

Abstract
Human pancreatic cancer is generally hypovascular in nature and rich in interstitium. These pathological barriers may contribute to the intractable nature of pancreatic cancer by binding the penetration of anticancer agents throughout the tumor tissue. The aim of the present study was to determine whether NK012 is an appropriate formulation for the treatment of hypovascular tumors. Among pancreatic tumor xenografts, PSN1 appeared to have the richest tumor vasculature and the least number of stromal cells and matrix. In contrast, Capan1 had the poorest tumor vasculature and most abundant stromal tissue. Fluorescence microscopy and high‐performance liquid chromatography analysis demonstrated that although NK012 accumulated and continued to be distributed for more than 48 h throughout the entire body of both tumors, CPT‐11 disappeared almost entirely from both tumors within 6 h. In addition, efficient sustained release of SN‐38 was maintained for more than 96 h in both tumors following administration of NK012. Following the administration of CPT‐11, SN‐38 was no longer detectable after 24 h in the Capan1 tumor or after 48 h in the PSN1 tumor. All tumors were eradicated in the mice treated with NK012 but not in those treated with CPT‐11. Because the antitumor activity of SN‐38 is time dependent, NK012, which combines enhanced distribution with sustained release of SN‐38 within tumors, may be ideal for the treatment of hypovascular tumors, such as pancreatic cancer. (Cancer Sci 2008; 99: 1258–1264)
Pancreatic cancer has one of the worst prognoses among cancers.( 1 ) The median survival of cases of advanced pancreatic cancer is only approximately one in two 1 year after systemic gemcitabine‐based chemotherapy.( 2 ) The recent success of molecular‐targeting agents has also had some impact on pancreatic cancer treatment. A recent phase III trial of gemcitabine alone versus gemcitabine plus erlotinib (a tyrosine kinase inhibitor) in patients with advanced pancreatic cancer showed that overall survival was significantly prolonged in the gemcitabine + erlotinib arm. However, median survival in the gemcitabine + erlotinib arm (6.24 months) was only 10 days longer than in the gemcitabine‐alone arm (5.91 months).( 3 ) There is therefore an urgent need to develop modalities by which cytotoxic drugs can exert their significant antitumor activity to their full potential and reasonably prolong the overall survival of patients with advanced pancreatic cancer. There may be several reasons why pancreatic cancer is intractable. It is conceivable that anticancer agents are not delivered efficiently enough to the pancreatic cancer cells to kill them. Pancreatic cancer tissue is generally hypovascular in nature,( 4 , 5 ) and is rich in stromal cells and extracellular matrix, and these pathological barriers may hinder efficient penetration of the anticancer agents throughout the entire body of the pancreatic cancer.
The role of drug delivery systems (DDS) is to selectively deliver cytotoxic drugs to tumor tissues while lessening their distribution to normal tissues in order to reduce their side effects.( 6 , 7 , 8 ) However, it is conceivable that satisfactory drug delivery cannot be achieved in cancers having very few tumor vessels and an abundant collagen‐rich interstitium. Therefore, a more sophisticated DDS may be needed for efficient delivery of drugs to such types of cancer as pancreatic cancer.
SN‐38, a biologically active metabolite of irinotecan hydrochloride (CPT‐11), has potent antitumor activity but has not been used clinically because of its water insolubility. NK012, a successful drug formulation composed of SN‐38‐incorporating polymeric micelles, has been developed recently, and the remarkable antitumor effects of NK012 against the human small cell lung cancer SBC‐3, especially the vascular endothelial growth factor (VEGF)‐secreting SBC‐3–VEGF tumor, has been demonstrated.( 8 )
In the present study, we clarified the relationship between the tumor vasculature and interstitium using several human pancreatic xenografts, and evaluated the therapeutic effect of NK012 in a hypovascular and hypervascular pancreatic tumor.
Materials and Methods
Drugs and cells. SN‐38 and NK012 were synthesized by Nippon Kayaku (Tokyo, Japan). CPT‐11 was purchased from Yakult (Tokyo, Japan). The human pancreatic cancer cell lines Panc1, PSN1, BxPC3, and Capan1 were purchased from American Type Culture Collection (Rockville, MD, USA).
