

Abstract
YKL‐40 is a 40 kDa secreted glycoprotein belonging to the family of ‘mammalian chitinase‐like proteins’, but without chitinase activity. YKL‐40 has a proliferative effect on fibroblasts, chondrocytes and synoviocytes, and chemotactic effect on endothelium and vascular smooth muscle cells. Elevated YKL‐40 levels are found in serum of patients with diseases characterized by inflammation, fibrosis and tissue remodeling. Several studies have reported that high serum YKL‐40 levels in patients with cancer are associated with poor prognosis. YKL‐40 expression is strongly elevated in serum and biopsy material from glioblastomas patients. We investigated the expression of YKL‐40 in three human malignant glioma cell lines exposed to different types of stress. Whereas a polymerase chain reaction transcript was detectable in all three cell lines, only U87 produced measurable amounts of YKL‐40 protein. In U87, hypoxia and ionizing radiation induced a significant increase in YKL‐40 after 24–48 h. The hypoxic induction of YKL‐40 was independent of HIF1. Etoposide, ceramide, serum depletion and confluence all led to elevated YKL‐40. Inhibition of p53 augmented the YKL‐40 expression indicating that YKL‐40 is attenuated by p53. In contrast, both basic fibroblast growth factor and tumor necrosing factor‐α repressed YKL‐40. These are the first data on regulation of YKL‐40 in cancer cells. Diverse types of stress resulted in YKL‐40 elevation, which strongly supports an involvement of YKL‐40 in the malignant phenotype as a cellular survival factor in an adverse microenvironment. (Cancer Sci 2005; 96: 183–190)
YKL‐40 a mammalian member of ‘chitinase‐like proteins’, is a secreted 40 kDa glycoprotein named after its molecular weight and the three N‐terminal amino acids, tyrosin (Y), lysine (K) and leucine (L). 1 , 2 Other names for YKL‐40 are human cartilage glycoprotein‐39 (HC gp‐39), (2) 38‐kDa heparin binding glycoprotein (Gp38k), 3 , 4 , 5 Chitinase 3‐like protein (CHI3L1), 6 , 7 Breast regressing protein‐39, (8) and Chondrex. (9) The gene for YKL‐40 (CHI3L1) 6 , 7 is located on chromosome 1q31‐q32. The crystallographic three‐dimensional structure of human 10 , 11 and goat YKL‐40 (12) are described, but the site and mode of binding to cell surface receptors is unknown. Although homologous in sequence to bacterial and fungal chitinases, and with high chitin binding ability, YKL‐40 fails to degrade chitin due to a single amino acid substitution in the proposed catalytic site. 2 , 13 The function of YKL‐40 is still elusive, but high expression is found during non‐pathological processes such as mammary involution, and in pathological conditions characterized by inflammation, tissue destruction and development of fibrosis, suggesting a major role of YKL‐40 in inflammation and tissue remodeling. 8 , 9 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 YKL‐40 messenger ribonucleic acid (mRNA) and protein expression is found in differentiated macrophages in vitro, and in a subset of macrophages in vivo under pathological conditions such as rheumatoid arthritis, osteoarthritis, giant cell arteritis and arteriosclerosis. 6 , 17 , 18 , 19 , 20 , 21 Expression is also seen in synovial cells, chondrocytes and neutrophils. 2 , 18 , 22 , 25 , 26 YKL‐40 stimulates proliferation of connective tissue cells, including skin and lung fibroblasts, chondrocytes and synovial cells, and it modulates expression of chemokines and metalloproteases in inflammatory fibroblasts. 27 , 28 , 29 The porcine variant of YKL‐40 (Gp38k) is expressed by vascular smooth muscle cells (4) and functions as a chemotactic factor for human umbilical vein endothelial cells, (5) as well as an adhesion and migration factor for vascular smooth muscle cells; (30) two processes involved in angiogenesis.
Concordantly, high serum concentrations of YKL‐40 are found in patients with pathologies, such as rheumatoid arthritis, osteoarthritis, giant cell arteritis, bacterial infections, inflammatory bowel diseases and liver fibrosis. 9 , 14 , 15 , 16 , 21 , 23 , 24 Strong evidence also point to a role of YKL‐40 in cancer. First, microarray gene analyses have identified the YKL‐40 gene to be one of the most highly over‐expressed genes in high‐grade malignant gliomas, 31 , 32 , 33 in papillary thyroid carcinomas, (34) and in extraskeletal myxoid chondrosarcoma. (35) Furthermore, YKL‐40 is expressed by murine mammary tumors initiated by neu/ras oncogenes but not by c‐myc or int‐2 oncogenes. (8)
Second, elevated serum YKL‐40 levels (compared to healthy subjects) are found in subgroups of patients with different types of solid tumors, including several types of primary or advanced adenocarcinomas (breast, colorectal, ovarian), small cell lung cancer (SCLC) and glioblastoma, and elevated serum YKL‐40 is related to short recurrence‐free interval and short survival. 33 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 In patients with gliomas, serum YKL‐40 was positively correlated to tumor grade and burden. (33)
Regulation studies of YKL‐40 expression are sparse. Transcriptional regulation of YKL‐40 during human macrophage differentiation has been described. (7) There are probably two independent transcription start sites and the promoter sequence contained binding sites for several known factors and specific binding of nuclear PU.1, Sp1, Sp3, USF, AML‐1 and C/EBP proteins. It was found that the Sp1‐family transcription factors seem to have a predominating role in controlling YKL‐40 promoter activity. (7)
Our general hypothesis is that YKL‐40 is a cellular survival factor. Therefore, we investigated to what degree various types of physiological stress, mimicking the adverse microenvironment of solid tumors during growth or therapy, influences the expression of YKL‐40.
Methods and Materials
Reagents. Cobalt chloride (CoCl2), N‐acetyl cysteine, tumor necrosing factor‐alpha (TNFα), basic fibroblast growth factor (bFGF) and interleukin (IL)‐6 were obtained from Sigma Aldrich. Cyclic pifithrin‐α and C2 ceramide was obtained from Calbiochem. All cell culture reagents were obtained from Invitrogen unless other suppliers are listed.
