

Abstract
The development of a diagnostic method for predicting the therapeutic efficacy or toxicity of anticancer drugs is a critical issue. We carried out a gene expression analysis to identify genes whose expression profiles were correlated with the sensitivity of 30 human tumor xenografts to 5‐fluorouracil (5‐FU)‐based drugs (tegafur + uracil [UFT], tegafur + gimeracil + oteracil [S‐1], 5′‐deoxy‐5‐fluorouridine [5′‐DFUR], and N4‐pentyloxycarbonyl‐5′‐deoxy‐5‐fluorocytidine [capecitabine]), as well as three other drugs (cisplatin [CDDP], irinotecan hydrochloride [CPT‐11], and paclitaxel) that have different modes of action. In the present study, we focused especially on the fluoropyrimidines. The efficacy of all anticancer drugs was assayed using human tumor xenografts in nude mice. The mRNA expression profile of each of these xenografts was analyzed using a Human Focus array. Correlation analysis between the gene expression profiles and the chemosensitivities of seven drugs identified 39 genes whose expression levels were correlated significantly with multidrug sensitivity, and we suggest that the angiogenic pathway plays a pivotal role in resistance to fluoropyrimidines. Furthermore, many genes showing specific correlations with each drug were also identified. Among the candidate genes associated with 5‐FU resistance, the dihydropyrimidine dehydrogenase mRNA expression profiles of the tumors showed a significant negative correlation with chemosensitivity to all of the 5‐FU based drugs except for S‐1. Therefore, the administration of S‐1 might be an effective strategy for the treatment of high dihydropyrimidine dehydrogenase‐expressing tumors. The results of the present study may enhance the prediction of tumor response to anticancer drugs and contribute to the development of tailor‐made chemotherapy. (Cancer Sci 2006; 97: 510 – 522)
Many chemotherapeutic agents have been used to treat cancer patients; however, the emergence of drug resistance has prevented successful treatment in many cases. A large population of cancer patients suffers from the adverse effects of chemotherapy without achieving any benefit in terms of a good response. Differences in the efficacy of anticancer drugs among patients have been associated with variations in polymorphisms and gene expression profiles in cancer cells,( 1 , 2 , 3 ) and predicting tumor response based on valid markers is important because patients who are unlikely to respond to a treatment can avoid the adverse effects of unsuccessful treatments and be placed on alternative regimens. Furthermore, a maximal response during the course of the first regimen is important to avoid the acquisition of drug resistance. Hence, the development of tailor‐made chemotherapy regimens, which would select a suitable regimen for each patient based on biological features (including genomic factors and gene expression profiles), is a very critical issue.
Combination therapy is now a standard treatment for cancer patients. The rationale for combination chemotherapy is to use suitable anticancer drugs that are active on different cell populations of cancer tissue, thereby increasing the possibility that more cancer cells will be killed. Although various drug combinations have been evaluated recently in clinical trials,( 4 , 5 , 6 ) some combinations may not only decrease the prognosis, but may induce the adverse effects of the treatment. Hence, identifying genes that contribute to single or multiple drug resistance is important for selecting the optimal drug combination for each patient.
5‐fluorouracil is one of the most commonly used anticancer drugs in chemotherapy against various solid tumors.( 7 ) 5‐FU has two main modes of action that are realized through its active metabolites: FdUMP (5‐fluoro‐2′‐deoxyuridine‐5′‐monophosphate) and FUTP (fluorouridine‐5′‐triphosphate). FdUMP inhibits TS (Thymidylate synthase) by forming a covalent ternary complex with 5,10‐methylentetrahydrofolate that subsequently suppresses DNA synthesis, whereas FUTP is incorporated into RNA, resulting in the distortion of gene expression.( 8 , 9 ) DPD (dihydropyrimidine dehydrogenase), which is both an initial and a rate‐limiting catabolic enzyme of 5‐FU, has been reported to play an important role in the pharmacokinetics of 5‐FU,( 10 ) and 80% of 5‐FU is catabolized rapidly into inactive metabolites by DPD in the liver. Furthermore, DPD not only inactivates 5‐FU, but also produces fluoroacetate and fluorohy‐droxypropionic acid, which have been reported to induce cardiotoxicities and neurotoxicities.( 10 ) To resolve this problem, oral fluoropyrimidine derivatives were developed in the form of 5‐FU prodrugs (e.g. tegafur, 5′‐DFUR and capecitabine)( 11 ) and both prodrugs and DPD inhibitors (e.g. S‐1, UFT).( 12 , 13 ) S‐1 and UFT are classified as DIF (DPD inhibitory fluoropyrimidines) drugs. UFT is a combination drug consisting of 1 M tegafur and 4 M uracil that selectively inhibits the degradation of 5‐FU by DPD. Recently, a clinical study on the use of UFT confirmed that adjuvant chemotherapy with UFT effectively prolonged the survival periods of patients with resected adenocarcinoma of the lung.( 14 ) S‐1 is a newly developed DIF that consists of 1 M tegafur, 0.4 M gimeracil (a potent DPD inhibitor) and 1 M oteracil (an oratate phosphoribosyltransferase inhibitor) to protect against gastrointestinal toxicity. It showed a high clinical efficacy when used in patients with unresectable advanced gastric,( 15 ) colorectal,( 16 ) breast( 17 ) and non‐small‐cell lung cancers.( 18 ) However, some factors other than DPD might play important roles in the efficacy of 5‐FU, but few studies have examined markers to predict the antitumor effects of various 5‐FU‐based drugs using genome‐wide expression analysis.
Microarray technology has been used widely for global gene expression analysis, and several studies have examined the comprehensive gene expression profiles for predicting the response of cancer cells to anticancer drugs.( 19 , 20 , 21 ) This technology has enabled us to identify new target genes that play a key role in drug efficacy, and has provided fundamental information for overcoming drug resistance.
In the present study, we carried out gene expression analysis to identify genes whose expression profiles were correlated with the sensitivity of 30 human tumor xenografts to 5‐FU based drugs (DIF: UFT, S‐1; non‐DIF: 5′‐DFUR, capecitabine). Furthermore we also examined some drugs (CDDP, CPT‐11, and paclitaxel) that have different mechanisms of action, because these drugs have already been used in combination therapies with 5‐FU based drugs, or may be used in the future. We have identified gene sets that showed a significant correlation with tumor sensitivity to each drug as well as candidate genes involved in multidrug resistance, and applied an ontological approach to extract genes that may be predictive markers of drug efficacy.
Materials and Methods
Nude mice and human tumor xenografts
Six gastric carcinoma xenografts (AZ‐521, SC‐2, ST‐40, 4‐1ST, SC‐4 and OCUM‐2MD3), six colon carcinoma xenografts (KM12C, HCT‐15, KM20C, COL‐1, KM12C/FU and CO‐3), six breast carcinoma xenografts (MC‐5, H‐31, MC‐2, MX‐1, MDA‐MB‐435SHM and MDA‐MD‐231), seven lung carcinoma xenografts (GT3TKB, LC‐11, Lu‐99, LX‐1, LC‐6, Lu‐134 and Lu‐130) and five pancreatic carcinoma xenografts (PAN‐3, PAN‐4, PAN‐12, H‐48 and BxPC‐3) were used in this study. KM12C and KM20C were kindly provided by Dr Kiyoshi Morikawa of the National Cancer Institute (Tokyo, Japan). KM12C/FU was established as described previously.( 22 ) MDA‐MB‐435SHM was established from an in vivo xenograft.( 23 ) LX‐1 and MX‐1 were kindly provided by Dr K. Inoue of the Cancer Chemotherapy Center (Tokyo, Japan). H‐31 and H‐48 were kindly provided by Dr Tetsuo Taguchi of the Research Institute for Microbial Diseases, Osaka University (Osaka, Japan). AZ‐521 and MDA‐MB‐231 were purchased from the Human Science Research Resource Bank (Osaka, Japan) and the American Tissue Culture Collection (Manassas, VA, USA), respectively. HCT‐15 and BxPc‐3 were purchased from Dainippon Pharm‐aceutical Company (Tokyo, Japan). The other lines were provided by the Central Institute for Experimental Animals (Kawasaki, Japan). Male BALB/c‐nu/nµ nude mice (5 weeks old; 18–20 g) were purchased from CLEA Japan, (Tokyo, Japan). The mice were maintained under specific pathogen‐free conditions, and were provided with sterile food and water ad libitum. Each human tumor xenograft (2‐mm cubic fragment) or cultured cell line was implanted subcutaneously into nude mice.
Chemicals
UFT, S‐1 and capecitabine were synthesized in our laboratory. 5′‐DFUR, CDDP, CPT‐11 and paclitaxel were purchased from Nippon Roche (Tokyo, Japan), BristolMyers Squibb (Tokyo, Japan), Yakult Honsha KK (Tokyo, Japan) and Wako Pure Chemicals (Osaka, Japan), respectively. [6‐14C]‐5‐FU (1.85 GBq/mmol) and [6‐3H]‐FdUMP (625 GBq/mmol) were obtained from Moravek Biochemicals (Brea, CA, USA). All other reagents were commercially available and of the highest quality.