Panc1, PSN1, and Capan1 were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, streptomycin, and l‐glutamine (Sigma, St Louis, MO, USA) in atmosphere of 5% CO2 at 37°C. BxPC3 were maintained in RPMI‐1640 supplemented with 10% fetal bovine serum, streptomycin, and l‐glutamine (Sigma) in an atmosphere of 5% CO2 at 37°C.
Experimental mouse model. Female BALB/c nude mice, 6 weeks old, were purchased from CLEA Japan (Tokyo, Japan). Mice were inoculated subcutaneously in the flank with 1 × 107 cells/300 µL phosphate‐buffered saline (PBS). All animal procedures were carried out in compliance with the guidelines for the care and use of experimental animals, laid down by the Committee for Animal Experimentation of the National Cancer Center; these guidelines meet the ethical standards required by law and also comply with the guidelines for the use of experimental animals in Japan.
Immunohistochemical study of various human pancreatic tumor xenografts. When the tumor volume reached 300 mm3, tumors were excised from the mice and used for immunohistochemical analysis. To stain the blood vessels, the tissues were embedded in Optimal Cutting Temperature Compound (Sakura Finetechnochemical, Tokyo, Japan) and frozen at –80°C until use. Six micrometer‐thick tumor sections were prepared using a cryostatic microtome, Tissue‐Tek Cryo3 (Sakura Finetechnochemical), and then air dried for 1 h. The sections were soaked in 10% formalin for 15 min, and washed three times with 0.2 M PBS. The sections were then rinsed with ultrapure water. Endogeneous peroxidase was blocked with a 0.3% hydrogen peroxide solution in 100% methanol for 20 min. The sections were then rinsed three times with PBS for 3 min each. Non‐specific protein binding was blocked with 5% skim milk (BD, Franklin Lakes, NJ, USA) in PBS for 30 min at room temperature. After draining off the skim milk solution, a polyclonal antibody against factor VIII (Zymed Laboratories, South San Francisco, CA, USA) was added at a dilution of 1:50, followed by incubation for 1 h and three rinses with PBS for 5 min each. Biotinylated antirabbit IgG was added at a dilution of 1:50, followed by incubation for 1 h. The sections were rinsed three times with PBS, and Vectastain Elite ABC Reagent (Vector Laboratories, Burlingame, CA, USA) was added for 30 min. The sections were rinsed again three times with PBS and incubated with the 3,3′‐diaminobenzidine tetrahydrochloride (DAB+) Liquid System (Dako, Glostrup, Denmark) for 30 s. Finally, the sections were rinsed and counterstained with hematoxylin solution. For staining of type I, III, and IV collagen, tissues were fixed with 4% formalin, and the paraffin sections were prepared by the Tokyo Histopathologic Laboratory (Tokyo, Japan). First, the sections were soaked three times for 5 min each in xylene, and then three times for 3 min each in ethanol to remove the paraffin. The sections were then rinsed with ultrapure water and endogeneous peroxidase was blocked with a 0.3% hydrogen peroxide solution in 100% methanol for 20 min, followed by three rinses for 5 min with PBS. Then, Proteinase K (Dako) was added. After the sections were rinsed three times for 5 min each with 0.2 M PBS, non‐specific protein binding was blocked with a 1% normal goat serum (Dako) solution in PBS for 30 min at room temperature. Then, after three rinses for 5 min each with PBS, polyclonal rabbit anti type I, III, and IV collagen antibodies (Dako) were added at dilutions of 1:500 (type I collagen), 1:10 000 (type III collagen), and 1:2000 (type IV collagen), followed by incubation for 1 h. The slides were rinsed in PBS and incubated for 30 min with Envision/HRP (Dako) directed against the primary antibody. After further rinsing, the sections were incubated with the DAB+ Liquid System (Dako) for 30 s. Then, after a final rinse, the sections were counterstained with hematoxylin solution.
In vitro growth assay. The growth‐inhibitory effects of NK012, SN‐38, and CPT‐11 were examined using the WST8 assay. Cell suspensions (5000 cells/100 µL) were seeded into a 96‐well microliter plate, which was incubated for 24 h at 37°C. Then, after removal of the medium, 100 µL of medium containing various concentrations of each drug was added to the wells, which were then incubated for 48 h at 37°C. After removal of the medium, 10 µL of WST8 solution and 90 µL of medium were added to the wells, followed by incubation for 4 h at 37°C. The growth‐inhibitory effect of each drug was assessed spectrophotometrically (SpectraMax 190; Molecular Devices, Sunnyvale, CA, USA).