Cell lines and treatments. The glioblastoma cell lines U87, U118 and U373 were obtained from American Type Culture Collection and cultured at 37°C, 5% carbon dioxide and 95–98% humidity in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS). The cell lines were tested free of mycoplasma. Twenty‐four hours prior to treatment, cells were seeded at 105 cells/mL. Immediately prior to treatment, the media was changed. After treatment, a 250 µL media sample was collected from individual dishes at different time points and frozen at −80°C with 40 µL of Complete protease inhibitor (Roche Diagnostics) for later enzyme‐linked immunosorbent assay. The cells were trypsinized, nigrosin stained and counted. Cells exposed to the same treatment were pooled, rinsed in ice cold phosphate‐buffered saline and frozen at −80°C for later RNA or protein isolation. For radiation, the cells were given a single dose of ionizing radiation of 0, 2, 5, 10 or 20 Gy (Stabilipan; Siemens). For hypoxia treatment, the cells were transferred to an incubator with a hypoxic environment, with 0.1% oxygen in an InVivo 400 Hypoxia Cabinet (Ruskinn Technology). For treatment with CoCl2 (0, 100 and 250 µM), etoposide (0, 0.16, 0.31, 0.63, 1.25 and 2.5 µM), C2 ceramide (0, 5, 10 and 20 µM) or IL‐6 (1, 10, 100 or 500 U/mL), cells were allowed to plate overnight (O/N) before treatment. When treated with bFGF and TNFα, cells were allowed to plate O/N in media containing 10% FBS. The cells were rinsed and given fresh media with 0.1% FBS with TNFα (10 ng/mL) or bFGF (25 ng/mL). Cyclic pifithrin‐α, a p53 inhibitor, (45) was given at 0, 100 nM or 1 µM 30–60 min prior to radiation. A commercial siRNA duplex against p53 was obtained from Dharmacon. Semi‐confluent cells were transfected with either buffer or siRNA (100 nM) by Lipofectamine2000 (Invitrogen) as described by the manufacturer for 6 h, left O/N and trypsinized for irradiation in 10 mL of media. After irradiation the cells were reseeded and cells and media was harvested 48 h later. For N‐acetyl cysteine (NAC) treatment (0, 5 and 25 mM) cells were treated 24 h before irradiation and continuously hereafter until harvest 48 h after irradiation.
Enzyme‐linked immunosorbent assay. YKL‐40 protein secreted by the cells was measured in the collected media samples by a two‐site sandwich‐type enzyme‐linked immunosorbent assay (ELISA) (Quidel Corporation). (9)
This assay uses a streptavidin‐coated microplate well, a biotinylated‐Fab monoclonal mouse antibody against human YKL‐40 (capture antibody), and an alkaline phosphatase‐labeled polyclonal rabbit antibody against human YKL‐40 (detection antibody). Bound enzyme activity is detected with p‐nitrophenyl phosphate as substrate. The sensitivity of the ELISA is 8 µg/l, and the intra‐ and interassay coefficient of variations are <3.6% and <5.3%, respectively. YKL‐40 content in the conditioned media was quantified per number of cells. Each time point/dose tested was analyzed in triplicate. No detectable YKL‐40 was measured in Dulbecco's minimal essential medium (DMEM) containing 10% FBS. Vascular endothelial growth factor (VEGF) protein secreted by the cells was measured in the media by ELISA (R & D System).
Reverse‐transcriptase polymerase chain reaction and Northern blots. Total RNA was extracted with TRizol (Invitrogen) or with RNeasy minikit (Qiagen, Germany) as described by the manufacturer. cDNA was generated by ThermoScript cDNA kit (Invitrogen) as described by the manufacturer. Total RNA (1 µg) was used for each reaction with polyT primers. All reverse transcriptase‐polymerase chain reaction (RT‐PCR) fragments were amplified using the AmpliTaqGold System (Applied Biosystems, USA) under the following conditions: 95°C for 5 min, followed by 35 cycles of 95°C for 30 s, a 1 min annealing step at 60°C, and a 1 min elongation step at 72°C. All PCR were finalized by a 10 min elongation step at 72°C. A control reaction with GADPH primers (sense 5′AACGGATTTGGTCGTATTGGGC3′ and antisense 5′‐TAAGCAGTTGGTGGTGCAGG3′) was performed to verify the quality of the cDNA before PCR with primers for human YKL‐40 (493‐bp product: sense 5′‐CCTGCTCAGCGCAGCACTGT‐3′ and antisense 5′‐GCTTTTGACGCTTTCCTGGTC.
For Northern blots, 10 µg total RNA was separated on a 1% agarose gel containing 2% formaldehyde and blotted to a Gene Screen plus membrane by capillary transfer. A 493 bp YKL‐40 probe was generated by RT‐PCR on cDNA from the osteosarcoma cell line MG‐63 (American Type Culture Collection), a well known producer of YKL‐40, with the above mentioned primers and conditions. The size and purity was analyzed on a 1.2% agarose and identity was verified by sequencing (MWG‐Biotech AG).
The YKL‐40 probe was labeled with P32 by random prime using a Rediprime DNA labeling kit (Amersham, Pharmacia Biotech). The probe was hybridized O/N at 42°C to membranes prehybridized for at least 1 h in 5% dextran sulfate, 50% formamide, 1% sodium dodecylsulfate, 1 M NaCl and 100 µg/mL salmon sperm DNA. Following hybridization, the membranes were washed under high stringency conditions and RNA expression was visualized on a STORM 840 PhosphoImager (Molecular Dynamics Inc.). After stripping, the membranes were probed for 28S to ensure equal loading. The 28S probe was a commercial oligo (Amersham, Pharmacia Biotech) that was labeled by 5′end labeling kit (Amersham, Pharmacia Biotech Inc.).
Western blotting. Protein was extracted from cell pellets with a lysis buffer (50 mM Tris [pH 7.5]), 150 mM NaCl, 1% NP‐40, 0.1% SDS, 1% (w/v) Natrium‐deoxycholat, 1 mM Na3VO4, 1 mM dithithreitol (DTT), Complete protease inhibitors (Roche Diagnostics), and homogenized by sonication. Protein concentration was determined by BCA protein assay (Pierce). 30–90 µg of protein was separated on NuPage Bis‐Tris gels (Invitrogen) at 150 V and transferred to nitrocellulose membranes by semidry blotting. Protein transfer was confirmed by ponceau dye staining. Western detection was performed after blocking in 5% dry milk in TRIS buffered saline with 0.1% tween‐20 (TBST). The membrane incubated O/N at 4°C with primary antibody in TBST (p53, Pharmingen, 15791 A, 2 µg/mL or Oncogene, OP43, 0,1 µg/mL; P21, Oncogene, Ab‐1, 1:500; hypoxia inducible factor (HIF) 1α, Transduction Laboratories, 1 µg/mL, Tubulin, Sigma T‐9026, 1:80 000) and horse radish peroxidase (HRP) coupled secondary antibody (Dako) for 1 h before detection with ECL Plus (Amersham, Pharmacia Biotech). The Western blots were visualized by a STORM 840 blue fluorescence scanner (Molecular Dynamics Inc.).
Statistical analysis was performed using t‐test.
Results
Three glioma cell lines were analyzed for YKL‐40 mRNA and protein expression by RT‐PCR and ELISA.
In all three cell lines, a transcript was detected (Fig. 1), whereas YKL‐40 protein was found in measurable amounts in conditioned media from U87 cells only (Fig. 2a).
Figure 1.
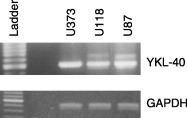
Reverse transcriptase‐polymerase chain reaction (RT‐PCR) of cDNA from U87, U118 and U373 cells with primers specific for YKL‐40. The PCR products were separated on a 1.2% agarose gel and visualized by ethidium bromide staining. A ladder was included as a size control and a separate RT‐PCR with primers for the housekeeping gene glyceraldehyde‐3‐phosphate dehydrogenase was performed to verify the quality of the cDNA.
Figure 2.
