

Abstract
Methylation is an important silencing mechanism of breast and ovarian cancer susceptibility gene 1 (BRCA1) expression in sporadic ovarian cancer. However, the role of BRCA1 methylation in chemotherapy in sporadic ovarian cancer and the related pathways have not been understood completely. This study has determined the roles of BRCA1 hypermethylation in chemotherapy of sporadic ovarian cancer and its related signaling pathways. We used bisulfite sequencing, real‐time polymerase chain reaction, and western blotting to check the methylation state and expression levels of BRCA1 of the following cell lines: platinum‐sensitive human ovarian cancer cell line COC1, platinum‐resistant cell line COC1/DDP, SKOV‐3, and 5‐Aza‐dC treated COC1. The cisplatin sensitivity of ovarian cancer cells was examined by MTS (methyl‐thiazol tetrazolium) assay. Tumorigenicity in vivo and DDP‐based chemosensitivity were compared among the above cells. Phosphatidylinositol 3′‐kinase (PI3K)–Akt pathway activation in ovarian cancer cells was studied by western blotting. The frequency of BRCA1 methylation in the COC1 cell line was higher than in COC1/DDP and SKOV‐3 cell lines, whereas the mRNA and protein expression of BRCA1 were lower than in the COC1/DDP and SKOV‐3 cell lines. DNA demethylation decreased the chemosensitivity of COC1 cells and partially increased the expression levels of BRCA1. The activation of the PI3K‐Akt pathway was low in ovarian cancer cells. Our results indicate that hypermethylation of BRCA1 might play an important role in the chemosensitivity of ovarian cancer, and that the PI3K–Akt pathway is not involved in this response. (Cancer Sci 2010)
Epithelial ovarian cancer (EOC) is one of the main causes of death among gynecological malignancies,( 1 ) and cisplatin is one of the most effective and commonly used chemotherapeutics for the treatment of ovarian cancer. However, the development of cisplatin‐based resistance is a key concern that limits the successful clinical application of this therapy.( 2 ) Although the precise mechanism underlying the development of platinum resistance in ovarian cancer patients is currently unknown, CPG‐island (CGI) methylation, which is strongly associated with aberrant gene silencing and ovarian tumorigenesis, is more prominent after the onset of chemoresistance,( 3 , 4 , 5 , 6 , 7 ) and may be used for the prediction of response to chemotherapy in breast cancer.( 8 , 9 )
Breast and ovarian cancer susceptibility gene 1 (BRCA1), a multi‐functional tumor suppressor protein, was identified on the basis of its linkage to familial breast and ovarian cancer.( 10 ) All the functions of BRCA1 have not yet been determined, but it is known to play roles in DNA damage repair, cell‐cycle checkpoint control, transcriptional regulation, and maintenance of genomic stability. Furthermore, BRCA1 promoter hypermethylation is an important positive regulator of BRCA1 expression. Aberrant DNA methylation may contribute to the disruption of key biological pathways during the progression of ovarian cancer to the drug‐resistant phenotype.
The signaling pathways of Fanconi‐anemia (FANC‐BRCA), p38/JNK, and ataxia telangiectasia mutated (ATM)/ATM‐ and Rad3‐related (ATR) were involved in BRCA1‐related DNA damage response.( 11 , 12 , 13 ) However, there is little knowledge of the pathway related to BRCA1 hypermethylation in chemotherapy of EOC. The phosphatidylinositol 3′‐kinase (PI3K)–Akt signaling pathway plays a role in the development of EOC and sensitivity toward cisplatin‐based chemotherapy.( 14 , 15 , 16 ) Increased AKT kinase activity has been reported in about 40% of breast and ovarian cancers;( 17 ) moreover, BRCA1 is a negative regulator of phosphorylated AKT (pAKT), and BRCA1 deficiency activates the AKT pathway.( 18 ) Hence, further research on the activation of the PI3K–Akt pathway in ovarian cancer cells is required to deepen the understanding of this epigenetic change and its role in chemosensitivity. The present study was designed to detect whether the methylation status of BRCA1 is associated with the cisplatin‐based chemosensitive response and whether it involves the PI3K–Akt pathway.
Materials and Methods
Cell culture. The SKOV‐3 human ovarian cancer cell line was obtained from American Type Culture Collection (Manassas, VA, USA). The human breast cancer cell line MCF‐7 was obtained from the Institute of Cell Biology (Shanghai, China). Human ovarian cancer cell lines COC1 and COC1/DDP were obtained from the China Center for Type Culture Collection (Wuhan, China). Cells were cultured in RPMI‐1640 medium supplemented with 100 units/mL penicillin/streptomycin and 10% fetal bovine serum (FBS) and maintained in an incubator at 37°C under a humidified atmosphere of 5% CO2. COC1/DDP cells were cultured in RPMI‐1640 medium containing 0.5 μg/mL cisplatin (Sigma, St. Louis, MO, USA) to maintain the drug‐resistant phenotype. All cells were obtained from the cell bank that performed cell line characterizations, and were passaged in our laboratory for <6 months after receipt.