Distribution studies of CPT‐11 and NK012 in the tumors by fluorescence microscopy. Nude mice bearing PSN1, as a hypervascular tumor model, or Capan1, as a hypovascular tumor model, were used for studying the distribution of NK012 and CPT‐11, when the tumors reached 300 mm3 in volume. The maximum tolerated dose (MTD) of NK012 (30 mg/kg) or CPT‐11 (66.7 mg/kg) was injected intravenously into the mice. At 1, 6, 24, or 48 h after the injection of NK012 or CPT‐11, the mice were administered fluorescein‐labeled Lycopersicon esculentum lectin (100 µL/mouse) (Vector Laboratories) for the purpose of visualizing the tumor blood vessels. The tumors were then excised and embedded in Optimal Cutting Temperature Compound and frozen at –80°C before 6 µm‐thick sections were prepared using Tissue‐Tek Cryo3. The frozen sections were examined under a fluorescence microscope, Biorevo (Keyence, Osaka, Japan), at an excitation wavelength of 377 nm and emission wavelength 447 nm in order to evaluate the distribution of CPT‐11 and NK012 within the tumor tissues. Because formulations containing SN‐38 bound via ester bonds possess a particular fluorescence, both CPT‐11 and NK012 were detected under the same fluorescence conditions.
Distribution studies of free SN‐38, CPT‐11, and NK012 in the tumors by high‐performance liquid chromatography. When PSN1 and Capan1 tumors reached 300 mm3 in volume, NK012 (30 mg/kg) or CPT‐11 (66.7 mg/kg) was administered intravenously to the mice. At 1, 6, 24, 72, or 96 h after the injection of NK012 and CPT‐11, each tumor was excised. The tumor tissues were rinsed with physiological saline, mixed with 0.1 M glycine‐HCl buffer (pH 3.0) in methanol at 5% (w/w) and homogenized. To detect free SN‐38 and CPT‐11, the tumor samples (100 µL) were mixed with 20 µL of 1 mM phosphoric acid in methanol (1:1), 40 µL ultrapure water, and camptothecin was used as the internal standard (10 ng/mL for free SN‐38, 15 ng/mL for CPT‐11). The samples were vortexed vigorously for 10 s and filtered through Ultrafree‐MC Centrifugal Filter Devices (Millipore, Bedford, MA, USA). Reverse‐phase high‐performance liquid chromatography (HPLC) was conducted at 35°C on a Mightysil RP‐18 GP column (150 × 4.6 mm; Kanto Chemical, Tokyo, Japan). The samples were injected into an Alliance Water 2795 HPLC system (Waters, Milford, MA, USA) equipped with a Waters 2475 multi λ fluorescence detector. The detector was set at 365 and 430 nm (excitation and emission wavelengths, respectively) for CPT‐11, and at 365 and 540 nm for SN‐38.
For polymer‐bound SN‐38 detection, SN‐38 was released from the conjugate as described previously.( 8 ) In brief, 100 µL tissue samples were diluted with 20 µL methanol (50%[w/w]) and 20 µL NaOH (0.7 M). The samples were incubated for 15 min at room temperature. After incubation, 20 µL HCl (0.7 M) and 60 µL of internal standard solution was added to the samples, and then the hydrolysate was filtered. The filtrate was applied to the HPLC system.
Polymer‐bound SN‐38 was determined by subtraction of non‐polymer‐bound SN‐38 from the total SN‐38 in the hydrolysate.
Antitumor activity of NK012 and CPT‐11 against Capan1 or PSN1 xenografts. When the tumor volume reached approximately 300 mm3 in volume, the mice were divided randomly into test groups consisting of five mice per group (day 0). The drugs were administered on days 0, 4, and 8 by intravenous injection into the tail vein. NK012 was given at a dose of 30 mg/kg (MTD) and CPT‐11 was given at a MTD of 66.7 mg/kg as indicated in the optimal schedule reported previously.( 9 )
The length (L) and width (W) of the tumor mass were measured every 3 days. The tumor volume (TV) was calculated as follows: TV = (L × W2) × 0.5233.
Statistical analysis. Student's t‐test was used for the statistical analyses. P < 0.05 was considered to denote statistical significance.