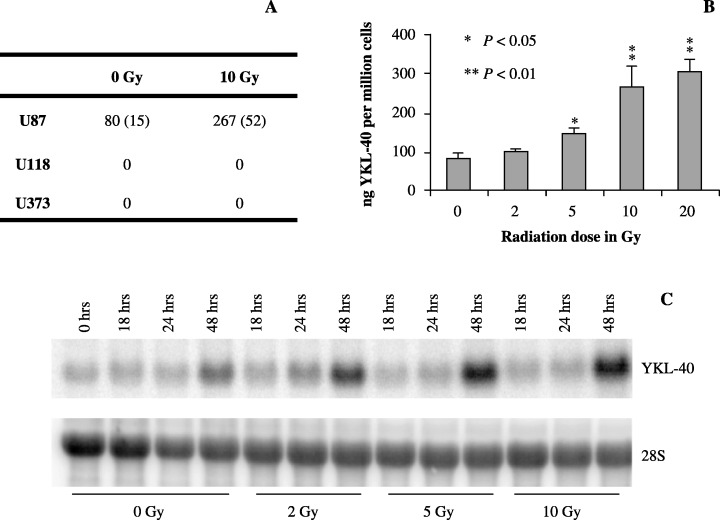
(a) U87, U118 and U373 cells were irradiated with 10 Gy. After 48 h, conditioned cell culture media was collected and YKL‐40 protein levels were determined by enzyme‐linked immunosorbent assay and divided by the cell number. Experiments were performed in triplicates. Mean (ng/million cells) and standard deviation (SD) are shown. (b) U87 cells were irradiated with 0, 2, 5, 10 and 20 Gy and harvested as in (a). Mean and SD are shown. (c) In a parallel experiment total ribonucleic acid was extracted at different time points after 0, 2, 5 and 10 Gy and analyzed by Northern blotting with a YKL‐40 probe. The stripped membrane was probed for 28S to verify equal loading.
Ionizing radiation induces YKL‐40 expression. U87, U118 and U373 cells were irradiated with 10 Gy, and YKL‐40 was measured in the media after 48 h. An increase in secreted YKL‐40 at 48 h after irradiation was found in U87, but not in U118 or U373 cells (Fig. 2a). The U87 cell lines was subjected to doses of 0, 2, 5, 10 and 20 Gy of irradiation, and the increase in YKL‐40 after irradiation was found to be dose dependent and significant at 5, 10 and 20 Gy (Fig. 2b). To ensure that this was not a consequence of YKL‐40 accumulating in the media after cell death, the up‐regulation was confirmed at mRNA level by Northern blotting, showing an increase in YKL‐40 mRNA after exposure of cells to 2, 5 and 10 Gy of ionizing radiation (Fig. 2c). Elevated YKL‐40 mRNA was not seen until 48 h after irradiation, indicating that the expression of YKL‐40 is a secondary response to ionizing radiation downstream of other signals. An increased YKL‐40 mRNA expression was also found in non‐irradiated cells as a result of high cell density, which in itself increases YKL‐40 production in U87 cells. A dose of 10 Gy reduced the cell number by approximately 50% at 48 h, relative to non‐irradiated control cells.
YKL‐40 expression is induced by hypoxia, independently of hypoxia inducible factor 1. During severe hypoxia (0.1%) the level of YKL‐40 in the media of U87 cells was significantly upregulated in a time dependent manner after 24 and 48 h of hypoxia (Fig. 3a). The hypoxia induced YKL‐40 production at 24 and 48 h was verified on mRNA level by Northern blotting (Fig. 3b). As with ionizing radiation, no effect of hypoxia on YKL‐40 expression was seen in U118 and U373 cells (data not shown).
Figure 3.
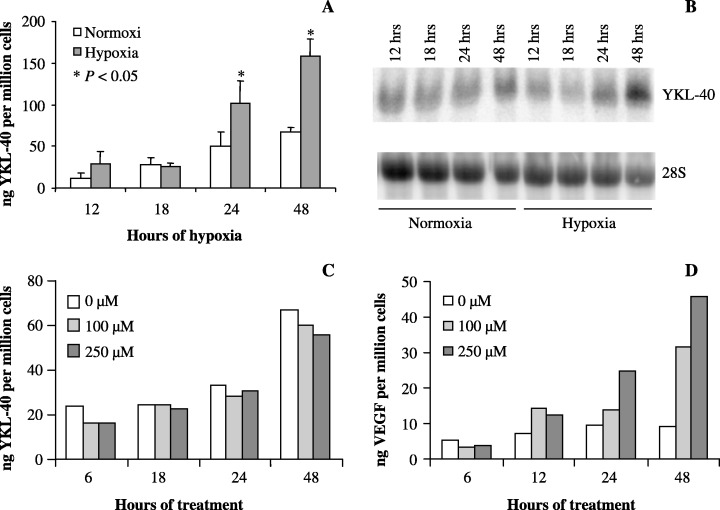
(a,b) U87 cells were exposed to hypoxia (0.1%) or normoxia. At different time points conditioned cell culture media and cells were collected for (a) enzyme‐linked immunosorbent assay (ELISA), and (b) total ribonucleic acid (RNA) for Northern blot. (a) The YKL‐40 protein level was correlated to cell number. Experiments were performed in triplicates. Mean and standard deviation (SD) are shown. (b) YKL‐40 messenger RNA was analyzed by Northern blotting with a YKL‐40 probe. The stripped membrane was probed for 28S to verify equal loading. (c,d) Cells were treated with cobalt chloride at 100 µM and 250 µM to induce ‘chemical hypoxia’. At different time points, conditioned cell culture media was collected for determination of YKL‐40 protein levels by ELISA (c), and vascular endothelial growth factor protein levels by ELISA (d).
HIF1 is a well‐characterized transcription factor responsible for upregulation of many genes in response to hypoxia. 46 , 47 , 48 , 49 A strong upregulation of HIF1α was observed after 4 h in response to hypoxia and a subsequent increase in VEGF after 8 h (data not shown). To investigate if the upregulation of YKL‐40, seen under hypoxic conditions, was mediated by HIF1, U87 cells were treated with cobalt chloride. Cobalt chloride induces ‘chemical hypoxia’ by preventing the normoxic degradation of the HIF1α subunit through an inhibitory binding of cobalt to prolyl‐4‐hydroxylase, which targets the HIF1α subunit of HIF1 for degradation. Consequently genes under transcriptional control of HIF1 are upregulated. (50) We found no increase in YKL‐40 protein levels in conditioned media of U87 cells following treatment with cobalt chloride, whereas VEGF levels in the media increased dose dependently, which confirmed the expected stabilization of the HIF1α in the HIF1 complex (Fig. 3c,d). Therefore HIF1 does not mediate the hypoxic upregulation of YKL‐40.
Etoposide induces YKL‐40 expression. U87 cells were treated with etoposide, which induces DNA double strand breaks through inhibition of topoisomerase II. The U87 cells had a mortality of approximately 50% after 48 h of treatment with 0.31 µM etoposide. YKL‐40 protein content in conditioned media increased significantly. This increase was confirmed on the mRNA level by Northern blotting (Fig. 4).
Figure 4.