DNA extraction and bisulfite modification. Genomic DNA was extracted from cultured cells by using QIAamp DNA kits (Qiagen, Germany). Sodium bisulfite reactions were performed with the EpiTect Bisulfite kit (Qiagen, Hilden, Germany). The methylation status of a DNA sequence can be determined using sodium bisulfite. Incubation of target DNA with sodium bisulfite converts the unmethylated cytosine residues into uracil and it does not react with the methylated cytosines. Therefore, bisulfite treatment yields different DNA sequences for methylated and unmethylated DNA.
Analysis of BRCA1 promoter methylation and bisulfite genomic sequencing. The previously known primers for BRCA1 can be used for analyzing the most significantly methylated loci. Real‐time polymerase chain reaction (PCR) with bisulfite‐modified DNA was performed using previously known primer sequences for the methylated (M) and unmethylated (U) reactions. Primer sequences for the M and U DNAs were as follows: M forward, TCGTGGTAACGGAAAAGCGC; M reverse, AA AT CTCA AC GAACTCACGCCG; U forward, TTG GTT TTT GTG GTA ATG GAAAAGTGT; and U reverse, CAAAAAATCTCAACAAACTC‐ACACCA. The sense primer of the methylated reaction begins at 1543 base pairs (bp) and that of the unmethylated reaction begins at 1536 bp of the GenBank sequence U37574. The methylated product is 75 bp long and the unmethylated product is 86 bp. This region intercepts the main transcription start site at 1581 bp.( 19 )
The total volume of the PCR mixture was 20 μL (Takara, Dalian, China). Each PCR cycle included initial denaturation at 95°C for 10 s and 45 cycles with the following profile: 5 s at 95°C and 30 s at 62°C. The PCR products were then subjected to bisulfite sequencing. The gene expression level was normalized using endogenous control gene GAPDH, and the relative gene‐expression level was determined using 2(–delta delta C (T)) (ΔΔCT) methods. DNA from human placenta tissue was treated in vitro with SssI bacterial methylase (New England Biolabs, Beverly, MA, USA) and used as positive control for methylated alleles. DNA from MCF‐7 cells was used as a negative control for methylated genes.
The resultant 75‐bp PCR product including 8 CpG dinucleotides was gel purified and cloned into TOPO TA Cloning vector (Invitrogen, Carlsbad, CA, USA). Ten recombinant clones were isolated using a Pmd18‐T (Takara) and sequenced using ABI automated DNA sequencer. The methylation status of individual CpG sites was determined by comparison with the sequence from known BRCA1 sequences. The number of methylated CpGs at each specific site was divided by the number of clones analyzed (n = 10) to obtain a value that represented the percentage of methylation at each site, as previously described.( 20 )
RNA isolation and real‐time PCR. Total RNA was prepared using TRIzol (Molecular Research Center, Cincinnati, OH, USA). We used 2 μg aliquots of total RNA for amplification by using the PrimeScript RT reagent (Takara). The primers used for cDNA amplification were as follows: BRCA1 forward, ACAGCTGTCTGGTGCTTCT GTG and reverse, CATTGTC‐CTCTGTCCAGGCATC and GAPDH forward, GA GT CA AC GG AT TT GGTCGT and reverse, TTGATTT TGGAGGGATCTCG. The expected fragment lengths of BRCA1 and GAPDH were 106 and 238 bp, respectively. The mRNA expression of BRCA1 was analyzed using TotalLab software by estimating the grayscale ratio of BRCA1 expression level to GAPDH expression level. Real‐time PCR was also used to evaluate BRCA1 expression using SYBR Premix ExTaq (Takara). All reactions were performed according to the manufacturer’s protocols. The annealing temperature for these primer sets was 60°C.
Western blotting analysis. The cells were first rinsed with PBS, and then scraped and lysed in ice‐cold radioimmunoprecipitation assay (RIPA) buffer on ice for 30 min. The total protein content was measured using Bio‐Rad protein assay reagent according to the manufacturer’s protocol. Equal amounts of proteins were separated on 6–15% sodium dodecylsulfate–polyacrylamide gel electrophoresis gels and subsequently transferred to nitrocellulose membranes. After estimating enzyme activities by using a microplate reader, the membranes were treated with 1% bovine serum albumin in 1 × Tris‐buffered saline for 1 h at room temperature and incubated with various primary antibodies (anti‐BRCA1, 1:50; anti‐AKT, 1:1000; anti‐P‐AKT, 1:1000; Cell Signaling Technology, Danvers, MA, USA), and GAPDH (Invitrogen) at 4°C overnight. The membranes were washed three times with Tris‐buffered saline (TBS) and then incubated with peroxidase‐linked secondary antibody (1:10000) for 1 h at room temperature. The signals were developed using an enhanced chemiluminescence kit (Pierce, Rockford, IL, USA), scanned, and analyzed with Image‐Pro Plus 6.0. The relative expression of target proteins was presented as the ratio of the amount of protein to that of β‐actin.