Results
Density of collagen and the number of tumor blood vessels in the various human pancreatic tumor xenografts. We examined the density of collagen in four pancreatic cancer xenografts (Fig. 1a). Type I collagen was present in the greatest abundance in Capan1 and was least abundance in PSN1. The density of type I collagen in Panc1 and BxPC3 was in second and third place, respectively.
Figure 1.
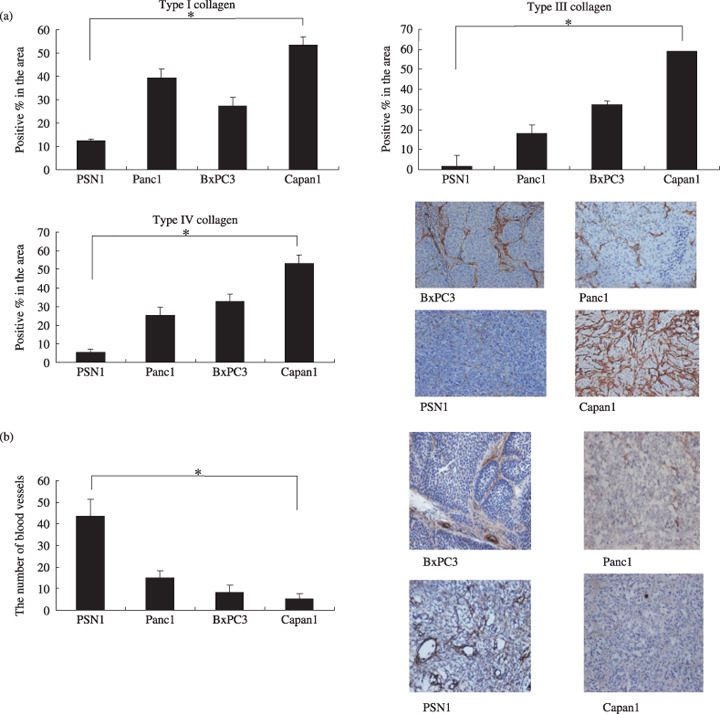
Examination of the amount of stroma and the number of tumor blood vessels in four human pancreatic cancer xenografts. (a) The amount of stroma in the four xenografts BxPC3, Panc1, PSN1, and Capan1. Immunohistochemical staining was conducted in order to determine the distribution of type I, III, and IV collagen in the tumors. The area occupied by each of type I collagen (left, upper), type III collagen (right, upper), and type IV collagen (left, bottom) was quantified. A representative immunostained image for type IV collagen is shown (right, bottom). (b) The number of blood vessels in the four xenografts. After immunostaining with anti‐factor VIII antibody, the number of tumor blood vessels in each of the xenografts was counted. *P < 0.05. Bar = SD.
Capan1 exhibited the highest density of type III collagen, and BxPC3 and PSN1 were in second and fourth place, respectively, with respect to the density of type III collagen. The distribution of type IV collagen tended to be similar to that of type I and III collagen.
We also examined the number of tumor blood vessels (Fig. 1b). The PSN1 tumor possessed the largest number of blood vessels among the tumors. In contrast, the Capan1 xenografts had the smallest number of tumor blood vessels.
We have summarized the results on collagen density and blood vessel number obtained in our study for each pancreatic xenograft. Capan1 was the most collagen‐rich tumor, and the density of collagen was lowest in PSN1. In contrast, tumor blood vessels were most abundant in PSN1 and least abundant in Capan1. Therefore, we decided to use Capan1 as a hypovascular tumor model and PSN1 as a hypervascular tumor model.
In vitro cytotoxic effects of NK012, SN‐38, and CPT‐11 against the Capan1 and PSN1 cell lines. The 50% inhibitory concentration (IC50) values of NK012 for the two cell lines, Capan1 (Fig. 2a) and PSN1 (Fig. 2b), ranged from 0.001 to 0.1 µM. NK012 exhibited a remarkably higher cytotoxic effect against both of the cell lines compared with CPT‐11. In contrast the cytotoxic effect of SN‐38 was similar to that of NK012. The IC50 value of each drug against PSN1 was almost similar to that of Capan1.
Figure 2.

(a) Capan1 and (b) PSN1 cells were exposed to the indicated concentrations of each drug for 72 h. The growth inhibition curves for NK012 ( ), SN‐38 (
), SN‐38 ( ), and CPT‐11 (
), and CPT‐11 ( ) are shown.
) are shown.