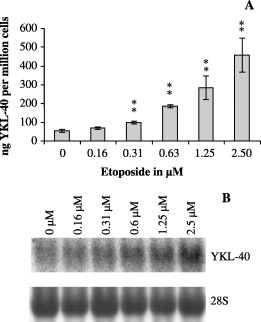
U87 cells were treated with etoposide (0.16, 0.31, 0.63, 1.25 and 2.5 µM, or vehicle only [0.1% DMSO]) for 48 h. (a) YKL‐40 was measured in the conditioned media by enzyme‐linked immunosorbent assay and divided by the cell number. Experiments were performed in triplicates. Mean and standard deviation are shown. (b) In a parallel experiment total ribonucleic acid was extracted and analyzed by Northern blotting with a YKL‐40 probe. The stripped membrane was probed for 28S to verify equal loading.
Ceramide induces YKL40 expression. Ceramide is an intracellular messenger, which has previously been found elevated after irradiation and by hypoxia and etoposide treatment. 51 , 52 , 53 , 54 , 55 U87 cells were treated with a dose range of C2 ceramide; a cell permeable analog of ceramide. We found a dose dependent increase in YKL‐40 mRNA expression after 72 h of treatment (Fig. 5b). The dose dependent increase in secreted YKL‐40 was significant with 10 and 20 µM C2 ceramide (Fig. 5a). C2 Ceramide is a known inducer of apoptosis (52) and a dose dependent toxicity, with 25% less cells at 20 µM C2 ceramide compared to control treatment was observed.
Figure 5.
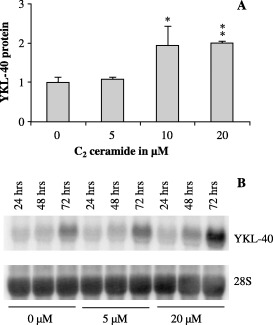
U87 cells were treated with C2 ceramide (5 µM or 20 µM, or vehicle only [0.1% dimethyl sulfoxide]) for 24, 48 and 72 h. (a) YKL‐40 was measured in the conditioned media by enzyme‐linked immunosorbent assay after 72 h of treatment and normalized to 0 µM. Experiments were performed in triplicates. Mean and standard deviation are shown. (b) Cells were collected after 24, 48 and 72 h of treatment, and YKL‐40 messenger ribonucleic acid was analyzed by Northern blotting with an YKL‐40 probe. The stripped membrane was probed for 28S to verify equal loading.
The tumor suppressor p53 has an inhibitory effect on YKL‐40 expression. The increase in YKL‐40 by irradiation and hypoxia is seen only in U87 cells, but not in U118 and U373 cells. U87 cells are p53 proficient, whereas U118 and U373 are p53 deficient. (56) P53 is a tumor suppressor, known to be involved in damage responses after irradiation, hypoxia and etoposide treatment acting upstream of ceramide signaling. 55 , 57 , 58 , 59 We hypothesized that the difference in YKL‐40 expression in U87 compared to U118 and U373 could be caused by the possible p53 response in U87 cells induced by cellular stress. We analyzed p53 function in U87, U118 and U373, 24 h after 10 Gy of ionizing irradiation (Fig. 6a). P53 was increased in U87 cells after irradiation. The increased levels of functional p53 are demonstrated by an increase in downstream p21. The high levels of p53 in U118 and U373 cell lines, both with and without irradiation, is most likely caused by a failure to degrade p53 through hDM2 ubiquitin ligase loop. (60) No difference in p21 levels were found in U118 and U373 after irradiation, reflecting that they have mutated (non‐functional) p53 protein as described in the literature. (56) Therefore, recognizing that only U87 cells have functional p53, we continued our investigations with this cell line. We repressed p53 function in U87 cells prior to irradiation (10 Gy) with either a small molecule p53 inhibitor, cyclic pifithrin‐α, or with siRNA against p53. In both cases, additional elevations in YKL‐40 mRNA (Fig. 6e,g) and secreted protein (Fig. 6d,f) were found, thus demonstrating that p53 has an inhibitory effect on YKL‐40. The inhibition of p53 function by cyclic pifithrin‐α and with siRNA against p53 was verified by Western blotting against p53, and the downstream protein p21 (Fig. 6b,c). It should be noted that while siRNA treatment against p53 causes down‐regulation of both p53 and p21, cyclic pifithrin‐α caused only a decrease in p53 levels, whereas no effect could be seen on p21 levels. This is in agreement with the original characterization of p53 inhibition with cyclic pifithrin‐α. (45) Cyclic pifithrin‐α caused no toxicity, neither before nor after irradiation, while siRNA against p53 resulted in approximately 25% less cells at 0 Gy, and no further increase in toxicity after 10 Gy of irradiation.
Figure 6.
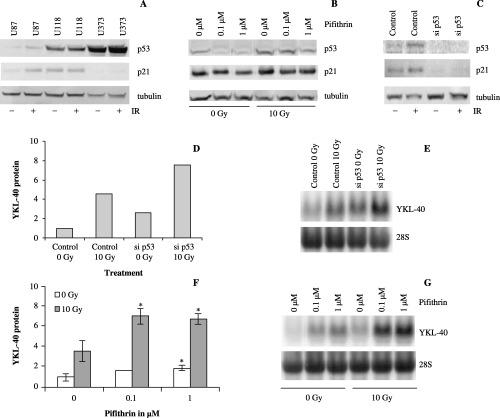
(a) U87, U118 and U373 cells were irradiated (10 Gy). Protein from irradiated and control cells was used for Western blotting with p53 and p21 antibodies. Equal loading was confirmed by tubulin antibody. (b) Confirmation of siRNA treatment against p53. Protein from siRNA treated U87 cells and controls with and without irradiation (10 Gy) was used for Western blotting with p53 and p21 antibodies. Equal loading was confirmed by tubulin antibody. (c) Similar to (b) with protein from cyclic pifithrin‐α treated U87 cells. (d) U87 cells were treated with siRNA against p53 (si p53) and ionizing radiation (10 Gy). Controls were treated with siRNA buffer only. YKL‐40 was measured in conditioned media by enzyme‐linked immunosorbent assay (ELISA) 48 h after ionizing radiation. The experiment was performed twice and the mean is shown. YKL‐40 levels were normalized to control (0 Gy). (e) YKL‐40 mRNA from the same experiment was analyzed by Northern blotting with an YKL‐40 probe. The stripped membrane was probed for 28S to verify equal loading. (f) U87 cells were treated with the small molecule p53 inhibitor cyclic pifithrin‐α, 1 h prior to ionizing radiation (10 Gy). Control cells were treated with vehicle (0.1% dimethyl sulfoxide) only. Media was harvested 48 h after irradiation for YKL‐40 protein measurement by ELISA. YKL‐40 levels are normalized to control (0 Gy). Experiments were performed in triplicates. Mean and standard deviation are shown. (g) The increases in YKL‐40 levels were confirmed on RNA levels by Northern blotting with YKL‐40 and 28S probes.
Antioxidant treatment enhances YKL‐40 expression. Reactive oxygen species (ROS) are generated by ionizing radiation and hypoxia. 61 , 62 Furthermore, ceramide signaling is documented to function downstream of ROS. 54 , 55 , 63 , 64 Cells were treated with the antioxidant NAC. A dose of 25 mM of NAC was significantly toxic and the increase in YKL‐40 protein level was significant both with and without irradiation (Fig. 7a). The increase in YKL‐40 mRNA is evident at both 5 and 25 mM, both with and without radiation. (Fig. 7b).
Figure 7.