Demethylation of COC1 cells with 5‐Aza‐Dc. COC1 cells were seeded at a density of 5 × 105 cells on a 100‐mm plate. After 24 h, the cells were treated with 10 μM of 5‐Aza‐dC (Sigma). The medium was changed after 72 h of treatment and the cells were cultured for another 48 h before harvesting for analysis of their methylation status, cell apoptosis, mRNA, and protein levels. The 5‐Aza‐dC was dissolved in PBS.
Methyl‐thiazol tetrazolium (MTS) assay. The methyl‐thiazol tetrazolium (MTS) assay (Promega, Madison, WI, USA) was used to measure cell viability. Briefly, 1 × 104 cells/well were seeded in 96‐well plates containing complete medium. After incubation for 48 h, the cells were rinsed twice and incubated with serum‐free medium supplemented with the platinum. After incubation for the indicated times, 20 μL of MTS solution was added and the cells were incubated for a further 1–4 h. The optical density was measured at 490 nm on a microplate reader. The half maximal inhibitory concentration (IC50) value was assessed by increasing the concentration of cisplatin.
Cell apoptosis. COC1 cells were treated with demethylating agents for the indicated times; subsequently, the cells were harvested, rinsed, fixed in 70% ethanol, and stained with 50 mg/mL propidium iodide. The fluorescence intensity was determined using FACScan. The percentage of apoptotic cells was calculated by determining the number of sub‐G1 cells.
In vivo studies. Twenty female Balb/c nu/nu mice (4–6 weeks of age) were obtained from the Chinese Academy of Sciences. The mice were randomly assigned into one of the following four groups (n = 5): (i) mice treated with COC1; (ii) mice treated with COC1/DDP; (iii) mice treated with COC1+5‐Aza‐Dc; and (iv) mice treated with 0.1 mL of PBS. The tumor model was established in nude mice by dorsal subcutaneous injection with COC1, COC1/DDP, and COC1+5‐Aza‐Dc cells, respectively. Tumors were harvested for dorsal xenotransplant when the diameter of tumors was up to 1 cm. After 2 weeks, intraperitoneal injection of cisplatin was administered at a dose of 5 mg/kg and twice a week. The tumors and body weight were measured twice a week. Animals were killed after 2 weeks. The tumor sizes were measured using the formula A × B 2 × 0.5236 (A, length; B, width; all measured in millimeters).( 20 )
Statistics. The results are presented as mean ± SE. Data were analyzed using one‐way anova and the unpaired Student’s t‐test was applied for the comparison of data among the groups. A P‐value of <0.05 was considered as statistically significant. All in vitro experiments were repeated independently at least three times, while the in vivo experiments were repeated twice.
Results
Hypermethylation of the CpG island of BRCA1 was detected in chemosensitive human ovarian cancer COC1 cells which resulted in loss of BRCA1 expression, thereby increasing the sensitivity of cells to chemotherapy in vitro and in vivo. Methylation of the CpG island of BRCA1 in four epithelial ovarian cancer cell lines (sensitive human ovarian cancer cell lines COC1, COC1 treated with 5‐Aza‐dC, acquired platinum‐resistant cell lines COC1/DDP, and inherent resistant cell lines SKOV‐3) was analyzed by real‐time PCR (Fig. 1a) and bisulfite sequencing (Fig. 1b). Real‐time PCR and western blotting analysis were performed to investigate whether the low BRCA1 mRNA and protein expression was coordinated with promoter hypermethylation (Fig. 2).
Figure 1.