Antitumor activity analysis of NK012 and CPT‐11 using Capan1‐ and PSN1‐bearing nude mice. Antitumor activity was observed in mice treated with NK012 at a dose of 30 mg/kg/d and CPT‐11 at a dose of 66.7 mg/kg/d in vivo (Fig. 3). Although CPT‐11 exerted a significant antitumor effect compared with the control group in mice bearing the Capan1 tumor, the tumor volume continued to increase consistently. However, in the mice treated with NK012, the tumor volume started to shrink on day 8, and the disappeared completely by day 28 in all treated mice bearing the Capan1 tumor.
Figure 3.

Antitumor effect of NK012 and CPT‐11. NK012 (), CPT‐11 ( ), or saline () was administered intravenously. When the mean tumor volumes reached 300 mm3 (on day 0), NK012 (30 mg/kg/day) or CPT‐11 (66.7 mg/kg/day) was administered on days 0, 4, and 8. Each group consisted of five mice. (a) Capan1 tumor; (b) PSN1 tumor. *P < 0.05 (NK012 vs CPT11); **P < 0.05 (NK012 vs saline).
), or saline () was administered intravenously. When the mean tumor volumes reached 300 mm3 (on day 0), NK012 (30 mg/kg/day) or CPT‐11 (66.7 mg/kg/day) was administered on days 0, 4, and 8. Each group consisted of five mice. (a) Capan1 tumor; (b) PSN1 tumor. *P < 0.05 (NK012 vs CPT11); **P < 0.05 (NK012 vs saline).
In contrast to the observations for the Capan1 tumor, mice bearing the PSN1 tumor treated with CPT‐11 showed a slight reduction in tumor volume from day 4 to 12. However, after day 12 the tumor volume began to increase again. On the other hand, the tumor disappeared completely in all mice bearing the PSN1 tumor treated with NK012.
Distribution studies of CPT‐11 and NK012 in the solid Capan1 and PSN1 tumors. With the purpose of evaluating drug distribution and accumulation over time, sections of the tumor treated with NK012 or CPT‐11 were examined by fluorescence microscopy. Also, we examined the number of tumor blood vessels. In sections of the Capan1 tumor treated with CPT‐11, maximum drug accumulation was observed within 1 h of the injection of CPT‐11 (Fig. 4a). At 6 h after the injection, the fluorescence originating from CPT‐11 had almost entirely disappeared. Subsequently, no accumulation of CPT‐11 was observed within the tumor tissues. However, in sections of the Capan1 tumor treated with NK012, fluorescence from NK012 began to appear around tumor blood vessels at 1 h after the intravenous injection and lasted until 48 h. After 6 h, the fluorescent area began to increase and the maximum fluorescence area was observed at 24 h after the injection. Similar results were obtained for the PSN1 tumor (Fig. 4b).
Figure 4.

Distribution of NK012 or CPT‐11 in the (a) Capan1 and (b) PSN1 tumor xenografts. Mice bearing Capan1 or PSN1 tumors were injected with NK012 (30 mg/kg/day) or CPT‐11 (66.7 mg/kg/day). The tumor tissues were excised at 1, 6, 24, and 48 h after the intravenous injection of NK012 or CPT‐11. Each mouse was administered an injection of fluorescein‐labeled Lycopersicon esculentum lectin just before being killed, for detecting the tumor blood vessels. The frozen sections were examined under a fluorescence microscope at an excitation wavelength of 377 nm and emission wavelength of 447 nm. The same fluorescence conditions can be applied for visualizing NK012 and CPT‐11 fluorescence. Free SN‐38 can not be detected under these fluorescence conditions.
These microscopic observations were confirmed quantitatively by measuring the amount of SN‐38 that could be extracted from each of the solid tumors by reverse‐phase HPLC. Only slight conversion from CPT‐11 to SN‐38 was seen from 1 to 24 h in the Capan1 tumor and from 1 to 48 h in the PSN1 tumor, and no SN‐38 was detected thereafter. In contrast, SN‐38 released from NK012 continued to be detected in both tumors from 1 to 96 h after the injection of NK012 (Fig. 5).
Figure 5.