Examination of antitumor activity
When the estimated tumor volume (0.5 × length × width2) reached 100–300 mm3, the tumor‐bearing mice were allocated randomly to a test group (day 0, n = 5). UFT, S‐1, 5′‐DFUR and capecitabine were administered orally once a day from day 1 to day 14 (q.d.), as per the reported maximal tolerated dose.( 24 ) The maximal tolerated doses of the other drugs used in each schedule were determined in a pre‐experiment (data not shown). The RTV (relative tumor volume) was calculated on day 15 as follows: tumor volume on day 15/tumor volume on day 0. The antitumor effect (inhibition rate [%]) was calculated as follows: inhibition rate [%] = (1 − mean RTV of drug‐treated group/mean RTV of untreated group) × 100. The tumor growth inhibition rate value on day 15 was regarded as representing the antitumor effect. All animal experiments were carried out according to the Guidelines for the Welfare of Animals in Experimental Neoplasia.( 25 )
Extraction of total RNA and genechip hybridizations
Total RNA was extracted from each xenograft using the RNeasy mini kit (Qiagen, Chatsworth, CA, USA), according to the manufacturer's instructions. The total RNA yields and purity were determined spectrophotometrically by measuring the absorbance of aliquots at 260 and 280 nm. cDNA and biotinylated cRNA were synthesized according to the standard protocols provided by Affymetrix (Santa Clara, CA, USA). Briefly, 5–10 µg of total RNA was reverse transcribed with a cDNA synthesis kit (Invitrogen, Carlsbad, CA, USA) in the presence of an oligo dT‐T7 primer. After phenol–chloroform extraction and ethanol precipitation, the cDNA pellet was air dried and resuspended in 12 µL of RNase‐free water. Ten microliters were used for the in vitro transcription–amplification reaction in the presence of biotinylated nucleotides (Enzo Diagnostics, Farmingdale, NY, USA). Fifteen micrograms of biotinylated cRNA were then fragmented in a solution of 40 mM Tris–acetate (pH 8.1), 100 mM potassium acetate, and 30 mM magnesium acetate at 94°C for 35 min and hybridized to HG Focus GeneChip arrays (Affymetrix) containing probe sets that represent approximately 8500 transcripts. Chip hybridization, washing and staining were carried out according to Affymetrix‐recommended protocols.
Clustering analysis of drug sensitivity
We carried out a clustering analysis based on drug sensitivity. We then calculated the standard correlation coefficient between drug a and drug b using the following formula:
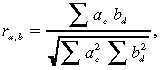 |
where r denotes the correlation of drug a and drug b based on their antitumor activity, a c represents the activity of drug a in xenograft c, and b d represents the activity of drug b in xenograft d.
Genechip
Automated processing of the image scans for the absolute expression analysis was done using Microarray Suite version 5.0 (Affymetrix). The software provided each transcript with a ‘detection call’, which predicted whether the gene was present at a level detectable by the array. The call specifies whether the transcript is detectable (P, present), undetectable (A, absent), or at the limit of detection (M, marginal). These data were then imported into GeneSpring software (Agilent, Palo Alto, CA, USA). We carried out a per‐chip (the expression of each probe set in each chip divided by the median of the chip) and a per‐gene (each gene divided by the mean of all the samples) normalization using the GeneSpring software. The normalized gene expression values were transformed logarithmically (log2). Genes for which the number of ‘present’ calls was less than half of the number of samples were dropped from the analysis. Furthermore, to prevent outlier values from biasing the correlation coefficient, we calculated the entropy, H, using the following formula:
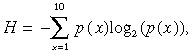 |
where p(x) is the probability that a value was within decile x of that gene expression profile. Genes whose entropy values were within the lowest 10% were dropped from further analysis. Finally, we selected 4144 genes for subsequent analysis.
Correlation analysis between gene expression and drug sensitivity
To investigate the correlation between gene expression and drug sensitivity, we calculated the Pearson correlation coefficients according to the following formula:
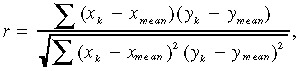 |
where x κ represents the log‐transformed expression value of gene x in the xenograft k, y k is the sensitivity to drug y in the xenograft k, and x mean represents the mean expression value. For this analysis, the difference between maximum and minimum drug sensitivity was fixed as 1. We selected genes with a significant correlation (P < 0.05) and whose absolute value of the slope of the regression line was larger than 1.5, where the difference in drug sensitivity between the most and the least sensitive xenograft was fixed as one.
Real‐time RT‐PCR
Real‐time RT‐PCR analysis was carried out using the Micro‐Fluidic CardsTM system (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions. In brief, 2.5 µg of total RNA was reverse transcribed using the High Capacity cDNA Archives Kit (Applied Biosystems) and MultiScribeTM reverse transcriptase. The reaction mixtures were incubated initially at 25°C for 10 min and subsequently at 37°C for 120 min. Quantitative PCR (TaqManTM) assays were carried out using the Micro‐Fluidic Cards system incorporating Assays‐On‐DemandTM (Applied Biosystems), a prevalidated library, into 384‐well Micro‐Fluidic Cards. cDNA samples (100 ng), along with 50 µL of 2 × PCR master mix, were loaded into each channel on the Micro‐Fluidic Card followed by a brief centrifugation (300 × g for 2 min at room temperature). The card was then sealed, and real‐time PCR and relative quantification were carried out using an ABI PRISM 7900 Sequence Detection System. The expression of each gene was normalized using β‐actin as a reference, and the relative expression levels were qualified using the ΔCt method (Applied Biosystems).( 26 )
DPD activity
The DPD enzymatic activity was measured using a method described by Takechi et al.( 27 ) Briefly, tumor tissues were sonicated in four volumes of homogenization buffer (20 mM potassium phosphate [pH 8.0] containing 1 mM EDTA and 1 mM β‐mercaptoethanol). Each homogenate was centrifuged at 105 000g for 1 h at 4°C, and its supernatant (cytosol) was collected. The enzyme reaction mixture, which contained 10 mM potassium phosphate (pH 8.0), 0.5 mM EDTA, 0.5 mM β‐mercaptoethanol, 2 mM dithiothreitol, 5 mM magnesium chloride, 20 µM [6‐14C] 5‐FU (American Radiolabeled Chemicals, St Louis, MO, USA), 100 µM NADPH, and 25 µL of the cytosol fraction in a final volume of 50 µL, was incubated at 37°C for 30 min. DPD activity was then determined by measuring the sum of the dihydrofluorouracil and 2‐fluoro‐β‐alanine produced from [6‐14C] 5‐FU. Supernatant aliquots (5 µL) were applied to thin‐layer chromatography plates (Silica gel 60F254; Merck, Darmstadt, Germany) and developed with a mixture of ethanol and 1 M ammonium acetate (5 : 1, v/v) according to a method described previously.( 28 )
TS contents
The TS content was determined as the quantity of [6‐3H]‐FdUMP binding activity in the cytosol of tumor tissue homogenates, based on the method described by Spears and colleagues,( 29 ) with minor modifications.
Results
Relationship of drug sensitivities
To evaluate the characteristics of each drug, we first carried out a hierarchical clustering analysis on seven drugs based on their antitumor effects on the 30 xenografts. The clustering analysis showed a weak negative correlation between the antitumor activities of the fluoropyrimidines and paclitaxel. Moreover, roughly two clusters were generated (Fig. 1). 5‐FU based drugs occupied the upper cluster, whereas the lower cluster consisted of non‐5‐FU derivatives. For the upper one, the dendrogram was divided into two groups: an ‘UFT/S‐1 cluster’ and a ‘5′‐DFUR/capecitabine cluster’. These results indicate that even among fluorinated pyrimidines, the pattern of antitumor activity differs between DIF and non‐DIF drugs.
Figure 1.
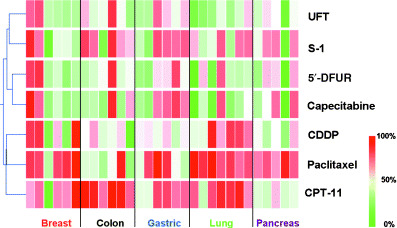
Two‐dimensional hierarchical clustering of each antitumor effect in the 30 xenografts. The inhibition rate of the relative tumor volume was regarded as representing antitumor activity. Red, represents a high sensitivity to the drug; green, resistant.
Correlations between gene expression and drug sensitivity
To screen for genes that may be associated with drug sensitivity, we carried out a correlation analysis based on the Pearson correlation coefficient between the expression profiles of the 4144 genes and the antitumor activities of the anticancer drugs in the 30 xenografts. We used all xenografts to screen for genes that would account for differences in the efficacy of the drugs against various types of tumors. The correlation analysis showed that the expression profiles of 684 genes showed significant association with sensitivity to at least one of the drugs. Furthermore, as shown in Table 1, various genes whose expression profiles were significantly correlated with tumor sensitivity to each drug were extracted by the correlation analysis. Table 2 summarizes the top 20 genes with the highest Pearson's correlation coefficients. These genes have various functions, as shown in the gene lists. Although some genes exhibited a broad negative correlation with fluoropyrimidines, many genes that showed specific correlations with each drug were also seen. These results indicate that each of the 5‐FU‐based drugs has distinctive characteristics, despite having a common cytocidal mechanism, and suggests that unidentified factors other than DPD are likely involved in the regulation of drug efficacy.
Table 1.
