE19 and BIC‐1 cells was detected by reverse transcription–polymerase chain reaction (RT‐PCR) after transfection of the empty vector (e.v.) or WIF‐1 expression vector. The amplified fragment was 451 bp. A 180‐bp fragment of the glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) gene was used as a positive control for RNA quality and loading. C+, normal esophagus RNA; C–, H2O, used as a control during RT‐PCR.
Restoration of WIF‐1 suppresses EAC cell growth. We carried out a cell proliferation assay after transient transfection with a WIF‐1 expression vector in OE19 cells. One week after transfection, the proliferation of the empty vector transfected OE19 cells reached 725% (100% corresponded to the cell proliferation at day 0), whereas the proliferation of the WIF‐1 transfected cells was 381% (approximately 47% decrease) (Fig. 5a).
Figure 5.
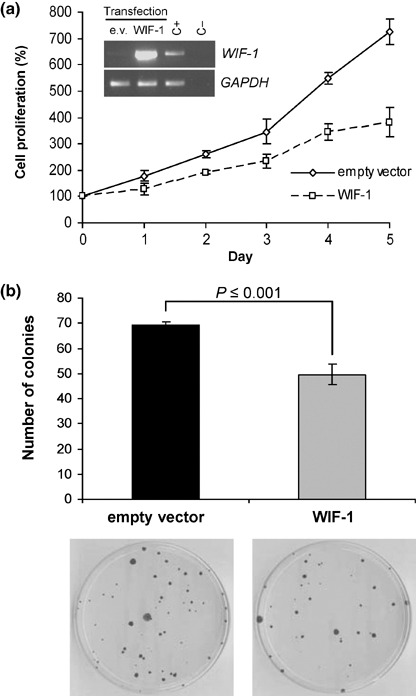
Growth suppression of OE19 cells by restoration of Wnt inhibitory factor‐1 (WIF‐1) expression. (a) Cell proliferation of OE19 cells after transfection of WIF‐1 expression vector or empty vector. Measurements were carried out 2 days after transfection (day 0) and repeated during the following five consecutive days. Results are means of sixplicates with error bars (standard deviation [SD]). Expression level of introduced WIF‐1 was detected by reverse transcription–polymerase chain reaction as for Fig. 4. (b) Colony formation assay using OE19 cells. The cells were transfected with empty vector or WIF‐1 expression vector, selected with G418 for 10 days and 2000 cells were plated in triplicate in 10‐cm dishes. Selection was maintained for four additional weeks. The bar graph represents the average of colony numbers of the triplicated experiments. Error bars are SD.
In addition, after selection of G418‐resistant colonies, we found that the colony numbers of WIF‐1 transfected OE19 cells significantly decreased compared to that of empty vector transfected cells (P ≤ 0.001) (Fig. 5b). Taken together, our data indicate that restoration of WIF‐1 suppresses EAC cell growth.
Restoration of WIF‐1 sensitizes EAC cells to the chemotherapy drug cisplatin. Lastly, we exposed the cells to cisplatin, a drug commonly used for the treatment of EAC patients, after transient transfection of a WIF‐1 expression vector. After 1 week of transfection and exposure to 0.1 µg/mL cisplatin, the cell proliferation of the empty vector transfected cells was higher than that of WIF‐1 transfected cells (646%vs 271%) (Fig. 6a). Most interestingly, WIF‐1 restoration conjugated to cisplatin treatment decreased cell growth (approximately 62%) more than WIF‐1 restoration or cisplatin treatment alone (47% and 11%, respectively) (P < 0.05), suggesting that WIF‐1 might sensitize EAC cells to cisplatin treatment (Fig. 6b).
Figure 6.
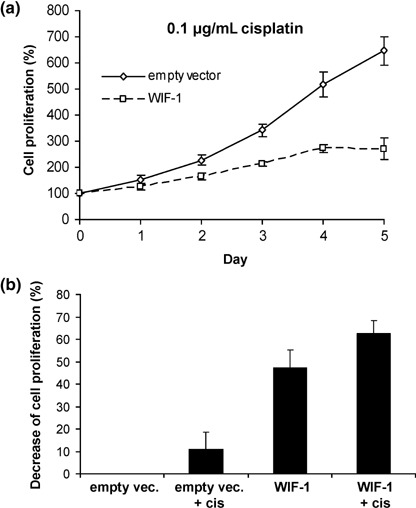
Growth suppression of OE19 cells by restoration of Wnt inhibitory factor‐1 (WIF‐1) expression and treatment with the chemotherapy drug cisplatin. (a) Cell proliferation of OE19 cells after transfection of WIF‐1 expression vector or empty vector. Cells were subjected to cisplatin 2 days after transfection (day 0) and measurements of the cell proliferation were repeated during the following five consecutive days. Cisplatin was added fresh every day. Results are means of sixplicates with error bars (standard deviation). (b) Graphical representation of the cell proliferation decrease at day 5 for each treatment.