Tumor distribution of CPT‐11, NK012 (or polymer bound SN‐38), and free SN‐38 after administration of NK012 and CPT‐11 to mice bearing (a) Capan1 or (b) PSN1 xenografts. The time profiles of polymer‐bound SN‐38 (), free SN‐38 released from NK012 ( ), CPT‐11 (
), CPT‐11 ( ), and free SN‐38 converted from CPT‐11 () were obtained by high‐performance liquid chromatography analysis. The time points examined were 1, 6, 24, 48, 72, and 96 h after the administration of CPT‐11 or NK012.
), and free SN‐38 converted from CPT‐11 () were obtained by high‐performance liquid chromatography analysis. The time points examined were 1, 6, 24, 48, 72, and 96 h after the administration of CPT‐11 or NK012.
Discussion
Recently, several new formulations categorized as DDS have been approved in the field of oncological treatment, such as Doxil, a polyethylene glycol‐liposome incorporating adriamycin,( 10 , 11 ) and abraxane, a taxol coated with albumin.( 12 , 13 ) In addition, several clinical trials of drugs based on the DDS concept are now underway.( 14 , 15 , 16 ) Because such formulations possess a longer plasma area under the curve (AUC), liposomal drugs should have sufficient time to exit from tumor blood vessel and accumulate at reasonably high dose levels in the surrounding interstitium.
It has been reported that although polyethylene glycol (PEG) liposomes can be delivered efficiently to a solid tumor, free drug is not transferred sufficiently to cancer cells, particularly those that are distant from the tumor vessels, because the formulations are too large to move through the tumor interstitium.( 17 ) Also, it has been suggested that liposomes are too stable to allow the drug within to be released easily. Therefore, it has been speculated that PEG liposomes may not be so effective against cancers in which the tumor vessel network is irregular and loose because of an abundant collagen‐rich matrix. Some examples of such cancers include scirrhous cancer of the stomach and pancreatic cancer. In fact, Doxil is known to be clinically effective against ovarian and breast cancers, both of which are characterized by a high density of tumor microvessels, whereas it is not effective against stomach and pancreatic cancers.( 18 ) Therefore, it is conceivable that some special device is necessary for DDS drugs to exert their antitumor effect sufficiently even against hypovascular tumors such as pancreatic cancer.
In the present study, we characterized the tumor vessel and its interstitium using four kinds of human pancreatic xenografts. The results revealed that the number of tumor blood vessels was inversely related to the amount of collagen within the tumor tissues. Among the four cell lines, Capan1 was the poorest in tumor vasculature and richest in the amount of collagen within the tumor tissue. Conversely, PSN1 was the richest in tumor vasculature and poorest in the amount of collagen. Therefore, it may safely be said that Capan1 xenografts are most like human pancreatic cancer tissue in terms of the amount of interstitial tissue among the four cell lines tested.
We evaluated the in vitro cytotoxic effect of CPT‐11, SN‐38, and NK012 and the in vivo antitumor activity of CPT‐11 and NK012 against Capan1 tumors as a hypovascular tumor model and PSN1 tumors as a hypervascular tumor model. SN‐38 and NK012 exhibited a higher cytotoxic effect against the two cell lines compared with CPT‐11. Between SN‐38 and NK012, the cytotoxic effect of NK012 was almost similar or a little lower compared with that of SN‐38. As CPT‐11 itself is a prodrug and is converted to SN‐38, an active metabolite of CPT‐11, by carboxylesterases, the activity of CPT‐11 is dependent on the activity of the enzymes. It is speculated that the efficient sustained release of SN‐38 from NK012 allows the formulation to exert a similar cytotoxic effect to that of SN‐38. In the in vivo experiment, CPT‐11 showed significant antitumor activity against both PSN1 tumors as a hypervascular tumor model and Capan1 tumors as a hypovascular tumor model. A slight reduction in tumor size was observed from day 4 to 12 in the case of PSN1 tumors, but not Capan1 tumors. We suggest that the higher antitumor activity seen in PSN1 compared with Capan1 tumors is probably because of the greater accumulation of CPT‐11 in the PSN1 xenografts because of the more abundant vasculature. Surprisingly, NK012 could cause complete disappearance of both PSN1 and Capan1 tumor xenografts. Before conducting the experiment, we had anticipated that NK012 might exert stronger antitumor effects against PSN1 compared with Capan1, because such macromolecular drugs can accumulate more efficiently in the PSN1 xenografts because of the richer vasculature. Therefore, we then intensively examined the distribution of NK012 and CPT‐11 within the PSN1 or Capan1 xenografts by fluorescence microscopy and HPLC. In the analysis by fluorescence microscopy, NK012 appeared within and around the tumor blood vessels in both the PSN1 and Capan1 xenografts at 1 h after the injection. NK012 began to spread from the blood vessels within the tumor tissue of both xenografts. Fluorescence originating from NK012 increased to a maximum in both of the tumors by 24 h after the injection of NK012. Namely, NK012 was distributed throughout the entire body of both tumors at 24 h after the injection. Furthermore, fluorescence originating from NK012 was clearly and diffusely detected throughout both tumors.