Number of genes significantly correlated with antitumor activity to seven anticancer drugs
| Number of genes | |||
|---|---|---|---|
| Positive correlation | Negative correlation | Total | |
| UFT | 72 | 59 | 131 |
| S‐1 | 84 | 31 | 115 |
| Capecitabine | 128 | 64 | 192 |
| 5′‐DFUR | 110 | 85 | 195 |
| CDDP | 25 | 88 | 113 |
| CPT‐11 | 99 | 90 | 189 |
| Paclitaxel | 30 | 35 | 65 |
Correlation analysis between 4144 probe sets and antitumor activity for seven anticancer drugs across 30 xenografts were carried out to screen the genes that correlated with sensitivity to the drug sensitivity. Genes were selected that represented the significant correlation that satisfied the following criteria: P < 0.05, and the absolute value of the slope of the regression line was >1.5 where the difference of the drug sensitivity between the most and the least xenografts was fixed as 1. CDDP, cisplatin; CPT‐11, irinotecan hydrochloride; 5′‐DFUR, 5′‐deoxy‐5‐fluorouridine; S‐1, 1 M tegafur−0.4 M 5‐chloro‐2,4‐dihydroxypyridine−1 M potassium oxonate; UFT, 1 M tegafur−4 M uracil.
Table 2.
Summary of the top 20 genes significantly correlated with sensitivity to each drug
| Drug | Index † | Gene name | Slope ‡ | r § | P‐value | GenBank no. | Product |
|---|---|---|---|---|---|---|---|
| S‐1 | 202922_at | GCLC | 1.69 | 0.627 | <0.001 | BF676980 | Glutamate‐cysteine ligase, catalytic subunit |
| 207463_x_at | PRSS3 | 4.70 | 0.631 | <0.001 | NM_002771 | Mesotrypsin preproprotein | |
| 202609_at | EPS8 | 3.74 | 0.617 | <0.001 | NM_004447 | Epidermal growth factor receptor pathway substrate 8 | |
| 202831_at | GPX2 | 5.75 | 0.614 | <0.001 | NM_002083 | Gastrointestinal glutathione peroxidase 2 | |
| 218854_at | SART2 | −3.47 | −0.602 | <0.001 | NM_013352 | Squamous cell carcinoma antigen recognized by T cells 2 | |
| 203476_at | TPBG | −3.78 | −0.593 | 0.001 | NM_006670 | 5T4 oncofetal trophoblast glycoprotein | |
| 217794_at | DKFZp564J157 | 1.94 | 0.593 | 0.001 | NM_018457 | DKFZp564J157 protein isoform 1 | |
| 201425_at | ALDH2 | 3.64 | 0.586 | 0.001 | NM_000690 | Mitochondrial aldehyde dehydrogenase 2 precursor | |
| 208453_s_at | XPNPEP1 | 1.57 | 0.581 | 0.001 | NM_006523 | X‐prolyl aminopeptidase (aminopeptidase P) 1, soluble | |
| 205402_x_at | PRSS2 | 4.48 | 0.557 | 0.001 | NM_002770 | Protease, serine, 2 preproprotein | |
| 219115_s_at | IL20RA | 2.35 | 0.555 | 0.001 | NM_014432 | Interleukin 20 receptor, alpha | |
| 221016_s_at | TCF7L1 | −2.08 | −0.557 | 0.001 | NM_031283 | HMG‐box transcription factor TCF‐3 | |
| 202794_at | INPP1 | 1.83 | 0.552 | 0.002 | NM_002194 | Inositol polyphosphate‐1‐phosphatase | |
| 203832_at | SNRPF | 1.64 | 0.551 | 0.002 | NM_003095 | Small nuclear ribonucleoprotein polypeptide F | |
| 201829_at | NET1 | 1.71 | 0.545 | 0.002 | AW263232 | Neuroepithelial cell transforming gene 1 | |
| 204608_at | ASL | 1.86 | 0.546 | 0.002 | NM_000048 | Argininosuccinate lyase | |
| 220189_s_at | MGAT4B | 1.51 | 0.546 | 0.002 | NM_014275 | Mannosyl‐glycoprotein β‐1,4‐N‐acetylglucosaminyltransferase, isoenzyme B isoform 1 | |
| 209605_at | TST | 5.09 | 0.544 | 0.002 | D87292 | Rhodanese | |
| 211184_s_at | aie‐75 | 3.17 | 0.541 | 0.002 | AB006955 | AIE‐75 | |
| 202674_s_at | LMO7 | 1.97 | 0.540 | 0.002 | NM_005358 | LIM domain only 7 | |
| UFT | 205395_s_at | MRE11A | −3.68 | −0.749 | <0.001 | NM_005590 | Meiotic recombination 11 homolog A isoform 2 |
| 201312_s_at | SH3BGRL | −5.76 | −0.639 | <0.001 | NM_003022 | SH3 domain binding glutamic acid‐rich protein like | |
| 203752_s_at | JUND | 1.50 | 0.633 | <0.001 | NM_005354 | Jun‐D proto‐oncogene | |
| 204333_s_at | AGA | −1.91 | −0.632 | <0.001 | NM_000027 | Aspartylglucosaminidase precursor | |
| 217788_s_at | GALNT2 | −2.21 | −0.616 | <0.001 | NM_004481 | Polypeptide N‐acetylgalactosaminyltransferase 2 | |
| 218854_at | SART2 | −4.22 | −0.620 | <0.001 | NM_013352 | Squamous cell carcinoma antigen recognized by T cells 2 | |
| 209526_s_at | HRP‐3 | −4.47 | −0.581 | 0.001 | AB029156 | HRP‐3 | |
| 214600_at | TEAD1 | −2.17 | −0.580 | 0.001 | AW771935 | TEA domain family member 1 | |
| 205187_at | Smad5 | −2.34 | −0.567 | 0.001 | AF010601 | SMAD5 | |
| 212983_at | HRAS | −2.05 | −0.564 | 0.001 | NM_005343 | v‐Ha‐ras Harvey rat sarcoma viral oncogene homolog isofrom 1 | |
| 217759_at | TRIM44 | −1.89 | −0.554 | 0.002 | NM_017583 | DIPB protein | |
| 201481_s_at | PYGB | 6.03 | 0.552 | 0.002 | NM_002862 | Brain glycogen phosphorylase | |
| 203874_s_at | SMARCA1 | −3.09 | −0.540 | 0.002 | NM_003069 | SWI/SNF‐related matrix‐associated actin‐dependent regulator of chromatin a1 isoform a | |
| 201177_s_at | UBA2 | −1.68 | −0.538 | 0.002 | NM_005499 | SUMO‐1 activating enzyme subunit 2 | |
| 201540_at | FHL1 | −4.53 | −0.530 | 0.003 | NM_001449 | Four and a half LIM domains 1 | |
| 202602_s_at | HTATSF1 | −1.67 | −0.530 | 0.003 | NM_014500 | HIV TAT specific factor 1 | |
| 200821_at | LAMP2 | −1.78 | −0.524 | 0.003 | NM_013995 | Lysosomal‐associated membrane protein 2 precursor | |
| 214257_s_at | SEC22L1 | −1.93 | −0.524 | 0.003 | AA890010 | Hypothetical protein | |
| 202082_s_at | SEC14L1 | −1.74 | −0.518 | 0.003 | AV748469 | SEC14 (S. cerevisiae)‐like 1 | |
| 203953_s_at | CLDN3 | 8.32 | 0.517 | 0.003 | BE791251 | Claudin 3 | |
| 5′‐DFUR | 201481_s_at | PYGB | 7.46 | 0.686 | <0.001 | NM_002862 | Brain glycogen phosphorylase |
| 203586_s_at | ARF4L | −1.97 | −0.669 | <0.001 | NM_001661 | ADP‐ribosylation factor 4‐like | |
| 205395_s_at | MRE11A | −3.30 | −0.674 | <0.001 | NM_005590 | Meiotic recombination 11 homolog A isoform 2 | |
| 202620_s_at | PLOD2 | −5.49 | −0.644 | <0.001 | NM_000935 | Procollagen‐lysine, 2‐oxoglutarate 5‐dioxygenase 2 isoform b | |
| 214600_at | TEAD1 | −2.33 | −0.626 | <0.001 | AW771935 | TEA domain family member 1 | |
| 218854_at | SART2 | −4.30 | −0.634 | <0.001 | NM_013352 | Squamous cell carcinoma antigen recognized by T cells 2 | |
| 222065_s_at | FLII | −1.73 | −0.632 | <0.001 | AI830227 | Flightless I homolog (Drosophila) | |
| 204201_s_at | PTPN13 | −3.53 | −0.610 | <0.001 | NM_006264 | Protein tyrosine phosphatase, non‐receptor type 13 isoform 2 | |
| 204646_at | DPYD | −4.86 | −0.603 | <0.001 | NM_000110 | Dihydropyrimidine dehydrogenase | |
| 219255_x_at | IL17RB | 4.16 | 0.603 | <0.001 | NM_018725 | Interleukin 17B receptor isoform 1 precursor | |
| 210827_s_at | ESE‐1 | 3.79 | 0.599 | 0.001 | U73844 | ESE‐1a | |
| 200989_at | HIF1A | −1.61 | −0.592 | 0.001 | NM_001530 | Hypoxia‐inducible factor 1, α subunit isoform 1 | |
| 220147_s_at | C12orf14 | 1.69 | 0.592 | 0.001 | NM_021238 | Chromosome 12 open reading frame 14 | |
| 201528_at | RPA1 | −1.89 | −0.580 | 0.001 | BG398414 | Replication protein A1, 70 kDa | |
| 201540_at | FHL1 | −4.94 | −0.578 | 0.001 | NM_001449 | Four and a half LIM domains 1 | |
| 212983_at | HRAS | −2.09 | −0.577 | 0.001 | NM_005343 | v‐Ha‐ras Harvey rat sarcoma viral oncogene homolog isofrom 1 | |
| 219553_at | NME7 | −1.94 | −0.580 | 0.001 | NM_013330 | Nucleoside‐diphosphate kinase 7 isoform a | |