Discussion
Numerous studies have established the crucial role of the Wnt signaling pathway in the pathogenesis of many human cancers, including the finding of nuclear accumulation of β‐catenin early in the transition from BE to EAC.( 3 , 29 ) Likewise, overexpression of Wnt signaling components as well as downregulation of Wnt antagonist SFRP have been confirmed along the EAC carcinogenesis sequence.( 10 , 30 )
In the present study, we investigated the methylation status of one of the Wnt antagonists WIF‐1 and its role in EAC. We found that WIF‐1 is downregulated in EAC cell lines and that frequent dense hypermethylation of the WIF‐1 promoter region is correlated with its silencing. Similar to the cell lines, WIF‐1 promoter hypermethylation was also commonly found in EAC tissue samples. Taken together, our data suggest that aberrant hypermethylation of the WIF‐1 promoter is a frequent epigenetic event leading to WIF‐1 silencing in EAC.
The majority of EAC is diagnosed at an advanced stage, and is associated with poor prognosis. Only early detection of cancer or preneoplastic lesions will allow effective treatment resulting in decreased mortality. Currently it is not possible to predict which BE patients will progress through dysplasia to EAC. Because the WIF‐1 promoter is frequently methylated and occurs early during the neoplastic progression of BE (we found WIF‐1 promoter methylation in 81% of BE patients with EAC but in only 20% of the BE patients without EAC), WIF‐1 promoter hypermethylation could be used as a marker to distinguish progressive from non‐progressive BE. However, confirmation in a larger cohort of samples is needed before it can be used clinically. Interestingly, the SFRP1 promoter was hypermethylated in BE patients with EAC as well as in BE patients without EAC.( 31 ) Therefore, we hypothesize that SFRP1 and WIF‐1 might have distinct roles in EAC carcinogenesis. Epigenetic alterations of the SFRP1 promoter might be associated with dysregulation of differentiation, leading to BE; epigenetic alterations of the WIF‐1 promoter might be associated with EAC carcinogenesis.
We showed that restoration of WIF‐1 in EAC cell lines lacking WIF‐1 expression decreased the activity of the Wnt/β‐catenin signaling pathway. The situation in EAC tissue samples seems more controversial. To date, several studies have reported Wnt signaling activation, indicated by nuclear β‐catenin accumulation, only in the presence of low‐grade dysplasia.( 29 , 32 ) Nevertheless, we observed that promoter hypermethylation of the WIF‐1 gene had already occurred in BE samples. Although those studies were carried out by several groups using different tissue samples, the data might indicate either that downregulation of WIF‐1 alone is not sufficient for the activation of the pathway, or that other concurrent alterations (overexpression of specific Wnt ligands and/or mutations of downstream components) are necessary to activate the pathway during BE neoplastic progression. It is also possible that WIF‐1 is involved in other pathways. Recently, Ohigashi et al. showed that WIF‐1 downregulates the PI3K/Akt pathway,( 33 ) supporting this hypothesis. In addition, the WIF domain shares homology with the extracellular domain of the RYK family of receptor tyrosine kinases.( 13 , 34 )
Targeted inhibition of Wnt signaling has been shown to induce apoptosis and inhibit cancer cell growth.( 7 , 24 , 25 , 27 , 35 , 36 , 37 ) When we restored WIF‐1 expression in EAC cell lines in which WIF‐1 was methylation silenced, cell proliferation and colony formation were inhibited. Furthermore, we showed that restoration of WIF‐1 in cells lacking WIF‐1 expression sensitize the cancer cells to the chemotherapy drug cisplatin. Taken together, our findings suggest that restoration of WIF‐1 alone or in combination with traditional chemotherapy drugs might offer a new targeted therapy for the treatment of EAC patients.
In conclusion, this study highlights a new epigenetic event in the carcinogenesis of EAC. Aberrant WIF‐1 promoter hypermethylation appears to occur early in the neoplastic progression of BE. Methylation of WIF‐1 promoter was observed more frequently in BE patients with EAC than in BE patients who had not progressed to cancer. Therefore, epigenetic alteration of WIF‐1 could be used as a new diagnostic and predictive marker for increased EAC risk in BE patients. Finally, the high prevalence of WIF‐1 hypermethylation in EAC patients supports the potential development of novel therapies exploiting the restoration of WIF‐1 function.