However, fluorescence originating from CPT‐11 increased to a maximum at 1 h in both tumors after the injection of CPT‐11, indicating that maximum distribution of CPT‐11 was achieved in both tumors within 1 h of the injection. No or very slight fluorescence of CPT‐11 was observed in the tumors at 6 h after CPT‐11 injection. These observations were confirmed quantitatively by measuring the amount of SN‐38 extracted from both tumors by reverse‐phase HPLC. Only slight conversion to SN‐38 from CPT‐11 was seen from 1 to 24 h in the Capan1 tumor and from 1 to 48 h in the PSN1 tumor, and no SN‐38 was detected thereafter. SN‐38 released from NK012 continued to be detected in both tumors from 1 to 96 h after the injection of NK012. In both CPT‐11 and NK012, SN‐38 binds to each counter molecule via an ester bond, which confers blue fluorescence on CPT‐11 and NK012. Therefore, it is speculated that polymer‐bound SN‐38 can be distributed throughout the entire body of the tumor, regardless of the amount of interstitial tissue. We are unable to explain clearly how NK012 was distributed well even in hypovascular tumors. However, it is speculated that NK012 can move smoothly within the tumor interstitium because of its relatively small particle size (20 nm) compared with other DDS formulations, and because of its flexibility the formulation can pass through even narrow gaps within the interstitium. Previously, we reported that sustained release of 74% free SN‐38 occurred from NK012 under physiological conditions within 48 h.( 8 ) It is also important to remember that the antitumor activity of SN‐38 is time dependent.( 19 ) Taking all of these data together, it may be concluded that NK012 can selectively accumulate in pancreatic tumor xenografts, to be distributed effectively throughout the entire body of the tumor, including in hypovascular tumors, and shows sustained release for a prolonged period of time. Consequently, NK012 can exert more significant antitumor activity than CPT‐11, which is not an ideal formulation for realizing the time‐dependent actions of the drug.
In addition to our present study, there have been several efforts to enhance the accumulation of anticancer agents in tumors to obtain higher antitumor activities of drugs. For example, it has been reported that a transforming growth factor‐β inhibitor can enhance tumor vascular permeability to promote accumulation of macromolecules.( 20 ) Conversely, combined use of an antiangiogenic agent, such as an antibody to VEGF, with an anticancer agent could enhance the antitumor activity, probably by lowering the tumor vascular permeability with a consequent decrease in the interstitial fluid pressure so that the anticancer agents may accumulate more easily in the tumor.( 21 , 22 ) However, much remains to be clarified.
In the present paper, we have shown not only the superiority of the antitumor effect of NK012 compared with that of CPT‐11, but also propose that enhanced accumulation, distribution, and retention of DDS within the tumor tissue and the sustained release of anticancer agents from DDS particles are key elements for the treatment of hypovascular tumors. A phase I clinical trial is now underway. Not only the clinical usefulness of NK012, but also the new concept for antitumor actions described in this paper are intended to be verified in the near future through further preclinical and clinical studies.
Acknowledgments
We thank Mrs H. Miyatake and Mrs N. Mie for their technical assistance and Mrs K. Shiina for her secretarial assistance. This work was supported partly by a Grant‐in‐Aid from the 3rd Term Comprehensive Control Research for Cancer, Ministry of Health, Labor and Welfare (Y. Matsumura) and Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology (Y. Matsumura).