| 213330_s_at | STIP1 | −1.64 | −0.576 | 0.001 | BE886580 | Stress‐induced‐phosphoprotein 1 | |
| 201289_at | CYR61 | −5.85 | −0.572 | 0.001 | NM_001554 | Cysteine‐rich, angiogenic inducer, 61 | |
| 203953_s_at | CLDN3 | 9.18 | 0.572 | 0.001 | BE791251 | Claudin 3 | |
| Capecitabine | 201481_s_at | PYGB | 7.79 | 0.742 | <0.001 | NM_002862 | Brain glycogen phosphorylase |
| 218059_at | LOC51123 | 1.71 | 0.660 | <0.001 | NM_016096 | HSPC038 protein | |
| 218854_at | SART2 | −4.56 | −0.696 | <0.001 | NM_013352 | Squamous cell carcinoma antigen recognized by T cells 2 | |
| 201528_at | RPA1 | −2.01 | −0.641 | <0.001 | BG398414 | Replication protein A1, 70 kDa | |
| 200989_at | HIF1A | −1.68 | −0.637 | <0.001 | NM_001530 | Hypoxia‐inducible factor 1, α subunit isoform 1 | |
| 203586_s_at | ARF4L | −1.80 | −0.632 | <0.001 | NM_001661 | ADP‐ribosylation factor 4‐like | |
| 204073_s_at | C11orf9 | 3.24 | 0.618 | <0.001 | NM_013279 | Chromosome 11 open reading frame 9 | |
| 205395_s_at | MRE11A | −2.93 | −0.620 | <0.001 | NM_005590 | Meiotic recombination 11 homolog A isoform 2 | |
| 204201_s_at | PTPN13 | −3.38 | −0.605 | <0.001 | NM_006264 | Protein tyrosine phosphatase, non‐receptor type 13 isoform 2 | |
| 209620_s_at | ABCB7 | −2.09 | −0.605 | <0.001 | AB005289 | ATP‐binding cassette, subfamily B (MDR/TAP), member 7 | |
| 222065_s_at | FLII | −1.60 | −0.606 | <0.001 | AI830227 | Flightless I homolog (Drosophila) | |
| 213330_s_at | STIP1 | −1.62 | −0.590 | 0.001 | BE886580 | Stress‐induced‐phosphoprotein 1 (Hsp70/Hsp90‐organizing protein) | |
| 214600_at | TEAD1 | −2.12 | −0.590 | 0.001 | AW771935 | TEA domain family member 1 (SV40 transcriptional enhancer factor) | |
| 217848_s_at | PP | 1.96 | 0.592 | 0.001 | NM_021129 | Inorganic pyrophosphatase | |
| 32837_at | AGPAT2 | 1.96 | 0.589 | 0.001 | U56418 | Lysophosphatidic acid acyltransferase‐beta | |
| 204351_at | S100P | 8.87 | 0.581 | 0.001 | NM_005980 | S100 calcium binding protein P | |
| 205403_at | IL1R2 | 6.30 | 0.579 | 0.001 | NM_004633 | Interleukin 1 receptor, type II precursor | |
| 209160_at | c‐hluPGFS | 7.40 | 0.578 | 0.001 | AB018580 | HluPGFS | |
| 212983_at | HRAS | −2.02 | −0.578 | 0.001 | NM_005343 | v‐Ha‐ras Harvey rat sarcoma viral oncogene homolog isofrom 1 | |
| 36742_at | ZNFB7 | 2.67 | 0.577 | 0.001 | U34249 | Zinc finger protein | |
| CDDP | 208012_x_at | SP110 | −2.77 | −0.662 | <0.001 | NM_004509 | SP110 nuclear body protein isoform a |
| 201278_at | DAB2 | −2.45 | −0.636 | <0.001 | N21202 | Disabled homolog 2, mitogen‐responsive phosphoprotein | |
| 218070_s_at | GMPPA | −1.66 | −0.593 | 0.001 | NM_013335 | GDP‐mannose pyrophosphorylase A | |
| 201661_s_at | ACSL3 | −1.51 | −0.592 | 0.001 | NM_004457 | Acyl‐CoA synthetase long‐chain family member 3 | |
| 203423_at | RBP1 | 4.07 | 0.584 | 0.001 | NM_002899 | Retinol binding protein 1, cellular | |
| 202659_at | PSMB10 | −3.07 | −0.581 | 0.001 | NM_002801 | Proteasome beta 10 subunit proprotein | |
| 211429_s_at | MYCPBP | −7.04 | −0.577 | 0.001 | AF119873 | PRO2275 | |
| 217844_at | CTDSP1 | −1.74 | −0.575 | 0.001 | NM_021198 | CTD (carboxy‐terminal domain, RNA polymerase II, polypeptide A) small phosphatase 1 | |
| 201482_at | QSCN6 | −3.49 | −0.569 | 0.001 | NM_002826 | quiescin Q6 isoform a | |
| 202100_at | RALB | −2.02 | −0.561 | 0.001 | BG169673 | v‐ral simian leukemia viral oncogene homolog B | |
| 203228_at | PAFAH1B3 | 1.92 | 0.558 | 0.001 | NM_002573 | Platelet‐activating factor acetylhydrolase, isoform Ib, gamma subunit 29 kDa | |
| 204306_s_at | CD151 | −1.94 | −0.556 | 0.001 | NM_004357 | CD151 antigen | |
| 209761_s_at | SP110 | −1.91 | −0.555 | 0.001 | AA969194 | SP110 nuclear body protein | |
| 203964_at | NMI | −2.38 | −0.544 | 0.002 | NM_004688 | N‐myc and STAT interactor | |
| 1729_at | TRADD | −1.68 | −0.533 | 0.002 | NM_003789 | Tumor necrosis factor receptor type 1 associated protein | |
| 202733_at | P4HA2 | −1.58 | −0.533 | 0.002 | NM_004199 | Procollagen‐proline, 2‐oxoglutarate 4‐dioxygenase,alpha polypeptide II | |
| 204001_at | SNAPC3 | 1.52 | 0.531 | 0.003 | NM_003084 | Small nuclear RNA activating complex, polypeptide 3,50 kDa | |
| 221523_s_at | RAGD | 2.92 | 0.519 | 0.003 | AL138717 | Ras‐related GTP binding D | |
| 201887_at | IL13RA1 | −2.13 | −0.515 | 0.004 | NM_001560 | Interleukin 13 receptor, alpha 1 precursor | |
| 202863_at | SP100 | −2.50 | −0.514 | 0.004 | NM_003113 | Nuclear antigen Sp100 | |
| CPT11 | 202870_s_at | CDC20 | 2.08 | 0.694 | <0.001 | NM_001255 | Cell division cycle 20 |
| 203832_at | SNRPF | 2.26 | 0.657 | <0.001 | NM_003095 | Small nuclear ribonucleoprotein polypeptide F | |
| 205085_at | ORC1L | 1.64 | 0.751 | <0.001 | NM_004153 | Origin recognition complex, subunit 1 | |
| 205412_at | ACAT1 | 3.01 | 0.730 | <0.001 | NM_000019 | Acetyl‐coenzyme A acetyltransferase 1 precursor | |
| 206653_at | POLR3G | 2.60 | 0.664 | <0.001 | BF062139 | Polymerase (RNA) III (DNA directed) polypeptide G (32 kDa) | |
| 208967_s_at | adk2 | 1.60 | 0.723 | <0.001 | U39945 | Adenylate kinase 2 | |
| 210567_s_at | SKP2 | 2.13 | 0.661 | <0.001 | BC001441 | S‐phase kinase‐associated protein 2, isoform 2 | |
| 212136_at | ATP2B4 | −3.26 | −0.744 | <0.001 | AW517686 | ATPase, Ca++ transporting, plasma membrane 4 | |
| 214177_s_at | PBXIP1 | −1.68 | −0.714 | <0.001 | AI935162 | Pre‐B‐cell leukemia transcription factor interacting protein 1 | |
| 215127_s_at | RBMS1 | −2.43 | −0.663 | <0.001 | AL517946 | RNA binding motif, single stranded interacting protein 1 | |
| 217988_at | CCNB1IP1 | 2.50 | 0.680 | <0.001 | NM_021178 | Cyclin B1 interacting protein 1 isoform a | |
| 220892_s_at | PSAT1 | 3.14 | 0.697 | <0.001 | NM_021154 | Phosphoserine aminotransferase isoform 2 | |
| 204127_at | RFC3 | 1.62 | 0.640 | <0.001 | BC000149 | Replication factor C 3, isoform 1 | |
| 205909_at | POLE2 | 1.64 | 0.640 | <0.001 | NM_002692 | DNA polymerase epsilon subunit 2 | |
| 200078_s_at | ATP6V0B | 1.86 | 0.634 | <0.001 | BC005876 | ATPase, H+ transporting, lysosomal 21 kDa, V0 subunit c″ | |
| 202705_at | CCNB2 | 1.55 | 0.622 | <0.001 | NM_004701 | Cyclin B2 | |
| 204244_s_at | ASK | 1.80 | 0.624 | <0.001 | NM_006716 | Activator of S phase kinase | |
| 204559_s_at | LSM7 | 1.91 | 0.632 | <0.001 | NM_016199 | U6 snRNA‐associated Sm‐like protein LSm7 | |
| 206752_s_at | DFFB | 2.58 | 0.631 | <0.001 | NM_004402 | DNA fragmentation factor, 40 kDa, beta polypeptide isoform 1 | |
| 216321_s_at | NR3C1 | −6.46 | −0.624 | <0.001 | X03348 | β‐Glucocorticoid receptor | |
| Paclitaxel | 201272_at | AKR1B1 | 3.88 | 0.581 | 0.001 | NM_001628 | Aldo‐keto reductase family 1, member B1 |
| 205659_at | HDAC9 | 3.14 | 0.577 | 0.001 | NM_014707 | Histone deacetylase 9 isoform 3 | |
| 204867_at | GCHFR | −2.41 | −0.566 | 0.001 | NM_005258 | GTP cyclohydrolase I feedback regulatory protein | |
| 206247_at | MICB | 2.72 | 0.532 | 0.003 | NM_005931 | MHC class I polypeptide‐related sequence B | |