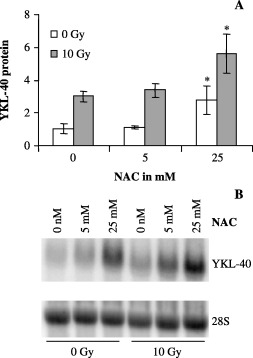
U87 cells were exposed to N‐acetyl cysteine (NAC) (5 µM and 25 µM) and ionizing radiation (10 Gy). Cells were treated with NAC 24 h prior to irradiation. Conditioned cell culture media and cells were collected for (a) enzyme‐linked immunosorbent assay and (b) Northern blot 48 h after irradiation. (a) Experiments were performed in triplicates and YKL‐40 levels were normalized to control, 0 Gy. Mean and standard deviation are shown. (b) YKL‐40 messenger ribonucleic acid was analyzed by Northern blotting with an YKL‐40 probe. The stripped membrane was probed for 28S to verify equal loading.
Serum depletion enhances YKL‐40 expression while TNFα and bFGF repress YKL‐40 expression. The YKL‐40 response to treatment with TNFα and bFGF was investigated. To avoid interference from growth factors present in serum, these investigations were performed at 0.1% FBS. Serum depletion (0.1%) in itself upregulated YKL‐40 mRNA and YKL‐40 protein levels. Band treatment with TNFα and bFGF prevented this upregulation of YKL‐40 at 0.1% FBS (Fig. 8a,b). IL‐6 treatment had no effect on YKL‐40 expression levels (data not shown).
Figure 8.
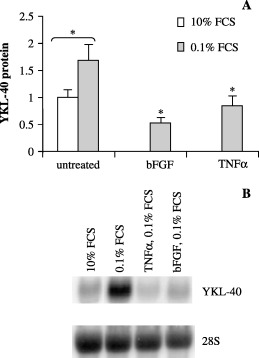
U87 cells were treated with 0.1% and 10% fetal bovine serum (FBS), as well as with tumor necrosing factor‐α (10 ng/mL) and basic fibroblast growth factor (25 ng/mL) in 0.1% FBS media for 72 h. Conditioned cell culture media and cells were collected for (a) enzyme‐linked immunosorbent assay and (b) Northern blot. (a) Experiments were performed in triplicates and YKL‐40 levels were normalized to untreated controls. Mean and standard deviation are shown. (b) YKL‐40 messenger ribonucleic acid was analyzed by Northern blotting with an YKL‐40 probe. The stripped membrane was probed for 28S to verify equal loading.
Discussion
We found increased YKL‐40 expression in U87 cells exposed to an array of stress stimuli in the form of ionizing radiation, hypoxia, cellular confluence, etoposide, ceramide treatment, p53 inhibition, antioxidant treatment and serum depletion (Table 1). The increase in YKL‐40 expression was found both on RNA levels and protein levels, which implies that YKL‐40 is regulated on mRNA level either through transcriptional control or mRNA stabilization.
Table 1.
Summary of factors that can regulate YKL‐40 expression in U87 cells
| Elevated YKL‐40 expression | Repressed YKL‐40 expression |
|---|---|
| Hypoxia | Tumor necrosing factor‐α |
| Ionizing radiation | Basic fibroblast growth factor |
| Confluence | |
| Etoposide | |
| Ceramide | |
| P53 inhibition | |
| Antioxidant treatment | |
| Serum depletion |
Hypoxia, chronic as well as acute, is an important constituent of the microenvironment of malignant tumors, and alterations in regulation of HIF1 and its downstream pathways are important determinants of the malignant phenotype. (46) The enhanced expression of YKL‐40, found during severe hypoxia, was not reproduced by stimulation with cobalt chloride, which induces ‘chemical hypoxia’ through stabilization of HIF1. (50) Therefore, the increase in YKL‐40 expression caused by hypoxia is highly unlikely to be mediated by HIF1. This is in accordance with the observation that a strong increase in HIF1α expression occurs as early as after 4 h of hypoxia with a subsequent increase in VEGF protein in the media after 8 h. In contrast, the increase in YKL‐40 expression occurred later and is first significantly present after 24 h and more pronounced at 48 h after initiation of hypoxia.
YKL‐40 is expressed in vivo by arthritic chondrocytes and macrophages in inflamed synovial membrane, and high levels of YKL‐40 are found in synovial fluid of patients with active rheumatoid arthritis. (18) It is known that the microenvironment of the arthritic joint is relatively ischemic and hypoxic, 65 , 66 and thus hypoxic upregulation of YKL‐40 expression in these situations is plausible.
The cellular response to irradiation is manifold and includes reactions such as DNA damage, ceramide signaling, p53 stabilization, generation of ROS and growth arrest. 51 , 52 , 59 , 61 , 67 Cells exposed to hypoxia share many of these responses. 54 , 57 , 58 , 62
U87 cells exposed to etoposide have elsewhere been shown to induce p53 stabilization as well as ROS and ceramide generation, (55) which would suggest an effect on YKL‐40 expression. The moderate increase in YKL‐40 levels on mRNA level could, in part, be explained by the continuous treatment with etoposide for 48 h as opposed to the instant DNA damage induced by irradiation. YKL‐40 is a late signal, and early cellular responses (e.g. DNA repair mechanisms) perhaps must be completed before YKL‐40 induction occurs.
Treatment with C2 ceramide led to elevated YKL‐40 levels. This response did not occur until after 72 h of treatment, whereas the responses to irradiation, hypoxia and etoposide were seen earlier (at 24 and 48 h). This difference might reflect a lower signaling efficiency of the C2 ceramide compared to endogenously generated ceramide, or it might indicate that ceramide is not part of the same pathways, but rather in itself initiates an YKL‐40 response after long term exposure.
As a result of an effective p53 signaling block in U87 cells by siRNA or cyclic pifithrin‐α, increased levels of YKL‐40 occurred both with and without irradiation. This strongly suggests that p53 stabilization exerts an inhibitory influence on YKL‐40 production. The different types of stress stimuli that we have applied (Table 1) can induce p53 stabilization, 55 , 59 , 68 which therefore must wear off before the YKL‐40 response occurs. Concordantly, one could speculate that p53 stabilization is responsible for the rather late increase in YKL‐40 following irradiation. Alternatively, p53 could be part of a balance between pro‐ and anti‐YKL‐40 stimuli, where the inhibitory signal from p53 is eventually overridden by stronger YKL‐40 stimulating signals. Protection against ROS, in this case by NAC treatment, produced a similar effect, reflecting that p53 and ROS could be in the same pathway upstream of YKL‐40 after irradiation.
Since p53 down regulation in U87 led to significantly increased levels of YKL‐40, the lack of YKL‐40 response in two other tested human glioma cell lines, U118 and U373, is unlikely to be a direct effect of their mutated non‐functional p53. (56)
We also observed that both bFGF and TNFα could block the YKL‐40 elevation induced by serum depletion. Effects of TNFα on YKL‐40 expression have previously been investigated in chondrocytes and synovial cells. 22 , 25 In early passages of human cultured chondrocytes, TNFα inhibited YKL‐40 protein expression, suggesting that the suppression by TNFα might be a general phenomenon in YKL‐40 producing cells. (22) In the same experiment, bFGF was found to have no or little stimulating effect on YKL‐40 protein expression in primary chondrocyte cultures or passage 1 chondrocytes; contrary to what we found in U87 cells. (22) No effect of TNFα on YKL‐40 protein expression was found in synovial cells. (25) The different response in YKL‐40 expression after stimulation with TNFα and bFGF possibly reflects the difference in cell types, TNFα and bFGF receptor status, and activated converging signaling pathways.