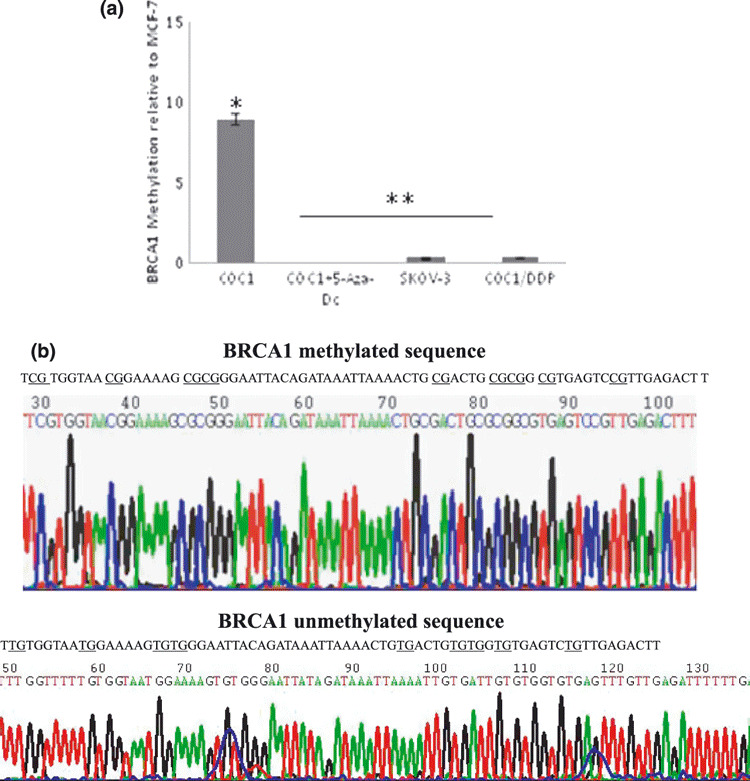
Methylation analysis of breast and ovarian cancer susceptibility gene 1 (BRCA1) in human ovarian cell lines. (a) Methylation analysis of BRCA1 in human ovarian cell lines COC1, COC1+5‐Aza‐dC, SKOV‐3, and COC1/DDP. Data are expressed as the mean ± SD. *P < 0.05 versus COC1+5‐Aza‐Dc, SKOV‐3, and COC1/DDP cells; **P > 0.05 among the COC1+5‐Aza‐Dc cells, SKOV‐3, and COC1/DDP cells. (b) Sequencing analysis of methylated and unmethylated BRCA1. After sodium bisulfite, cytosines in the methylated sequence remained unchanged, but cytosines conversed to uracils in the unmethylated sequence.
Figure 2.
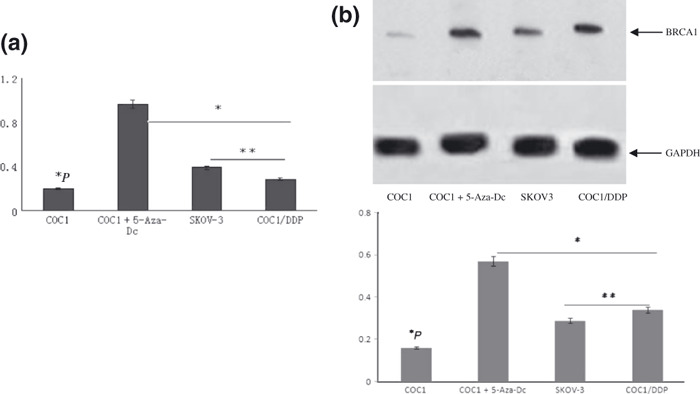
The expression levels of breast and ovarian cancer susceptibility gene 1 (BRCA1) mRNA and protein in ovarian cancer cells. (a) The expression levels of BRCA1 mRNA by real‐time PCR. GAPDH (glyceraldehyde‐ 3‐phosphate dehydrogenase) was used as the internal control. *P < 0.05 versus COC1+5‐Aza‐Dc, SKOV‐3, and COC1/DDP cells; **P > 0.05. (b) Expressions of BRCA1 protein by western blot analysis. *P < 0.05 versus COC1+5‐Aza‐Dc, SKOV‐3, and COC1/DDP cells; **P > 0.05.
The frequency of BRCA1 methylation in the chemosensitive cell line was higher than that in chemoresistant cells (Fig. 1a). However, the frequency did not differ significantly between the acquired and inherent resistant cell lines. Simultaneously, after treatment with 5‐Aza‐dC, the frequency of BRCA1 methylation was obviously decreased.
Bisulfite sequencing analyses showed that incubation with sodium bisulfite did not react with the methylated cytosines in the target BRCA1 DNA sequence (Fig. 1b, left), while it converted the unmethylated cytosine residues into uracil and the uracil turned into thymine after PCR amplification (Fig. 1b, right).
The BRCA1 mRNA expression ratio to GAPDH was lower in COC1 cells than that in COC1/DDP and SKOV‐3 cells (BRCA1, P < 0.001) (Fig. 2a). Aberrant protein expression, which was in accordance with the results of RT‐PCR, was significantly lower than promoter hypermethylation (BRCA1, P = 0.008) of the gene (Fig. 2b).
Fluorescence‐activated cell sorting (FACS) and MTS methods were used to compare cisplatin sensitivity in COC1 and COC1/DDP cells (Fig. 3). As seen in Figure 3(a), COC1 cells showed a significant increase in the apoptosis rate compared to cells treated with demethylation agent. Furthermore, the approximate IC50 value of ovarian cell lines COC1 was 4.5148 μM, while the values for resistant cell lines COC1/DDP and SKOV‐3 were 11‐fold and 13‐fold higher, respectively. However, no significant difference was found in the IC50 value of COC1/DDP and SKOV‐3 cell lines (Fig. 3b).
Figure 3.