| 204981_at | SLC22A18 | −2.99 | −0.530 | 0.003 | NM_002555 | Tumor suppressing subtransferable candidate 5 | |
| 201012_at | ANXA1 | 3.48 | 0.516 | 0.004 | NM_000700 | Annexin I | |
| 201564_s_at | FSCN1 | 3.95 | 0.517 | 0.004 | NM_003088 | Fascin 1 | |
| 207717_s_at | PKP2 | −2.32 | −0.515 | 0.004 | NM_004572 | Plakophilin 2 isoform 2b | |
| 210264_at | GPR35 | −1.98 | −0.515 | 0.004 | AF089087 | G protein‐coupled receptor | |
| 202722_s_at | GFPT1 | −1.75 | −0.514 | 0.004 | NM_002056 | Glucosamine‐fructose‐6‐phosphate aminotransferase | |
| 205443_at | SNAPC1 | 1.79 | 0.512 | 0.004 | NM_003082 | Small nuclear RNA activating complex, polypeptide 1, 43 kDa | |
| 201746_at | TP53 | −1.67 | −0.506 | 0.004 | NM_000546 | Tumor protein p53 | |
| 204527_at | MYO5A | 1.97 | 0.504 | 0.005 | NM_000259 | Myosin VA (heavy polypeptide 12, myoxin) | |
| 201540_at | FHL1 | 3.43 | 0.503 | 0.005 | NM_001449 | Four and a half LIM domains 1 | |
| 203423_at | RBP1 | 2.66 | 0.498 | 0.005 | NM_002899 | Retinol binding protein 1, cellular | |
| 204404_at | SLC12A2 | −2.63 | −0.497 | 0.005 | NM_001046 | Solute carrier family 12, member 2 | |
| 213757_at | EIF5A | 2.92 | 0.492 | 0.006 | AA393940 | Eukaryotic translation initiation factor 5 A | |
| 209016_s_at | KRT7 | 4.56 | 0.488 | 0.006 | BC002700 | Keratin 7 | |
| 203953_s_at | CLDN3 | −6.15 | −0.480 | 0.007 | BE791251 | Claudin 3 | |
| 35148_at | TJP3 | −2.87 | −0.478 | 0.008 | AC005954 | Tight junction protein 3 (zona occludens 3) |
Index: Affimetrix probe set ID, a unique identifier that can be used for GenBank accession numbers and consensus gene sequences.
‡ Slope: slope of the regression line between gene expression level and drug sensitivity.
r: Pearson's correlation coefficient. CDDP, cisplatin; CPT‐11, irinotecan hydrochloride; 5′‐DFUR, 5′‐deoxy‐5‐fluorouridine; S‐1, 1 M tegafur−0.4 M 5‐chloro‐2,4‐dihydroxypyridine−1 M potassium oxonate; UFT, 1 M tegafur−4 M uracil.
Analysis of functional genes affecting antitumor activity
To screen the genes that may serve as predictive markers of antitumor activity, we examined genes that were associated with multiple drug sensitivities, and classified them ontologically into key pathways suggested or previously shown to play a role in drug metabolism or resistance. First, we selected 179 genes whose expression profiles were correlated with sensitivity to more than two 5‐FU‐based drugs. Next, we selected 25 genes that were closely correlated with more than two drugs other than fluoropyrimidines. We found three genes that were common to these gene lists, and selected 210 genes by combining these lists. We then selected genes that could be ontologically classified into key pathways or functions thought to be related to drug efficacy. The Kyoto Encyclopedia of Genes and Genomes, Simplified Gene Ontology (GeneSpring version 6.1), and Gene Ontology Consortium were used to investigate the biological processes and important pathways (such as transforming growth factor‐α/MAPK/Wnt signaling, cell proliferation, cell adhesion, oncogenes, nucleotide sugars metabolism, and pyrimidine metabolism). Finally, we identified 39 genes using an ontological approach (Fig. 2). In addition to the genes that have been shown previously to play important roles in drug resistance, we found many genes that have not been reported to be associated with drug sensitivity and that may serve as putative predictive markers of chemosensitivity. The expression level of DPD, a well‐known predictor of resistance to 5‐FU, showed a broad negative correlation with the 5‐FU‐based drugs, except S‐1. Furthermore, a significant negative correlation between TS expression and sensitivity to non‐DIF (5′‐DFUR and capecitabine) was seen, but TS expression was not correlated with sensitivity to DIF. The expression level of AKR1B1 has been reported to be associated with multiple drug sensitivity.( 20 , 30 ) In the present study, AKR1B1 expression was correlated with four of the seven drugs. CYR61, which is involved in angiogenesis and mediates diverse roles in cellular development,( 31 ) showed significant associations with six drugs, including all of the 5‐FU‐based drugs. Finally, we visualized the relationships between gene expression and drug sensitivity (Fig. 3). In this network, the connection between drugs and genes whose association was not previously known can be seen. Some genes associated with sensitivity to 5‐FU derivatives showed a significant inverse correlation with sensitivity to paclitaxel.
Figure 2.

Correlation between drug sensitivity and the expression profiles of 39 genes that were significantly correlated with multidrug sensitivity, and classified into key pathways. Red, a positive correlation; green, a negative correlation.
Figure 3.
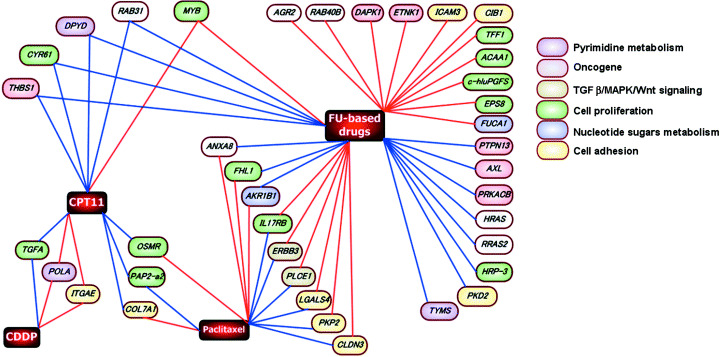
The association between gene expression profiles correlated with antitumor acticity of multiple drugs. The nodes represent genes whose expression profiles were significantly correlated with drug sensitivity. The red connecting lines indicate a positive correlation, and the blue connecting lines indicate a negative correlation. The nodes between genes and FU‐based drugs means whose expression profile were correlated with sensitivity to more than two fluoropyrimidine drugs. The color of each node represents the ontology or pathway based on the KEGG and GOC. Detailed information on the selected ge is presented in Fig. 2.
Validation of genechip experiments using real‐time RT‐PCR
To verify the genechip expression data more quantitatively, we carried out real‐time RT‐PCR using the same RNA as that used in the genechip analysis. To validate the genechip data, we verified the mRNA level of topic genes mainly associated with pyrimidine metabolism, folate metabolism, and some genes selected by the correlation analysis carried out in this study (CYR61, MYB). As shown in Table 3, although the expression data for most of the genes (57 out of 65 genes) that we examined by real‐time RT‐PCR were significantly correlated with the expression data obtained by the genechip analysis, the data for eight genes were not significantly correlated. Next, we checked the number of ‘detection calls’ in the genechip data in all of the genes whose expression was verified by real‐time RT‐PCR. Six of seven genes (AK5, ENTPD1, FOLR2, TNSF6, GPR44 and MTHFR) whose expression was not correlated had less than 1 or 0 ‘present’ calls. This result indicates that the expression of these genes may be difficult to detect using the genechip system. The remaining gene (UMPS) had a ‘present’ call in each of the xenografts. Although the reason for the discrepancy in the expression data is uncertain, the observation that the coefficient of variance of UMPS among 30 xenografts was the lowest among all of the genes examined by real‐time RT‐PCR may be related to the discrepancy. Overall, these results indicate that the majority of the gene expression data obtained using the genechip system was reliable.
Table 3.