So far, our findings suggest that YKL‐40 expression is enhanced at a late stage of stress (possibly after transient p53 stabilization), when the cells are committed to either survival or apoptosis.
Ling and Recklies have recently shown that stimulation of fibroblasts and chondrocytes with IL‐1β and TNFα elicit an inflammation‐like cytokine response, which is suppressed in the presence of YKL‐40. (29) They suggested that YKL‐40 has a physiological role in limiting the catabolic effects in inflammatory situations. (29) YKL‐40 stimulation of fibroblasts leads to AKT phosphorylation through the PI‐3K pathway. Since the PI‐3K pathway, and in particular the phosphorylation of AKT, has been strongly associated with cell survival, it has been suggested that YKL‐40 protects the cells from undergoing apoptosis. (27) Taken together with the functional observation of YKL‐40 involvement in fibroblast proliferation, (27) the possible role of YKL‐40 in angiogenesis, 4 , 5 , 30 and the clinical studies showing increased serum concentrations of YKL‐40 in patients with cancer diseases and poor prognosis, there is circumstantial evidence for a physiological function of YKL‐40 in inflammation, tissue destruction and subsequent remodeling.
The present documentation of a regulated increase in YKL‐40 expression in the human glioblastoma cell line U87, 24–72 h after diverse stress induction, strongly supports a function for YKL‐40 in cellular survival or adaptation to key elements of the adverse microenvironment of cancers such as inflammation, hypoxia, and nutrient deprivation, as well as to the immediate effects of radiotherapy and cytostatic chemotherapy.
Acknowledgments
Pia Knudsen and Tonni Løve Hansen are thanked for excellent technical assistance. Quidel Corporation, San Diego, CA, USA provided YKL‐40 ELISA kits. The study was supported by grants from The Danish Cancer Society, The Danish Medical Research Council, and ‘Sagfører Valdemar Beck og hustru, Marie Beck, f. Thiedemanns mindelegat’.
References
- 1. Johansen JS, Williamson MK, Rice JS, Price PA. Identification of proteins secreted by human osteoblastic cells in culture. J Bone Miner Res 1992; 7: 501–12. [DOI] [PubMed] [Google Scholar]
- 2. Hakala BE, White C, Recklies AD. Human cartilage gp‐39, a major secretory product of articular chondrocytes and synovial cells, is a mammalian member of a chitinase protein family. J Biol Chem 1993; 268: 25803–10. [PubMed] [Google Scholar]
- 3. Millis AJ, Hoyle M, Reich E, Mann DM. Isolation and characterization of a Mr = 38,000 protein from differentiating smooth muscle cells. J Biol Chem 1985; 260: 3754–61. [PubMed] [Google Scholar]
- 4. Shackelton LM, Mann DM, Millis AJ. Identification of a 38‐kDa heparin‐binding glycoprotein (gp38k) in differentiating vascular smooth muscle cells as a member of a group of proteins associated with tissue remodeling. J Biol Chem 1995; 270: 13076–83. [DOI] [PubMed] [Google Scholar]
- 5. Malinda KM, Ponce L, Kleinman HK, Shackelton LM, Millis AJ. Gp38k, a protein synthesized by vascular smooth muscle cells, stimulates directional migration of human umbilical vein endothelial cells. Exp Cell Res 1999; 250: 168–73. [DOI] [PubMed] [Google Scholar]
- 6. Rehli M, Krause SW, Andreesen R. Molecular characterization of the gene for human cartilage gp‐39 (CHI3L1), a member of the chitinase protein family and marker for late stages of macrophage differentiation. Genomics 1997; 43: 221–5. [DOI] [PubMed] [Google Scholar]
- 7. Rehli M, Niller HH, Ammon C, Langmann S, Schwarzfischer L, Andreesen R, Krause SW. Transcriptional regulation of CHI3L1, a marker gene for late stages of macrophage differentiation. J Biol Chem 2003; 278: 44058–67. [DOI] [PubMed] [Google Scholar]
- 8. Morrison BW, Leder P. Neu and ras initiate murine mammary tumors that share genetic markers generally absent in c‐myc and int‐2‐initiated tumors. Oncogene 1994; 9: 3417–26. [PubMed] [Google Scholar]
- 9. Harvey S, Weisman M, O'Dell J, Scott T, Krusemeier M, Visor J, Swindlehurst C. Chondrex: new marker of joint disease. Clin Chem 1998; 44: 509–16. [PubMed] [Google Scholar]
- 10. Fusetti F, Pijning T, Kalk KH, Bos E, Dijkstra BW. Crystal structure and carbohydrate‐binding properties of the human cartilage glycoprotein‐39. J Biol Chem 2003; 278: 37753–60. [DOI] [PubMed] [Google Scholar]
- 11. Houston DR, Recklies AD, Krupa JC, Van Aalten DM. Structure and ligand‐induced conformational change of the 39‐kDa glycoprotein from human articular chondrocytes. J Biol Chem 2003; 278: 30206–12. [DOI] [PubMed] [Google Scholar]
- 12. Mohanty AK, Singh G, Paramasivam M, Saravanan K, Jabeen T, Sharma S, Yadav S, Kaur P, Kumar P, Srinivasan A, Singh TP. Crystal structure of a novel regulatory 40‐kDa mammary gland protein (MGP‐40) secreted during involution. J Biol Chem 2003; 278: 14451–60. [DOI] [PubMed] [Google Scholar]
- 13. Renkema GH, Boot RG, Au FL, Donker‐Koopman WE, Strijland A, Muijsers AO, Hrebicek M, Aerts JM. Chitotriosidase, a chitinase, and the 39‐kDa human cartilage glycoprotein, a chitin‐binding lectin, are homologues of family 18 glycosyl hydrolases secreted by human macrophages. Eur J Biochem 1998; 251: 504–9. [DOI] [PubMed] [Google Scholar]
- 14. Vind I, Johansen JS, Price PA, Munkholm P. Serum YKL‐40, a potential new marker of disease activity in patients with inflammatory bowel disease. Scand J Gastroenterol 2003; 38: 599–605. [DOI] [PubMed] [Google Scholar]
- 15. Punzi L, Podswiadek M, D’Inca R, Zaninotto M, Bernardi D, Plebani M, Sturniolo GC. Serum human cartilage glycoprotein 39 as a marker of arthritis associated with inflammatory bowel disease. Ann Rheum Dis 2003; 62: 1224–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johansen JS, Christoffersen P, Moller S, Price PA, Henriksen JH, Garbarsch C, Bendtsen F. Serum YKL‐40 is increased in patients with hepatic fibrosis. J Hepatol 2000; 32: 911–20. [DOI] [PubMed] [Google Scholar]