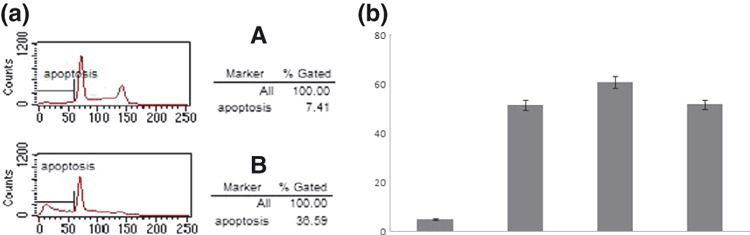
Apopotosis of cells COC1 and COC1 treated with 5‐ Aza‐Dc agent. (a) Apoptosis in COC1 cells and COC1 cells treated with 5‐ Aza‐Dc agent was detected by fluorescence‐activated cell sorting. (b) Different IC50 values of ovarian cancer cell reaction to platinum by MTS (methyl‐thiazol tetrazolium) assay.
In vivo studies on nude mice revealed that the COC1 cell‐treated group developed larger tumors than COC1/DDP‐treated groups (P < 0.01) (Table 1). Meanwhile, the tumors in the former group were more sensitive to cisplatin chemotherapy than those in the other two groups, and complete tumor regression was observed in three of the five mice administered with cisplatin. However, the total number of tumors in the three groups did not differ significantly. In our study, the metastases were limited to the pelvic region, which may be related to the subcutaneous xenotransplantation tumor models established instead of intraperitoneal inoculation (Fig. 4).
Table 1.
Tumorigenicity and its reaction to platinum‐based chemotherapy in female Balb/c nu/nu mice
| Clone | Number of nu/nu mice | Tumori‐genicity | Tumor volume | |
|---|---|---|---|---|
| Before chemotherapy | After chemotherapy | |||
| COC1 | 5 | 12 | 0.36 ± 0.0143* | 0.1212 ± 0.0931** |
| COC1+5‐Aza‐Dc | 5 | 8 | 0.184 ± 0.03863 | 0.1306 ± 0.00684 |
| COC1/DDP | 5 | 9 | 0.179 ± 0.00469 | 0.1574 ± 0.0238 |
*P < 0.05 versus mice treated with COC1 + 5‐Aza‐Dc and COC1/DDP before chemotherapy. **P < 0.05 versus mice treated with COC1 + 5‐Aza‐Dc and COC1/DDP after chemotherapy.
Figure 4.
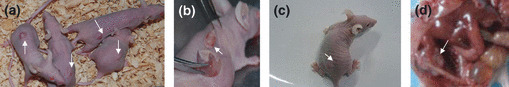
In vivo tumor‐bearing models. Subcutaneous xenotransplanted tumor model of ovarian carcinoma cells established in female Balb/c nu/nu mice (a–d). After dorsal subcutaneous injection with COC1, COC1+5‐Aza‐Dc, and COC1/DDP cells, respectively, tumorigenicity was found in each group (a). Tumors were alated for the xenotransplant tumor model when the diameter of tumors was up to 1 cm (b). The model was established successfully (c). Metastases in the pelvic region. Arrow shows the tumors (d).
5‐Aza‐dC‐induced DNA demethylation resulted in increase of BRCA1 and decreased cisplatin cytoxicity of cells in vitro and in vivo. To determine whether BRCA1 promoter methylation could be further linked to the loss of gene expression, we treated COC1 cells with the demethylating agent 5‐Aza‐dC. We found that the proliferation rate of COC1 cells was obviously decreased, while the mRNA and protein expression were increased (Fig. 2).
In addition, 5‐Aza‐dC decreased cisplatin cytoxicity. The effect of 5‐Aza‐dC on cisplatin‐based cytoxicity was analyzed by FACS and MTS assay. The IC50 value was significantly increased in COC1 cells treated with 5‐Aza‐dC (Fig. 3b). Fluorescence‐activated cell sorting (FACS) studies showed that the cisplatin‐induced apoptotic rate of 5‐Aza‐dC‐treated COC1 cells was significantly lower than that (36.02% ± 1.53%) of untreated COC1 cells (Fig. 3a).
To evaluate whether the re‐expression of BRCA1 by demethylation agent lowered tumorigenicity, we studied an in vivo tumor‐bearing model (Fig. 4). In our study, mice treated with COC1+5‐Aza‐dC developed smaller tumors than those treated with COC1 only; meanwhile, there was no significant decrease in tumor size after administration of cisplatin in the COC1+5‐Aza‐dC group, which was consistent with the observation of the COC1/DDP group (Table 1).