Validation of gene expression data obtained using the genechip system
| Gene | No. ‘present’ | r † | P‐value |
|---|---|---|---|
| ABCC1 | 30 | 0.727 | <0.001*** |
| NT5E | 26 | 0.933 | <0.001*** |
| THBS1 | 22 | 0.917 | <0.001*** |
| PTGS2 | 17 | 0.909 | <0.001*** |
| MYB | 20 | 0.887 | <0.001*** |
| UPP1 | 16 | 0.875 | <0.001*** |
| CDA | 12 | 0.866 | <0.001*** |
| ABCC3 | 17 | 0.861 | <0.001*** |
| CYR61 | 20 | 0.859 | <0.001*** |
| CNN3 | 26 | 0.858 | <0.001*** |
| TP53 | 27 | 0.854 | <0.001*** |
| ABCC4 | 18 | 0.846 | <0.001*** |
| ECGF1 | 28 | 0.842 | <0.001*** |
| GSTP1 | 30 | 0.826 | <0.001*** |
| ABCC2 | 2 | 0.817 | <0.001*** |
| GSTT1 | 7 | 0.807 | <0.001*** |
| DPYD | 21 | 0.795 | <0.001*** |
| GGH | 30 | 0.763 | <0.001*** |
| BRCA1 | 26 | 0.750 | <0.001*** |
| TYMS | 30 | 0.748 | <0.001*** |
| ATP7B | 28 | 0.733 | <0.001*** |
| SLCO2B1 | 6 | 0.729 | <0.001*** |
| SLC19A3 | 2 | 0.687 | <0.001*** |
| RPLP0 | 30 | 0.678 | <0.001*** |
| MTHFD2 | 30 | 0.660 | <0.001*** |
| E2F1 | 30 | 0.658 | <0.001*** |
| POLA | 24 | 0.654 | <0.001*** |
| PCNA | 30 | 0.639 | <0.001*** |
| SHMT2 | 30 | 0.628 | <0.001*** |
| POLB | 26 | 0.626 | <0.001*** |
| FOLR1 | 14 | 0.610 | <0.001*** |
| SHMT1 | 2 | 0.599 | <0.001*** |
| DCTD | 30 | 0.591 | <0.001*** |
| UNG | 30 | 0.584 | <0.001*** |
| DCK | 30 | 0.563 | 0.001** |
| RRM1 | 30 | 0.559 | 0.001** |
| TOP2A | 30 | 0.555 | 0.001** |
| VEGFB | 19 | 0.550 | 0.002** |
| GCLC | 30 | 0.539 | 0.002** |
| RRM2 | 30 | 0.519 | 0.003** |
| DUT | 30 | 0.483 | 0.007** |
| MTR | 29 | 0.476 | 0.008** |
| MFTC | 30 | 0.466 | 0.009** |
| GART | 30 | 0.462 | 0.010* |
| AMT | 8 | 0.457 | 0.011* |
| NME1 | 30 | 0.457 | 0.011* |
| CAD | 29 | 0.456 | 0.011* |
| DTYMK | 24 | 0.452 | 0.012* |
| TOP1 | 30 | 0.450 | 0.012* |
| ERCC1 | 20 | 0.450 | 0.013* |
| ATIC | 30 | 0.443 | 0.014* |
| LIG3 | 17 | 0.438 | 0.015* |
| ITPA | 25 | 0.423 | 0.020* |
| DHFR | 29 | 0.421 | 0.020* |
| CTPS | 30 | 0.417 | 0.022* |
| CTPS2 | 30 | 0.413 | 0.023* |
| MTHFD1 | 30 | 0.394 | 0.031* |
| POLD1 | 22 | 0.329 | 0.076 |
| AK5 | 1 | 0.314 | 0.091 |
| ENTPD1 | 0 | 0.180 | 0.341 |
| FOLR2 | 1 | 0.128 | 0.499 |
| TNFSF6 | 1 | 0.128 | 0.500 |
| GPR44 | 0 | 0.113 | 0.552 |
| UMPS | 30 | 0.078 | 0.680 |
| MTHFR | 1 | −0.026 | 0.892 |
r: Pearson's correlation coefficient.
P < 0.001.
P < 0.01.
P < 0.05.
Correlation between gene expression level and enzymatic activity
Among the genes that were screened in the global gene expression analysis, we focused on DPD and TS because these genes are associated with the molecular mechanism of 5‐FU and many reports have examined the relationship between the expression of these genes and the antitumor activity of fluoropyrimidines. We examined the enzymatic activity of DPD and the protein level of TS in 30 xenografts. As shown in Fig. 4, the DPD mRNA level was significantly correlated with activity. As for TS, a positive correlation was also confirmed between the level of mRNA and protein.
Figure 4.
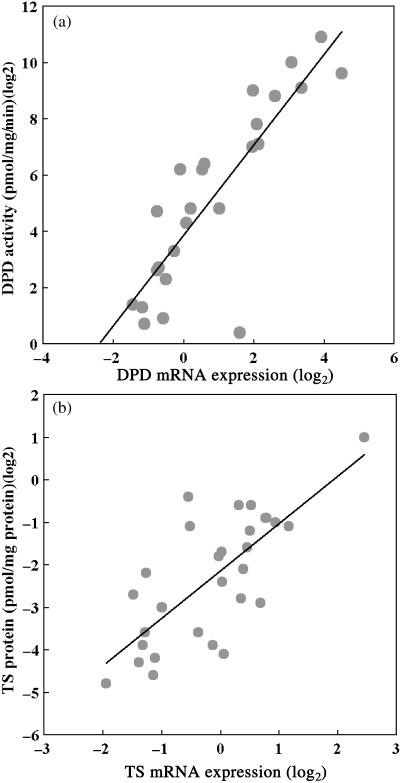
Correlation between mRNA expression and protein level. Scatter plots of protein expression levels against mRNA expression levels obtained by GeneChip system. Each symbol represents a xenograft. The Pearson correlation coefficients (r) and P value (P) are shown.
Discussion
Microarray technology has enabled us to determine the expression levels of thousands of genes in a single experiment. This technology is a very powerful tool for screening new target genes that have not been previously reported to be associated with drug resistance,( 32 , 33 ) or for investigating the global expression patterns of various tissues or cell lines. This technology has been successfully applied to shed light into complex phenomena, such as invasion and metastasis,( 34 , 35 ) and has provided novel insights into the mechanism of drug resistance and sensitivity.( 36 )
One of our goals is to establish a method for predicting the chemosensitivity of various types of tumors. In the present study, we carried out gene expression analysis to screen for genes whose expression profiles were significantly correlated with the sensitivity of 30 human tumor xenografts to seven anticancer drugs. There are some disadvantages to in evaluating antitumor activities using nude mouse models. One is that the characteristics of tumors in nude mouse transplants may not reflect the original tissues because the tumors are grown subcutaneously in nude mouse that may differ from the original environment. This may lead to some concern regarding the compatibility of the transplants to the parent tumor. However, this may not be such a concern in our study as we were investigating the correlation between gene expression in the tumors and chemosensitivity. There are possible differences in drug metabolism among species. The metabolic enzymes against each drug are unclear in the mouse model. However, cyp2a5 is the mouse homolog of human CYP2A6, the main enzyme responsible for the metabolism of tegafur, and these enzymes are highly expressed in the liver. We demonstrated previously that FT is metabolized to 5‐FU when it is incubated in mouse liver microsomes (Nagayama et al., unpublished data), and these data suggest the possibility that tegafur is metabolized by cyp2a5 in mouse liver. For other drugs we have no idea about the mouse internal systems. Even so, there are enormous differences in drug metabolism ability between each mouse, as is the case in humans. Drug delivery and metabolism are vastly subject to the effects of various factors, including sex, age and polymorphisms. However, the antitumor effect of each drug may reflect different gene expression profiles in each tumor as it could be assumed that drug metabolism capacity is approximately uniform in each mouse model. Furthermore, it was reported that antitumor activity in human tumor xenograft models tends to coincide well with clinical effects, although drug metabolism in the mouse may be different from that in humans.( 37 ) We therefore assume chemosensitivities in xenografts can be correlated with clinical effects.