References
- 1. Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin 2007; 57: 43–66. [DOI] [PubMed] [Google Scholar]
- 2. Burris HA 3rd, Moore MJ, Andersen J et al . Improvements in survival and clinical benefit with gemcitabine as first‐line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997; 15: 2403–13. [DOI] [PubMed] [Google Scholar]
- 3. Moore MJ, Goldstein D, Hamm J et al . Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007; 25: 1960–6. [DOI] [PubMed] [Google Scholar]
- 4. Hosoki T. Dynamic CT of pancreatic tumors. AJR Am J Roentgenol 1983; 140: 959–65. [DOI] [PubMed] [Google Scholar]
- 5. Sofuni A, Iijima H, Moriyasu F et al . Differential diagnosis of pancreatic tumors using ultrasound contrast imaging. J Gastroenterol 2005; 40: 518–25. [DOI] [PubMed] [Google Scholar]
- 6. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 1986; 46: 6387–92. [PubMed] [Google Scholar]
- 7. Muggia FM. Doxorubicin‐polymer conjugates: further demonstration of the concept of enhanced permeability and retention. Clin Cancer Res 1999; 5: 7–8. [PubMed] [Google Scholar]
- 8. Koizumi F, Kitagawa M, Negishi T et al . Novel SN‐38‐incorporating polymeric micelles, NK012, eradicate vascular endothelial growth factor‐secreting bulky tumors. Cancer Res 2006; 66: 10 048–56. [DOI] [PubMed] [Google Scholar]
- 9. Kawato Y, Furuta T, Aonuma M, Yasuoka M, Yokokura T, Matsumoto K. Antitumor activity of a camptothecin derivative, CPT‐11, against human tumor xenografts in nude mice. Cancer Chemother Pharmacol 1991; 28: 192–8. [DOI] [PubMed] [Google Scholar]
- 10. Muggia FM. Liposomal encapsulated anthracyclines: new therapeutic horizons. Curr Oncol Rep 2001; 3: 156–62. [DOI] [PubMed] [Google Scholar]
- 11. Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer 2005; 5: 161–71. [DOI] [PubMed] [Google Scholar]
- 12. Green MR, Manikhas GM, Orlov S et al . Abraxane, a novel Cremophor‐free, albumin‐bound particle form of paclitaxel for the treatment of advanced non‐small‐cell lung cancer. Ann Oncol 2006; 17: 1263–8. [DOI] [PubMed] [Google Scholar]
- 13. Gradishar WJ, Tjulandin S, Davidson N et al . Phase III trial of nanoparticle albumin‐bound paclitaxel compared with polyethylated castor oil‐based paclitaxel in women with breast cancer. J Clin Oncol 2005; 23: 7794–803. [DOI] [PubMed] [Google Scholar]
- 14. Matsumura Y, Hamaguchi T, Ura T et al . Phase I clinical trial and pharmacokinetic evaluation of NK911, a micelle‐encapsulated doxorubicin. Br J Cancer 2004; 91: 1775–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hamaguchi T, Matsumura Y, Suzuki M et al . NK105, a paclitaxel‐incorporating micellar nanoparticle formulation, can extend in vivo antitumour activity and reduce the neurotoxicity of paclitaxel. Br J Cancer 2005; 92: 1240–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Uchino H, Matsumura Y, Negishi T et al . Cisplatin‐incorporating polymeric micelles (NC‐6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br J Cancer 2005; 93: 678–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Unezaki S, Maruyama K, Hosoda J et al . Direct measurement of the extravasation of polyethylene glycol‐coated liposomes into solid tumour tissue by in vivo fluorescence microscopy. Int J Pharmacol 1996; 144: 11–17. [Google Scholar]
- 18. Tsukioka Y, Matsumura Y, Hamaguchi T, Koike H, Moriyasu F, Kakizoe T. Pharmaceutical and biomedical differences between micellar doxorubicin (NK911) and liposomal doxorubicin (Doxil). Jpn J Cancer Res 2002; 93: 1145–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kawato Y, Aonuma M, Hirota Y, Kuga H, Sato K. Intracellular roles of SN‐38, a metabolite of the camptothecin derivative CPT‐11, in the antitumor effect of CPT‐11. Cancer Res 1991; 51: 4187–91. [PubMed] [Google Scholar]
- 20. Kano MR, Bae Y, Iwata C et al . Improvement of cancer‐targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF‐β signaling. Proc Natl Acad Sci USA 2007; 104: 3460–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jain RK. Normalizing tumor vasculature with anti‐angiogenic therapy: a new paradigm for combination therapy. Nat Med 2001; 7: 987–9. [DOI] [PubMed] [Google Scholar]
- 22. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005; 307: 58–62. [DOI] [PubMed] [Google Scholar]