The PI3K pathway was not involved in methylation‐induced chemosensitivity of ovarian cancer cells. Phosphorylation of Akt at regulatory residues Thr‐308 and Ser‐473 activates it completely.( 19 ) Thus, to determine whether the PI3K pathway was responsible for BRCA1 methylation‐induced chemosensitivity in ovarian cancer cells, we performed western blotting to analyze the amount of total Akt and phosphorylated Akt at residues Ser473 and Thr308 in the four groups (COC1, COC1/DDP, SKOV‐3, and COC1+5‐Aza‐dC cells). Results showed that the total protein level of Akt was not significantly different among the above‐mentioned cell lines. Furthermore, a low level of Akt phosphorylation (both Thr‐308 and Ser‐473) was detected in EOC cells (P > 0.05) (Fig. 5).
Figure 5.
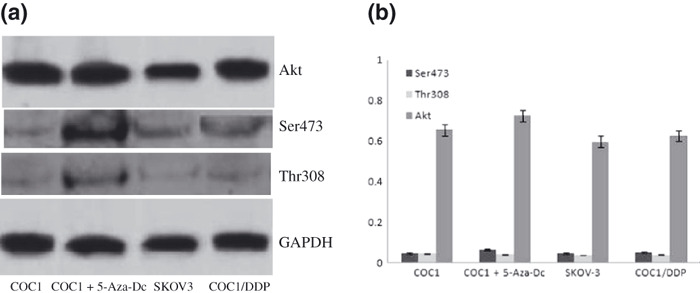
Total Akt and phosphorylation of Akt at residues Ser473 and Thr308 in ovarian cancer cell lines. Low levels of both p‐Ser473 and p‐Thr308 AKT expression were detected in each of the four groups. But total Akt protein was highly expressed in different ovarian cancer cells.
Discussion
Somatic mutation of the BRCA1 gene is rarely observed in sporadic ovarian cancer. Furthermore, hypermethylation of the BRCA1 promoter, which occurs in 15–70% of ovarian cancer patients,( 20 , 21 ) was not found in patients with germline mutations,( 22 ) and the BRCA1 promoter was not methylated in patients with high risk of germ‐line BRCA1 mutations. Frequently, tumor cells with methylated BRCA1 have lost the wild‐type BRCA allele( 23 , 24 ) and are unable to repair DNA cross‐links and DNA double‐strand breaks by homologous recombination, which probably explains the genomic instability and cancer predisposition of these cells.( 25 ) Consistent with this hypothesis, the results of in vivo studies showed that mice treated with COC1 cells (decreased BRCA1 expression) developed larger tumors than the others, and increasing the expression of BRCA1 by the demethylation agent developed smaller tumors. Survival of BRCA‐deficient cells is generally believed to be dependent upon dysfunction of the checkpoints, in which tumorigenic potential could be acquired through additional genomic rearrangements and gene mutations. But functions of BRCA1 methylation, such as DDP‐based chemosensitivity, in ovarian cancer remain to be clarified. Hence, we suggested that the methylation of the BRCA1 promoter is an important mechanism of gene silencing and a predictive marker of response to DNA damage caused by chemotherapy in sporadic EOC.( 26 )
Previous studies had reported that hypomethylation was detected in the majority of ovarian cancer cells;( 27 , 28 ) however, we found that the methylation frequency was higher in COC1 ovarian cancer cells than in SKOV‐3 and COC1/DDP cells, The COC1 cells were DDP‐sensitive, while SKOV‐3 and COC1/DDP cells were DDP‐resistant. This result may explain why hypomethylation was detected in SKOV‐3 cells, and suggested that DNA methylation may play an important role in chemosensitivity.
Epigenetic gene silencing mechanisms such as aberrant methylation may markedly reduce the BRCA1 mRNA level in cancer cells compared to its normal homologue. Consistent with this hypothesis, the results of our study showed that the BRCA1 mRNA and protein expression in methylated COC1 cells was lower than that in unmethylated SKOV‐3 and COC1/DDP cells. Furthermore, the demethylation state induced by 5‐Aza‐Dc increased cell proliferation and decreased the apoptosis. Taken together, these results suggest that hypermethylation of the BRCA1 promoter may be associated with the reduced levels of BRCA1 mRNA and protein.
Resistance to cisplatin‐based drugs occurs in the large majority of initially responsive tumors, resulting in complete chemoresistance and fatal disease. Cisplatin cytotoxity involves the formation of intra‐ and inter‐strand crosslinks that result in the stalling of DNA replication, S‐phase arrest, and replication fork collapse. The loss of BRCA1‐mediated DNA repair, cell‐cycle checkpoint regulation, and apoptosis and transcriptional control lead to the promotion of cell survival. Hence, the loss of BRCA1 expression could increase the sensitivity to drugs.
Transcriptional silencing of distinct DNA repair and apoptosis‐associated genes by hypermethylation of promoter CpG islands (CGIs)( 29 , 30 ) has now been associated with platinum drug resistance in numerous cancers, including ovarian carcinoma.( 31 , 32 ) On the basis of these previous observations, we performed MTS and apoptosis assays to analyze the cisplatin sensitivity and cisplatin resistance of the cell lines, and observed that the cisplatin‐sensitive cells exhibited higher frequency of methylation than both inherently and acquired platinum‐resistant cells. After treatment with a demethylating agent, the cells (COC1 cells) developed resistance to platinum. The results of in vivo studies were consistent with those of in vitro studies.