At first, we focused on the drug sensitivity profiles of the 30 xenografts. A clustering analysis based on drug sensitivity revealed that the 5‐FU‐based drugs could be divided into two clusters: DIF and non‐DIF drugs (Fig. 1). In addition to the DPD inhibitory effect, DIF drugs differ from capecitabine and 5′‐DFUR in terms of the mechanism of 5‐FU activation. Whereas capecitabine and 5′‐DFUR are converted to 5‐FU by an enzyme (TP), DIF drugs are activated to 5‐FU by CYP2A6 in the liver. These differences may reflect differences in chemosensitivity profiles. In candidate genes, the DPD mRNA expression profiles of the tumors were negatively correlated with chemosensitivity to UFT, 5′‐DFUR and capecitabine, except for S‐1 (Fig. 2). In the present study, sensitivity to UFT was correlated with DPD regardless of DIF. This discrepancy was thought to originate from the difference in DPD inhibitory activity between uracil and gemeracil. Gemeracil, which is present in S‐1, is 200‐fold more potent as a DPD inhibitor than uracil.( 38 ) The combination of 5‐FU and gimeracil for the treatment of tumors with high DPD has led to greater antitumor activity than treatment with 5‐FU alone.( 39 ) The superior antitumor activity of S‐1 in tumors with high DPD activity has been reported in vivo, ( 40 ) and in a clinical study.( 41 )
In the present study, we also identified some genes that showed a significant correlation with sensitivity to a specific drug. We applied an ontological approach to further characterize these gene lists as it would be difficult to examine the biological function of all of the genes in the list, Finally, we identified 39 genes that were correlated with sensitivity to either two or six drugs; these associations seem to be more important than those of other genes that showed a correlation to a specific drug. As shown in Fig. 2, among the 39 genes, some have already been to shown to be associated with drug sensitivity. TGFα activates the epidermal growth factor receptor.( 42 ) An antisense oligonucleotide against TGFα has been reported to enhance the effects of some anticancer drugs, including CDDP.( 43 ) Our results suggest that TGFα may contribute to the drug resistance of CDDP and CPT‐11. The expression of TS was associated with sensitivity to some fluoropyrimidines. TS is a key enzyme in the synthesis of DNA and is the target enzyme of 5‐FU. The relationship between overexpression of TS and 5‐FU resistance has been well characterized.( 44 ) The Ras‐related protein RAB40B, which is a member of the RAS oncogene family, showed a significant correlation with sensitivity to all fluoropyrimidines. In agreement with our study, the expression of RAB40B was downregulated in 5‐FU‐resistant colorectal cell lines.( 45 ) Caudin‐3 (CLDN3) can mediate cell adhesion and play a major role in tight junction‐specific obliteration of the intracellular surface. A low level of CLDN3 was associated with poor patient outcome.( 46 ) Galectin‐4 (LGALS4) is an S‐type lectin that is strongly underexpressed in colorectal cancer.( 47 ) Expression of LGALS4 was associated with multidrug sensitivities in a previous report.( 19 ) The role of these genes in drug sensitivities should be clarified. In addition, some genes ontologically categorized as being involved in cell adhesion, cell proliferation, or Wnt signaling, such as death‐associated protein kinase (DAPK1), v‐erb‐b2 erythroblastic leukemia vial oncogene (ERBB3), and intracellular adhesion molecule 3 (ITGAE3) may be a candidate target of the development of a new drug. CYR61, from the CCN gene family, is a secreted and matrix‐associated protein, which is known as an angiogenic inducer that can promote tumor growth vascularization.( 48 ) Its expression level is regulated by HIF1A under hypoxic conditions.( 31 ) CYR61 plays an important role in resistance to chemotherapeutic agent‐induced apoptosis by a mechanism involving the activation of the integrin/NF‐κB/XIAP signaling pathways.( 49 ) It has also been suggested that the expression level of CYR61 is associated with sensitivity to multiple drugs, including 5‐FU.( 20 ) In our study, the chemosensitivities of six drugs showed a significant association with the expression of CYR61. Furthermore, HIF1A expression exhibited a significant negative correlation with 5‐FU‐based drugs for S‐1 (r = 0.42, P = 0.021), UFT (r = 0.51, P = 0.004), 5′‐DFUR (r = 0.59, P = 0.001), and capecitabine (r = 0.64, P < 0.001). These results support the hypothesis that angiogenic factors might play an important role in resistance to fluoropyrimidines, suggesting that these genes might be useful as common predictive markers for sensitivity to 5‐FU‐based therapy. In addition, the development of drugs that disrupt the HIF1A pathway may lead to additional antitumor effects by targeting tumor‐infiltrating stromal cells, including tumor‐associated fibroblasts and endothelial cells. The fact that our analysis could pick up some genes that have been previously known to be related to drug sensitivity or resistance appears to support the validity of our results, and other genes, which have not been reported previously, may become novel target genes for therapeutic strategies. A validation study for some of these genes is ongoing.
Interestingly, some genes that showed a significant association with sensitivity to 5‐FU‐based drugs were inversely correlated with paclitaxel (Fig. 3). This result suggests that tumors resistant to 5‐FU‐based drugs may respond to paclitaxel therapy. In fact, the combination of S‐1 and paclitaxel has been shown to have potent antitumor and antimetastatic effects on refractory human breast cancer.( 23 ) This combination was also evaluated in a clinical trial and appeared to be tolerated well.( 50 )
Next, we carried out real‐time RT‐PCR for some of the genes and measured the TS and DPD protein levels to validate the expression data obtained by the genechip analysis. The expression data obtained using these two methods were closely correlated for most of the genes that were examined. As for these genes, we also confirmed strong correlations between protein level and mRNA levels. Collectively, these results indicate that the expression profiles obtained using the genechip system are reliable and underscore the importance of the identified genes in drug sensitivity. Our next step will be in vivo validation of the identified genes.
Most previous studies on drug resistance have focused on a limited number of genes with proven functional significance to specific drug sensitivity. Although an evaluation of the genome‐wide gene expression profile may be necessary to identify novel targets, it is difficult to interpret the biological importance of all of the selected genes using various statistical methods alone. In a typical microarray experiment, we are also faced with an extreme multiple testing situation and the possibility of statistical errors. Therefore, we applied an ontological approach to a list of genes that were statistically associated with drug sensitivity to eliminate genes that were unlikely to be related to drug efficacy, despite their statistical significance. The combination of a global gene expression analysis and an ontology analysis provided useful information on possible new gene candidates involved in drug resistance. These results will provide comprehensive genetic information linked to drug sensitivity and serve as a foundation for subsequent functional studies. The results may also enhance the prediction of tumor response to anticancer drugs and contribute to the development of tailor‐made chemotherapy. It has become apparent that tumor response to an anticancer drug cannot be predicted by a single factor and may be determined by a critical balance of various factors. In addition to gene expression profiling, a combination of various approaches, including analyses of polymorphisms, proteomes, metaboromes and genomics, may be applied to achieve a precise diagnosis of future cancer patients.
References
- 1. Golub TR, Slonim DK, Tamayo P et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 1999; 286: 531–7. [DOI] [PubMed] [Google Scholar]
- 2. Golub TR. Genome‐wide views of cancer. N Engl J Med 2001; 8: 601–2. [DOI] [PubMed] [Google Scholar]
- 3. Ulrich CM, Robien K, McLeod HL. Cancer pharmacogenetics: polymorphisms, pathways and beyond. Nat Rev Cancer 2003; 12: 912–20. [DOI] [PubMed] [Google Scholar]
- 4. Mayer F, Hartmann JT, Von Pawel J et al. A phase I study of oral uracil–ftorafur plus folinic acid in combination with weekly paclitaxel in patients with solid tumors. Ann Oncol 2002; 5: 755–9. [DOI] [PubMed] [Google Scholar]
- 5. Takahashi N, Suzuki H, Iwabuchi S, Yamazaki Y, Yanaga K. Third‐line chemotherapy with paclitaxel, irinotecan hydrochloride and cisplatin for recurrent gastric cancer: a case report. Hepatogastroenterology 2005; 61: 326–8. [PubMed] [Google Scholar]
- 6. Iwase H, Shimada M, Tsuzuki T et al. A phase II multicentric trial of S‐1 combined with 24 h infusion of cisplatin in patients with advanced gastric cancer. Anticancer Res 2005; 25: 1297–301. [PubMed] [Google Scholar]
- 7. Schipper DL, Wagener DJ. Chemotherapy of gastric cancer. Anticancer Drugs 1996; 7: 137–49. [DOI] [PubMed] [Google Scholar]
- 8. Langenbach RJ, Danenberg PV, Heidelberger C. Thymidylate synthetase: mechanism of inhibition by 5‐fluoro‐2′‐deoxyuridylate. Biochem Biophys Res Commun 1972; 6: 1565–71. [DOI] [PubMed] [Google Scholar]