Acknowledgments
This study was partially supported by a grant from the US National Institutes of Health (RO1 CA 093708–01A3), the Larry Hall and Zygielbaum Memorial Trust, the Kazan, McClain, Edises, Abrams, Fernandez, Lyons and Farrise Foundation and by a grant from the Swiss Cancer League/Oncosuisse (OCS 1638‐02‐2005). G.C. was supported by the Swiss National Science Foundation.
References
- 1. Flejou JF. Barrett's oesophagus: from metaplasia to dysplasia and cancer. Gut 2005; 54 (Suppl 1): i6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blot WJ, McLaughlin JK. The changing epidemiology of esophageal cancer. Semin Oncol 1999; 26: 2–8. [PubMed] [Google Scholar]
- 3. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 2004; 20: 781–810. [DOI] [PubMed] [Google Scholar]
- 4. Polakis P. Wnt signaling and cancer. Genes Dev 2000; 14: 1837–51. [PubMed] [Google Scholar]
- 5. Morin PJ, Sparks AB, Korinek V et al . Activation of beta‐catenin‐Tcf signaling in colon cancer by mutations in beta‐catenin or APC. Science 1997; 275: 1787–90. [DOI] [PubMed] [Google Scholar]
- 6. Uematsu K, He B, You L et al . Activation of the Wnt pathway in non small cell lung cancer: evidence of dishevelled overexpression. Oncogene 2003; 22: 7218–21. [DOI] [PubMed] [Google Scholar]
- 7. Uematsu K, Kanazawa S, You L et al . Wnt pathway activation in mesothelioma: evidence of Dishevelled overexpression and transcriptional activity of beta‐catenin. Cancer Res 2003; 63: 4547–51. [PubMed] [Google Scholar]
- 8. Weeraratna AT, Jiang Y, Hostetter G et al . Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 2002; 1: 279–88. [DOI] [PubMed] [Google Scholar]
- 9. Rhee CS, Sen M, Lu D et al . Wnt and frizzled receptors as potential targets for immunotherapy in head and neck squamous cell carcinomas. Oncogene 2002; 21: 6598–605. [DOI] [PubMed] [Google Scholar]
- 10. Clement G, Braunschweig R, Pasquier N, Bosman FT, Benhattar J. Alterations of the Wnt signaling pathway during the neoplastic progression of Barrett's esophagus. Oncogene 2006; 25: 3084–92. [DOI] [PubMed] [Google Scholar]
- 11. Behrens J, Von Kries JP, Kuhl M et al . Functional interaction of beta‐catenin with the transcription factor LEF‐1. Nature 1996; 382: 638–42. [DOI] [PubMed] [Google Scholar]
- 12. Giles RH, Van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta 2003; 1653: 1–24. [DOI] [PubMed] [Google Scholar]
- 13. Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci 2003; 116: 2627–34. [DOI] [PubMed] [Google Scholar]
- 14. Urakami S, Shiina H, Enokida H et al . Epigenetic inactivation of Wnt inhibitory factor‐1 plays an important role in bladder cancer through aberrant canonical Wnt/beta‐catenin signaling pathway. Clin Cancer Res 2006; 12: 383–91. [DOI] [PubMed] [Google Scholar]
- 15. Wissmann C, Wild PJ, Kaiser S et al . WIF1, a component of the Wnt pathway, is down‐regulated in prostate, breast, lung, and bladder cancer. J Pathol 2003; 201: 204–12. [DOI] [PubMed] [Google Scholar]
- 16. Ai L, Tao Q, Zhong S et al . Inactivation of Wnt inhibitory factor‐1 (WIF1) expression by epigenetic silencing is a common event in breast cancer. Carcinogenesis 2006; 27: 1341–8. [DOI] [PubMed] [Google Scholar]
- 17. Mazieres J, He B, You L et al . Wnt inhibitory factor‐1 is silenced by promoter hypermethylation in human lung cancer. Cancer Res 2004; 64: 4717–20. [DOI] [PubMed] [Google Scholar]
- 18. Batra S, Shi Y, Kuchenbecker KM et al . Wnt inhibitory factor‐1, a Wnt antagonist, is silenced by promoter hypermethylation in malignant pleural mesothelioma. Biochem Biophys Res Commun 2006; 342: 1228–32. [DOI] [PubMed] [Google Scholar]
- 19. Taniguchi H, Yamamoto H, Hirata T et al . Frequent epigenetic inactivation of Wnt inhibitory factor‐1 in human gastrointestinal cancers. Oncogene 2005; 24: 7946–52. [DOI] [PubMed] [Google Scholar]