Many pathways such as Fanconi‐anemia (FANC‐BRCA), p38/JNK, ATM/ATR, and PI3K‐Akt had been reported previously to be involved in the BRCA1‐related DNA damage response.( 11 , 12 , 13 ) We have performed western blotting to analyze phosphorylated ATM (Ser1981) and ATR (Ser428), and expression of c‐myc and β‐catenin protein in EOC cells (COC1, COC1/DDP, SKOV‐3, and COC1+5‐Aza‐dC cells). But there was no expression of the above proteins in the above cells.
The microarray analyses on the isogenic drug‐sensitive and ‐resistant pairs for global CGI methylation in ovarian cancer cells showed that the aberrant methylation‐mediated activation pathways PI3K/Akt, TGF‐β, and cell cycle progression, may contribute to the onset of chemoresistance in ovarian cancer cells.( 33 ) Moreover, AKT phosphorylates BRCA1,( 18 ) and BRCA1 deficiency leads to an increased nuclear localization of pAKT. Taken together, we proposed that the PI3K–Akt pathway could be involved in the methylation of the BRCA1, thereby inducing chemosensitivity in EOC. However, in the present study we detected low levels of Akt activation in both the methylated and the demethylated ovarian cancer cell lines, and the difference of phosphorylated Akt level was not significant between the COC1 and its homologue treated with 5‐Aza‐Dc, suggesting that the activation of the PI3K/Akt pathway may be unrelated to the methylation of the BRCA1.
In summary, the present study proposes that BRCA1 methylation may represent a new target for preventing acquired drug resistance and improving ovarian cancer treatment. Further studies about the pathway related to BRCA1 hypermethylation are ongoing in our laboratory.
Supporting information
Fig. S1. Value of COC1 cell reaction to platinum with different concentrations of 5‐Aza‐Dc by MTS (methyl‐thiazol tetrazolium) assay.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
This work was supported by grants from the Key Project of Science and Technology Commission of Shanghai Municipality (07JC14031).
References
- 1. Ozols RF, Bookman MA, Connolly DC. Focus on epithelial ovarian cancer. Cancer Cell 2004; 5: 19–24. [DOI] [PubMed] [Google Scholar]
- 2. Horváth V, Blanárová O, Svihálková‐Sindlerová L et al. Platinum(IV) complex with adamantylamine overcomes intrinsic resistance to cisplatin in ovarian cancer cells. Gynecol Oncol 2006; 102: 32–40. [DOI] [PubMed] [Google Scholar]
- 3. Li M, Balch C, Montgomery JS et al. Integrated analysis of DNA methylation and gene expression reveals specific signaling pathways associated with platinum resistance in ovarian cancer. BMC Med Genomics 2009; 2: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer 2004; 4: 143–53. [DOI] [PubMed] [Google Scholar]
- 5. Fojo T, Bates S. Strategies for reversing drug resistance. Oncogene 2003; 22: 7512–23. [DOI] [PubMed] [Google Scholar]
- 6. Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128: 683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Konstantinopoulos PA, Fountzilas E, Pillay K et al. Carboplatin‐induced gene expression changes in vitro are prognostic of survival in epithelial ovarian cancer. BMC Med Genomics 2008; 1: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. James G, Herman MD, Stephen B, Baylin MD. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 2003; 349: 2042–54. [DOI] [PubMed] [Google Scholar]
- 9. Lee MN, Tseng RC, Hsu HS et al. Epigenetic inactivation of the chromosomal stability control genes BRCA1, BRCA2, and XRCC5 in non‐small cell lung cancer. Clin Cancer Res 2007; 13: 832–8. [DOI] [PubMed] [Google Scholar]
- 10. Billack B, Monteiro AN. BRCA1 in breast and ovarian cancer predisposition. Cancer Lett 2005; 227: 1–7. [DOI] [PubMed] [Google Scholar]
- 11. Taniguchi T, Tischkowitz M, Ameziane N et al. Disruption of the Fanconi anemia‐BRCA pathway in cisplatin‐sensitive ovarian tumors[J]. Nat Med, 2003; 9(5): 568–74. [DOI] [PubMed] [Google Scholar]
- 12. Fan W, Jin S, Tong T et al. BRCA1 regulates GADD45 through its interactions with the OCT‐1 and CAAT motifs[J]. J Biol Chem, 2002; 277(10): 8061–7. [DOI] [PubMed] [Google Scholar]
- 13. Zhang J, Feng Z, Kim S et al. Chk2 phosphorylation of BRCA1 regulates DNA double‐strand break repair[J]. Mol Cell Biol 2004; 24(2): 708–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kissel CK, Schadendorf D, Röckmann H. The altered apoptotic pathways in cisplatin and etoposide‐resistant melanoma cells are drug specific. Melanoma Res 2006; 16(6): 527–35. [DOI] [PubMed] [Google Scholar]