- 9. Matsuoka H, Ueo H, Sugimachi K, Akiyoshi T. Preliminary evidence that incorporation of 5‐fluorouracil into RNA correlates with antitumor response. Cancer Invest 1992; 4: 265–9. [DOI] [PubMed] [Google Scholar]
- 10. Diasio RB. Clinical implications of dihydropyrimidine dehydrogenase inhibition. Oncology 1999; 13: 17–21. [PubMed] [Google Scholar]
- 11. Malet‐Martino M, Martino R. Clinical studies of three oral prodrugs of 5‐fluorouracil (capecitabine, UFT, S‐1): a review. Oncologist 2002; 7: 288–323. [DOI] [PubMed] [Google Scholar]
- 12. Fukushima M, Shimamoto Y, Kato T et al. Anticancer activity and toxicity of S‐1, an oral combination of tegafur and two biochemical modulators, compared with continuous i.v. infusion of 5‐fluorouracil. Anticancer Drugs 1998; 9: 817–23. [DOI] [PubMed] [Google Scholar]
- 13. Shirasaka T, Taguchi T. Preclinical and clinical practice of S‐1 in Japan. In: Rustum YH, ed. Fluoropyrimidines in Cancer Therapy. Totowa, NJ: Humana Press, 2002; 285–302. [Google Scholar]
- 14. Kato H, Ichinose Y, Ohta M et al. A randomized trial of adjuvant chemotherapy with uracil–tegafur for adenocarcinoma of the lung. N Engl J Med 2004; 350: 1713–21. [DOI] [PubMed] [Google Scholar]
- 15. Sakata Y, Ohtsu A, Horikoshi N, Sugimachi K, Mitachi Y, Taguchi T. Late phase II study of novel oral fluoropyrimidine anticancer drug S‐1 (1 M tegafur−0.4 M gimestat−1 M otastat potassium) in advanced gastric cancer patients. Eur J Cancer 1998; 34: 1715–20. [DOI] [PubMed] [Google Scholar]
- 16. Shirao K, Ohtsu A, Takada H et al. Phase II study of oral S‐1 for treatment of metastatic colorectal carcinoma. Cancer 2004; 100: 2355–61. [DOI] [PubMed] [Google Scholar]
- 17. Saeki T, Takashima S, Sano M et al. A phase II study of S‐1 in patients with metastatic breast cancer – a Japanese trial by the S‐1 Cooperative Study Group, Breast Cancer Working Group. Breast Cancer 2004; 11: 194–202. [DOI] [PubMed] [Google Scholar]
- 18. Furuse K, Kawahara M, Hasegawa K et al. Early phase II study of S‐1, a new oral fluoropyrimidine, for advanced non‐small‐cell lung cancer. Int J Clin Oncol 2001; 5: 236–41. [DOI] [PubMed] [Google Scholar]
- 19. Zembutsu H, Ohnishi Y, Tsunoda T et al. Genome‐wide cDNA microarray screening to correlate gene expression profiles with sensitivity of 85 human cancer xenografts to anticancer drugs. Cancer Res 2002; 62: 518–27. [PubMed] [Google Scholar]
- 20. Dan S, Tsunoda T, Kitahara O et al. An integrated database of chemosensitivity to 55 anticancer drugs and gene expression profiles of 39 human cancer cell lines. Cancer Res 2002; 62: 1139–47. [PubMed] [Google Scholar]
- 21. Park JS, Young Yoon S, Kim JM, Yeom YI, Kim YS, Kim NS. Identification of novel genes associated with the response to 5‐FU treatment in gastric cancer cell lines using a cDNA microarray. Cancer Letter 2004; 214: 19–33. [DOI] [PubMed] [Google Scholar]
- 22. Fukushima M, Fujioka A, Uchida J, Nakagawa F, Takechi T. Thymidylate synthase (TS) and ribonucleotide reductase (RNR) may be involved in acquired resistance to 5‐fluorouracil (5‐FU) in human cancer xenografts in vivo . Eur J Cancer 2001; 37: 1681–7. [DOI] [PubMed] [Google Scholar]
- 23. Nukatsuka M, Fujioka A, Nakagawa F et al. Antimetastatic and anticancer activity of S‐1, a new oral dihydropyrimidine‐dehydrogenase‐inhibiting fluoropyrimidine, alone and in combination with paclitaxel in an orthotopically implanted human breast cancer model. Int J Oncol 2004; 25: 1531–6. [PubMed] [Google Scholar]
- 24. Takechi T, Okabe H, Ikeda K et al. Correlations between antitumor activities of fluoropyrimidines and DPD activity in lung tumor xenografts. Oncol Report 2005; 14: 33–9. [PubMed] [Google Scholar]
- 25. United Kingdom Co‐ordinating Committee on Cancer Research (UKCCCR) guidelines for the welfare of animals in experimental neoplasia ( second edition). Br J Cancer 1998; 77: 1–10. [DOI] [PMC free article] [PubMed]
- 26. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 2001; 25: 402–8. [DOI] [PubMed] [Google Scholar]
- 27. Takechi T, Okabe H, Fujioka A, Murakami Y, Fukushima M. Relationship between protein levels and gene expression of dihydropyrimidine dehydrogenase in human tumor cells during growth in culture and in nude mice. Jpn J Cancer Res 1998; 89: 1144–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ikenaka K, Shirasaka T, Kitano S, Fujii S. Effect of uracil on metabolism of 5‐fluorouracil in vitro . Gann 1979; 70: 353–9. [PubMed] [Google Scholar]
- 29. Spears CP, Shahinian AH, Moran RG, Heidelberger C, Corbett TH. In vivo kinetics of thymidylate synthetase inhibition of 5‐fluorouracil‐sensitive and ‐resistant murine colon adenocarcinomas. Cancer Res 1982; 42: 450–6. [PubMed] [Google Scholar]
- 30. Dan S, Shirakawa M, Mukai Y et al. Identification of candidate predictive markers of anticancer drug sensitivity using a panel of human cancer cell lines. Cancer Sci 2003; 94: 1074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kunz M, Moeller S, Koczan D et al. Mechanisms of hypoxic gene regulation of angiogenesis factor Cyr61 in melanoma cells. J Biol Chem 2003; 278: 45 651–60. [DOI] [PubMed] [Google Scholar]
- 32. Scherf U, Ross DT, Waltham M et al. A gene expression database for the molecular pharmacology of cancer. Nat Genet 2000; 24: 236–44. [DOI] [PubMed] [Google Scholar]
- 33. Zhou Y, Gwadry FG, Reinhold WC. Transcriptional regulation of mitotic genes by camptothecin‐induced DNA damage: microarray analysis of dose‐ and time‐dependent effects. Cancer Res 2002; 62: 1688–95. [PubMed] [Google Scholar]
- 34. Pusztai L, Sotiriou C, Buchholz TA. Molecular profiles of invasive mucinous and ductal carcinomas of the breast: a molecular case study. Cancer Genet Cytogenet 2003; 141: 148–53. [DOI] [PubMed] [Google Scholar]
- 35. De Lange R, Burtscher H, Jarsch M, Weidle UH. Identification of metastasis‐associated genes by transcriptional profiling of metastatic versus non‐metastatic colon cancer cell lines. Anticancer Res 2001; 21: 2329–39. [PubMed] [Google Scholar]
- 36. Ross DT, Scherf U, Eisen MB et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet 2000; 24: 227–35. [DOI] [PubMed] [Google Scholar]
- 37. Tashiro T, Inaba M, Kobayashi T et al. Responsiveness of human lung cancer/nude mouse to antitumor agents in a model using clinically equivalent doses. Cancer Chemother Pharmacol 1989; 24: 187–92. [DOI] [PubMed] [Google Scholar]
- 38. Tatsumi K, Fukushima M, Shirasaka T, Fujii S. Inhibitory effects of pyrimidine, barbituric acid and pyridine derivatives on 5‐fluorouracil degradation in rat liver extracts. Jpn J Cancer Res 1987; 78: 748–55. [PubMed] [Google Scholar]
- 39. Takechi T, Fujioka A, Matsushima E, Fukushima M. Enhancement of the antitumour activity of 5‐fluorouracil (5‐FU) by inhibiting dihydropyrimidine dehydrogenase activity (DPD) using 5‐chloro‐2,4‐dihydroxypyridine (CDHP) in human tumour cells. Eur J Cancer 2002; 38: 1271–7. [DOI] [PubMed] [Google Scholar]
- 40. Fujiwara H, Terashima M, Irinoda T et al. Superior antitumor activity of S‐1 in tumours with a high dihydropyrimidine dehydrogenase activity. Eur J Cancer 2003; 39: 2387–94. [DOI] [PubMed] [Google Scholar]
- 41. Usuki H, Ishimura K, Yachida S et al. Dihydropyrimidine dehydrogenase (DPD) activity in gastric cancer tissue and effect of DPD inhibitory fluoropyrimidines. Gastric Cancer 2003; 6: 66–70. [DOI] [PubMed] [Google Scholar]
- 42. Massague J. Transforming growth factor‐α. A model for membrane‐anchored growth factors. J Biol Chem 1990; 15: 21 393–6. [PubMed] [Google Scholar]
- 43. De Luca A, Selvam MP, Sandomenico C et al. Anti‐sense oligonucleotides directed against EGF‐related growth factors enhance anti‐proliferative effect of conventional anti‐tumor drugs in human colon‐cancer cells. Int J Cancer 1997; 73: 277–82. [DOI] [PubMed] [Google Scholar]
- 44. Johnston PG, Lenz HJ, Leichman CG. Thymidylate synthase gene and protein expression correlate and are associated with response to 5‐fluorouracil in human colorectal and gastric tumors. Cancer Res 1995; 55: 1407–12. [PubMed] [Google Scholar]
- 45. Mariadason JM, Arango D, Shi Q et al. Gene expression profiling‐based prediction of response of colon carcinoma cells to 5‐fluorouracil and camptothecin. Cancer Res 2003; 63: 8791–812. [PubMed] [Google Scholar]
- 46. Heinzelmann‐Schwarz VA, Gardiner‐Garden M, Henshall SM et al. Overexpression of the cell adhesion molecules DDR1, Claudin 3, and Ep‐CAM in metaplastic ovarian epithelium and ovarian cancer. Clin Cancer Res 2004; 10: 4427–36. [DOI] [PubMed] [Google Scholar]
- 47. Rechreche H, Mallo GV, Montalto G, Dagorn JC, Iovanna JL. Cloning and expression of the mRNA of human galectin‐4, an S‐type lectin down‐regulated in colorectal cancer. Eur J Biochem 1997; 248: 225–30. [DOI] [PubMed] [Google Scholar]
- 48. Babic AM, Kireeva ML, Kolesnikova TV, Lau LF. CYR61, a product of a growth factor‐inducible immediate early gene, promotes angiogenesis and tumor growth. Proc Natl Acad Sci USA 1998; 95: 6355–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lin MT, Chang CC, Chen ST et al. Cyr61 expression confers resistance to apoptosis in breast cancer MCF‐7 cells by a mechanism of NF‐κB‐dependent XIAP up‐regulation. J Biol Chem 2004; 279: 24 015–23. [DOI] [PubMed] [Google Scholar]
- 50. Villalona‐Calero MA, Weiss GR, Burris HA et al. Phase I and pharmacokinetic study of the oral fluoropyrimidine capecitabine in combination with paclitaxel in patients with advanced solid malignancies. J Clin Oncol 1999; 17: 1915–25. [DOI] [PubMed] [Google Scholar]