- 17. Kirkpatrick RB, Emery JG, Connor JR, Dodds R, Lysko PG, Rosenberg M. Induction and expression of human cartilage glycoprotein 39 in rheumatoid inflammatory and peripheral blood monocyte‐derived macrophages. Exp Cell Res 1997; 237: 46–54. [DOI] [PubMed] [Google Scholar]
- 18. Volck B, Johansen JS, Stoltenberg M, Garbarsch C, Price PA, Ostergaard M, Ostergaard K, Lovgreen‐Nielsen P, Sonne‐Holm S, Lorenzen I. Studies on YKL‐40 in knee joints of patients with rheumatoid arthritis and osteoarthritis. Involvement of YKL‐40 in the joint pathology. Osteoarthritis Cartilage 2001; 9: 203–14. [DOI] [PubMed] [Google Scholar]
- 19. Baeten D, Boots AM, Steenbakkers PG, Elewaut D, Bos E, Verheijden GF, Berheijden G, Miltenburg AM, Rijnders AW, Veys EM, De Keyser F. Human cartilage gp‐39+,CD16+ monocytes in peripheral blood and synovium: correlation with joint destruction in rheumatoid arthritis. Arthritis Rheum 2000; 43: 1233–43. [DOI] [PubMed] [Google Scholar]
- 20. Boot RG, Van Achterberg TA, Van Aken BE, Renkema GH, Jacobs MJ, Aerts JM, De Vries CJ. Strong induction of members of the chitinase family of proteins in atherosclerosis: chitotriosidase and human cartilage gp‐39 expressed in lesion macrophages. Arterioscler Thromb Vasc Biol 1999; 19: 687–94. [DOI] [PubMed] [Google Scholar]
- 21. Johansen JS, Baslund B, Garbarsch C, Hansen M, Stoltenberg M, Lorenzen I, Price PA. YKL‐40 in giant cells and macrophages from patients with giant cell arteritis. Arthritis Rheum 1999; 42: 2624–30. [DOI] [PubMed] [Google Scholar]
- 22. Johansen JS, Olee T, Price PA, Hashimoto S, Ochs RL, Lotz M. Regulation of YKL‐40 production by human articular chondrocytes. Arthritis Rheum 2001; 44: 826–37. [DOI] [PubMed] [Google Scholar]
- 23. Nordenbaek C, Johansen JS, Junker P, Borregaard N, Sorensen O, Price PA. YKL‐40, a matrix protein of specific granules in neutrophils, is elevated in serum of patients with community‐acquired pneumonia requiring hospitalization. J Infect Dis 1999; 180: 1722–6. [DOI] [PubMed] [Google Scholar]
- 24. Johansen JS, Jensen HS, Price PA. A new biochemical marker for joint injury. Analysis of YKL‐40 in serum and synovial fluid. Br J Rheumatol 1993; 32: 949–55. [DOI] [PubMed] [Google Scholar]
- 25. Nyirkos P, Golds EE. Human synovial cells secrete a 39 kDa protein similar to a bovine mammary protein expressed during the non‐lactating period. Biochem J 1990; 269: 265–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Volck B, Price PA, Johansen JS, Sorensen O, Benfield TL, Nielsen HJ, Calafat J, Borregaard N. YKL‐40, a mammalian member of the chitinase family, is a matrix protein of specific granules in human neutrophils. Proc Assoc Am Physicians 1998; 110: 351–60. [PubMed] [Google Scholar]
- 27. Recklies AD, White C, Ling H. The chitinase 3‐like protein human cartilage glycoprotein 39 (HC‐gp39) stimulates proliferation of human connective‐tissue cells and activates both extracellular signal‐regulated kinase‐ and protein kinase B‐mediated signalling pathways. Biochem J 2002; 365: 119–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Ceuninck F, Gaufillier S, Bonnaud A, Sabatini M, Lesur C, Pastoureau P. YKL‐40 (cartilage gp‐39) induces proliferative events in cultured chondrocytes and synoviocytes and increases glycosaminoglycan synthesis in chondrocytes. Biochem Biophys Res Commun 2001; 285: 926–31. [DOI] [PubMed] [Google Scholar]
- 29. Ling H, Recklies AD. The chitinase 3‐like protein HC‐gp39 inhibits cellular responses to the inflammatory cytokines interleukin 1 and tumour necrosis factor‐alpha. Biochem J 2004; 380: 651–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nishikawa KC, Millis AJ. Gp38k (CHI3L1) is a novel adhesion and migration factor for vascular cells. Exp Cell Res 2003; 287: 79–87. [DOI] [PubMed] [Google Scholar]
- 31. Lal A, Lash AE, Altschul SF, Velculescu V, Zhang L, McLendon RE, Marra MA, Prange C, Morin PJ, Polyak K, Papadopoulos N, Vogelstein B, Kinzler KW, Strausberg RL, Riggins GJ. A public database for gene expression in human cancers. Cancer Res 1999; 59: 5403–7. [PubMed] [Google Scholar]
- 32. Markert JM, Fuller CM, Gillespie GY, Bubien JK, McLean LA, Hong RL, Lee K, Gullans SR, Mapstone TB, Benos DJ. Differential gene expression profiling in human brain tumors. Physiol Genomics 2001; 5: 21–33. [DOI] [PubMed] [Google Scholar]
- 33. Tanwar MK, Gilbert MR, Holland EC. Gene expression microarray analysis reveals YKL‐40 to be a potential serum marker for malignant character in human glioma. Cancer Res 2002; 62: 4364–8. [PubMed] [Google Scholar]
- 34. Huang Y, Prasad M, Lemon WJ, Hampel H, Wright FA, Kornacker K, LiVolsi V, Frankel W, Kloos RT, Eng C, Pellegata NS, De La Chapelle A. Gene expression in papillary thyroid carcinoma reveals highly consistent profiles. Proc Natl Acad Sci USA 2001; 98: 15044–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sjogren H, Meis‐Kindblom JM, Orndal C, Bergh P, Ptaszynski K, Aman P, Kindblom LG. Studies on the molecular pathogenesis of extraskeletal myxoid chondrosarcoma‐cytogenetic, molecular genetic, and cDNA microarray analyses. Am J Pathol 2003; 162: 781–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Johansen JS, Christensen IJ, Riisbro R, Greenall M, Han C, Price PA, Smith K, Brunner N, Harris AL. High serum YKL‐40 levels in patients with primary breast cancer is related to short recurrence free survival. Breast Cancer Res Treat 2003; 80: 15–21. [DOI] [PubMed] [Google Scholar]
- 37. Johansen JS, Cintin C, Jorgensen M, Kamby C, Price PA. Serum YKL‐40: a new potential marker of prognosis and location of metastases of patients with recurrent breast cancer. Eur J Cancer 1995; 31A: 1437–42. [DOI] [PubMed] [Google Scholar]
- 38. Jensen BV, Johansen JS, Price PA. High levels of serum HER‐2/neu and YKL‐40 independently reflect aggressiveness of metastatic breast cancer. Clin Cancer Res 2003; 9: 4423–34. [PubMed] [Google Scholar]