- 20. Lin YC, You L, Xu Z et al . Wnt signaling activation and WIF‐1 silencing in nasopharyngeal cancer cell lines. Biochem Biophys Res Commun 2006; 341: 635–40. [DOI] [PubMed] [Google Scholar]
- 21. Chan SL, Cui Y, Van Hasselt A et al . The tumor suppressor Wnt inhibitory factor 1 is frequently methylated in nasopharyngeal and esophageal carcinomas. Laboratory Invest 2007; 87: 644–50. [DOI] [PubMed] [Google Scholar]
- 22. Baisse B, Bian YS, Benhattar J. Microdissection by exclusion and DNA extraction for multiple PCR analyses from archival tissue sections. Biotechniques 2000; 28: 856–8, 860, 862. [PubMed] [Google Scholar]
- 23. Benhattar J, Clement G. Methylation‐sensitive single‐strand conformation analysis. a rapid method to screen for and analyze DNA methylation. Methods Mol Biol 2004; 287: 181–93. [DOI] [PubMed] [Google Scholar]
- 24. He B, Reguart N, You L et al . Blockade of Wnt‐1 signaling induces apoptosis in human colorectal cancer cells containing downstream mutations. Oncogene 2005; 24: 3054–8. [DOI] [PubMed] [Google Scholar]
- 25. He B, Lee AY, Dadfarmay S et al . Secreted frizzled‐related protein 4 is silenced by hypermethylation and induces apoptosis in beta‐catenin‐deficient human mesothelioma cells. Cancer Res 2005; 65: 743–8. [PubMed] [Google Scholar]
- 26. Clement G, Benhattar J. A methylation sensitive dot blot assay (MS‐DBA) for the quantitative analysis of DNA methylation in clinical samples. J Clin Pathol 2005; 58: 155–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee AY, He B, You L et al . Expression of the secreted frizzled‐related protein gene family is downregulated in human mesothelioma. Oncogene 2004; 23: 6672–6. [DOI] [PubMed] [Google Scholar]
- 28. He B, You L, Uematsu K et al . SOCS‐3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc Natl Acad Sci USA 2003; 100: 14133–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bian YS, Osterheld MC, Bosman FT, Fontolliet C, Benhattar J. Nuclear accumulation of beta‐catenin is a common and early event during neoplastic progression of Barrett esophagus. Am J Clin Pathol 2000; 114: 583–90. [DOI] [PubMed] [Google Scholar]
- 30. Zou H, Molina JR, Harrington JJ et al . Aberrant methylation of secreted frizzled‐related protein genes in esophageal adenocarcinoma and Barrett's esophagus. Int J Cancer 2005; 116: 584–91. [DOI] [PubMed] [Google Scholar]
- 31. Clement G, Braunschweig R, Pasquier N, Bosman FT, Benhattar J. Methylation of APC, TIMP3, and TERT: a new predictive marker to distinguish Barrett's oesophagus patients at risk for malignant transformation. J Pathol 2006; 208: 100–7. [DOI] [PubMed] [Google Scholar]
- 32. Seery JP, Syrigos KN, Karayiannakis AJ, Valizadeh A, Pignatelli M. Abnormal expression of the E‐cadherin–catenin complex in dysplastic Barrett's oesophagus. Acta Oncol 1999; 38: 945–8. [DOI] [PubMed] [Google Scholar]
- 33. Ohigashi T, Mizuno R, Nakashima J, Marumo K, Murai M. Inhibition of Wnt signaling downregulates Akt activity and induces chemosensitivity in PTEN‐mutated prostate cancer cells. Prostate 2005; 62: 61–8. [DOI] [PubMed] [Google Scholar]
- 34. Patthy L. The WIF module. Trends Biochem Sci 2000; 25: 12–13. [DOI] [PubMed] [Google Scholar]
- 35. You L, He B, Xu Z et al . An anti‐Wnt‐2 monoclonal antibody induces apoptosis in malignant melanoma cells and inhibits tumor growth. Cancer Res 2004; 64: 5385–9. [DOI] [PubMed] [Google Scholar]
- 36. Mazieres J, You L, He B et al . Inhibition of Wnt16 in human acute lymphoblastoid leukemia cells containing the t(1;19) translocation induces apoptosis. Oncogene 2005; 24: 5396–400. [DOI] [PubMed] [Google Scholar]
- 37. Mazieres J, You L, He B et al . Wnt2 as a new therapeutic target in malignant pleural mesothelioma. Int J Cancer 2005; 117: 326–32. [DOI] [PubMed] [Google Scholar]