- 15. Zhang X‐Y, Zhang H‐Y, Zhang P‐N, Lu X, Hong S. Elevated phosphatidylinositol 3‐kinase activation and its clinicopathological significance in cervical cancer. Eur J Obstet Gynecol Reprod Biol 2008; 139: 237–44. [DOI] [PubMed] [Google Scholar]
- 16. Zhang HY, Zhang PN, Sun H. Aberration of the PI3K/AKT/mTOR signaling in epithelial ovarian cancer and its implication in cisplatin‐based chemotherapy. Eur J Obstet Gynecol Reprod Biol 2009; 146: 81–6. [DOI] [PubMed] [Google Scholar]
- 17. Jordan NJ, Gee JMW, Barrow D, Wakeling AE, Nicholson RI. Increased constitutive activity of PKB/Akt in tamoxifen resistant breast cancer MCF‐7 cells. Breast Cancer Res Treat 2004; 87: 167–80. [DOI] [PubMed] [Google Scholar]
- 18. Xiang T, Ohashi A, Huang Y et al. Negative regulation of AKT activation by BRCA1. Cancer Res 2008; 68: 10040–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Esteller M, Silva JM, Dominguez G et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast cancer and ovarian tumors. J Natl Cancer Inst 2000; 92: 564–9. [DOI] [PubMed] [Google Scholar]
- 20. Zhong Q, Wen YJ, Yang HS et al. Efficient inhibition of cisplatin‐resistant human ovarian cancer growth and prolonged survival by gene transferred vesicular stomatitis virus matrix protein in nude mice. Ann Oncol 2008; 19: 1584–91. [DOI] [PubMed] [Google Scholar]
- 21. Quinn JE, Carser JE, James CR, Kennedy RD, Harkin DP. BRCA1 and implications for response to chemotherapy in ovarian cancer. Gynecol Oncol 2009; 113: 134–42. [DOI] [PubMed] [Google Scholar]
- 22. Narod SA, Foulkes WD. BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer 2004; 4: 665–76. [DOI] [PubMed] [Google Scholar]
- 23. Rathi A, Virmani AK, Schorge JO et al. Methylation profiles of sporadic ovarian tumors and nonmalignant ovaries from high‐risk women. Clin Cancer Res 2002; 8: 3324–31. [PubMed] [Google Scholar]
- 24. Birgisdottir V, Stefansson OA, Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG, Eyfjord JE. Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast Cancer Res 2006; 8(4): R38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moynahan ME. The cancer connection: BRCA1 and BRCA2 tumor suppression in mice and humans. Oncogene 2002; 21: 8994–9007. [DOI] [PubMed] [Google Scholar]
- 26. Scully R, Livingston DM. In search of the tumour‐suppressor functions of BRCA1 and BRCA2. Nature 2000; 408: 429–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chan KY, Ozçelik H, Cheung AN, Ngan HY, Khoo US. Epigenetic factors controlling the BRCA1 and BRCA2 genes in sporadic ovarian cancer. Cancer Res 2002; 62: 4151–6. [PubMed] [Google Scholar]
- 28. Teodoridis JM, Hall J, Marsh S et al. CpG island methylation of DNA damage response genes in advanced ovarian cancer. Cancer Res 2005; 65: 8961–7. [DOI] [PubMed] [Google Scholar]
- 29. Catteau A, Morris JR. BRCA1 methylation: a significant role in tumour development? Semin Cancer Biol 2002; 12: 359–71. [DOI] [PubMed] [Google Scholar]
- 30. Li M, Balch C, Montgomery JS et al. Integrated analysis of DNA methylation and gene expression reveals specific signaling pathways associated with platinum resistance in ovarian cancer. BMC Med Genomics 2009; 8: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karpinski P, Ramsey D, Grzebieniak Z, Sasiadek MM, Blin N. The CpG island methylator phenotype correlates with long‐range epigenetic silencing in colorectal cancer. Mol Cancer Res 2008; 6: 585–91. [DOI] [PubMed] [Google Scholar]
- 32. Wu Q, Lothe RA, Ahlquist T et al. DNA methylation profiling of ovarian carcinomas and their in vitro models identifies HOXA9, HOXB5, SCGB3A1, and CRABP1 as novel targets. Mol Cancer 2007; 10: 6–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wei SH, Chen CM, Strathdee G et al. Methylation microarray analysis of late‐stage ovarian carcinomas distinguishes progression‐free survival in patients and identifies candidate epigenetic markers. Clin Cancer Res 2002; 8: 2246–52. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Value of COC1 cell reaction to platinum with different concentrations of 5‐Aza‐Dc by MTS (methyl‐thiazol tetrazolium) assay.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item