- 39. Cintin C, Johansen JS, Christensen IJ, Price PA, Sorensen S, Nielsen HJ. Serum YKL‐40 and colorectal cancer. Br J Cancer 1999; 79: 1494–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cintin C, Johansen JS, Christensen IJ, Price PA, Sorensen S, Nielsen HJ. High serum YKL‐40 level after surgery for colorectal carcinoma is related to short survival. Cancer 2002; 95: 267–74. [DOI] [PubMed] [Google Scholar]
- 41. Dehn H, Hogdall EV, Johansen JS, Jorgensen M, Price PA, Engelholm SA, Hogdall CK. Plasma YKL‐40, as a prognostic tumor marker in recurrent ovarian cancer. Acta Obstet Gynecol Scand 2003; 82: 287–93. [DOI] [PubMed] [Google Scholar]
- 42. Hogdall EV, Johansen JS, Kjaer SK, Price PA, Christensen L, Blaakaer J, Bock JE, Glud E, Hogdall CK. High plasma YKL‐40 level in patients with ovarian cancer stage III is related to shorter survival. Oncol Report 2003; 10: 1535–8. [PubMed] [Google Scholar]
- 43. Dupont J, Tanwar MK, Thaler HT, Fleisher M, Kauff N, Hensley ML, Sabbatini P, Anderson S, Aghajanian C, Holland EC, Spriggs DR. Early detection and prognosis of ovarian cancer using serum YKL‐40. J Clin Oncol 2004; 22: 3330–9. [DOI] [PubMed] [Google Scholar]
- 44. Johansen JS, Drivsholm L, Price PA, Christensen IJ. High serum YKL‐40 level in patients with small cell lung cancer is related to early death. Lung Cancer 2004; 46: 333–40. [DOI] [PubMed] [Google Scholar]
- 45. Komarov PG, Komarova EA, Kondratov RV, Christov‐Tselkov K, Coon JS, Chernov MV, Gudkov AV. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999; 285: 1733–7. [DOI] [PubMed] [Google Scholar]
- 46. Semenza GL. Targeting HIF‐1 for cancer therapy. Nat Rev Cancer 2003; 3: 721–32. [DOI] [PubMed] [Google Scholar]
- 47. Ma J, Fei ZL, Klein‐Szanto A, Gallo JM. Modulation of angiogenesis by human glioma xenograft models that differentially express vascular endothelial growth factor. Clin Exp Metastasis 1998; 16: 559–68. [DOI] [PubMed] [Google Scholar]
- 48. Ambrosini G, Nath AK, Sierra‐Honigmann MR, Flores‐Riveros J. Transcriptional activation of the human leptin gene in response to hypoxia. Involvement of hypoxia‐inducible factor 1. J Biol Chem 2002; 277: 34601–9. [DOI] [PubMed] [Google Scholar]
- 49. Wang GL, Semenza GL. Purification and characterization of hypoxia‐inducible factor 1. J Biol Chem 1995; 270: 1230–7. [DOI] [PubMed] [Google Scholar]
- 50. Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C. elegans EGL‐9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001; 107: 43–54. [DOI] [PubMed] [Google Scholar]
- 51. Vit JP, Rosselli F. Role of the ceramide‐signaling pathways in ionizing radiation‐induced apoptosis. Oncogene 2003; 22: 8645–52. [DOI] [PubMed] [Google Scholar]
- 52. Kolesnick R, Fuks Z. Radiation and ceramide‐induced apoptosis. Oncogene 2003; 22: 5897–906. [DOI] [PubMed] [Google Scholar]
- 53. Yoshimura S, Banno Y, Nakashima S, Takenaka K, Sakai H, Nishimura Y, Sakai N, Shimizu S, Eguchi Y, Tsujimoto Y, Nozawa Y. Ceramide formation leads to caspase‐3 activation during hypoxic PC12 cell death. Inhibitory effects of Bcl‐2 on ceramide formation and caspase‐3 activation. J Biol Chem 1998; 273: 6921–7. [DOI] [PubMed] [Google Scholar]
- 54. Yoshimura S, Banno Y, Nakashima S, Hayashi K, Yamakawa H, Sawada M, Sakai N, Nozawa Y. Inhibition of neutral sphingomyelinase activation and ceramide formation by glutathione in hypoxic PC12 cell death. J Neurochem 1999; 73: 675–83. [DOI] [PubMed] [Google Scholar]
- 55. Sawada M, Nakashima S, Kiyono T, Nakagawa M, Yamada J, Yamakawa H, Banno Y, Shinoda J, Nishimura Y, Nozawa Y, Sakai N. p53 regulates ceramide formation by neutral sphingomyelinase through reactive oxygen species in human glioma cells. Oncogene 2001; 20: 1368–78. [DOI] [PubMed] [Google Scholar]
- 56. Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens AC, Van Meir EG. Frequent co‐alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol 1999; 9: 469–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Renton A, Llanos S, Lu X. Hypoxia induces p53 through a pathway distinct from most DNA‐damaging and stress‐inducing agents. Carcinogenesis 2003; 24: 1177–82. [DOI] [PubMed] [Google Scholar]
- 58. Koumenis C, Alarcon R, Hammond E, Sutphin P, Hoffman W, Murphy M, Derr J, Taya Y, Lowe SW, Kastan M, Giaccia A. Regulation of p53 by hypoxia: dissociation of transcriptional repression and apoptosis from p53‐dependent transactivation. Mol Cell Biol 2001; 21: 1297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fei P, El Deiry WS. P53 and radiation responses. Oncogene 2003; 22: 5774–83. [DOI] [PubMed] [Google Scholar]
- 60. Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 1997; 420: 25–7. [DOI] [PubMed] [Google Scholar]
- 61. Schmidt‐Ullrich RK, Dent P, Grant S, Mikkelsen RB, Valerie K. Signal transduction and cellular radiation responses. Radiat Res 2000; 153: 245–57. [DOI] [PubMed] [Google Scholar]
- 62. Kim JY, Park JH. ROS‐dependent caspase‐9 activation in hypoxic cell death. FEBS Lett 2003; 549: 94–8. [DOI] [PubMed] [Google Scholar]
- 63. Okamoto Y, Obeid LM, Hannun YA. Bcl‐xL interrupts oxidative activation of neutral sphingomyelinase. FEBS Lett 2002; 530: 104–8. [DOI] [PubMed] [Google Scholar]
- 64. Martin D, Salinas M, Fujita N, Tsuruo T, Cuadrado A. Ceramide and reactive oxygen species generated by H2O2 induce caspase‐3‐independent degradation of Akt/protein kinase B. J Biol Chem 2002; 277: 42943–52. [DOI] [PubMed] [Google Scholar]
- 65. Koch AE. The role of angiogenesis in rheumatoid arthritis: recent developments. Ann Rheum Dis 2000; 59 (Suppl. 1): i65–i71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mapp PI, Grootveld MC, Blake DR. Hypoxia, oxidative stress and rheumatoid arthritis. Br Med 1995; 51: 419–36. [DOI] [PubMed] [Google Scholar]
- 67. Watters D. Molecular mechanisms of ionizing radiation‐induced apoptosis. Immunol Cell Biol 1999; 77: 263–71. [DOI] [PubMed] [Google Scholar]
- 68. Koo HN, Jeong HJ, Hong SH, Choi JH, An NH, Kim HM. High molecular weight water‐soluble chitosan protects against apoptosis induced by serum starvation in human astrocytes. J Nutr Biochem 2002; 13: 245–9. [DOI] [PubMed] [Google Scholar]