

Abstract
Pharmacogenetic testing for UDP‐glucuronosyltransferase (UGT) 1A1*28, a promoter variant of the UGT1A1 gene, is now carried out clinically to estimate the risk of irinotecan‐associated toxicity. We studied the clinical significance of UGT1A1*6 and UGT1A1*27, two variants in exon 1 of the UGT1A1 gene that are found mainly in Asians. The study group comprised 46 Japanese patients who received various regimens of chemotherapy including irinotecan at doses from 50 to 180 mg/m2. Pharmacogenetic relationships were explored between the UGT1A1 genotype and the ratio of the area under the plasma concentration–time curve (AUC) of the active metabolite of irinotecan (SN‐38) to that of SN‐38 glucuronide (SN‐38G), used as a surrogate for UGT1A1 activity (AUCSN‐38/AUCSN‐38G). No patient was homozygous for UGT1A1*28, and none had UGT1A1*27. Two were heterozygous for UGT1A1*28. Two were homozygous and 15 heterozygous for UGT1A1*6, all of whom were wild type with respect to UGT1A1*28. Two patients were simultaneously heterozygous for UGT1A1*28 and UGT1A1*6, present on different chromosomes. The other 25 patients had none of the variants studied. The two patients simultaneously heterozygous for UGT1A1*28 and UGT1A1*6 and the two patients homozygous for UGT1A1*6 had significantly higher AUCSN‐38/AUCSN‐38G ratios than the others (P = 0.0039). Concurrence of UGT1A1*28 and UGT1A1*6, even when heterozygous, altered the disposition of irinotecan remarkably, potentially increasing susceptibility to toxicity. Patients homozygous for UGT1A1*6 should also be carefully monitored. UGT1A1 polymorphisms in the coding region of the UGT1A1 gene should be genotyped in addition to testing for UGT1A1*28 to more accurately predict irinotecan‐related toxicity, at least in Asian patients. (Cancer Sci 2006; 97: 1255–1259)
Irinotecan is a camptothecin analog with potent antitumor activity resulting from the inhibition of topoisomerase. This anticancer drug is now used widely to treat colorectal, lung and other types of cancer.( 1 , 2 ) Dose‐limiting toxicity of irinotecan includes severe leukopenia, neutropenia and diarrhea.( 3 , 4 ) Post‐marketing studies of approximately 14 000 patients with cancer who received irinotecan in Japan have estimated that the incidence of grade 3 or higher leukopenia is 23.8% for irinotecan monotherapy and 38.3% for irinotecan combined with other drugs, whereas that of grade 3 or higher diarrhea is approximately 10%, regardless of regimen.( 3 )
Genetic polymorphisms of UDP‐glucuronosyltransferase (UGT) 1A1, a key metabolizing enzyme of irinotecan, are important determinants of individual variations in susceptibility to toxicity.( 5 ) Irinotecan is a prodrug that is metabolized by carboxylesterase to its principal active metabolite, SN‐38. SN‐38 is subsequently conjugated mainly by UGT1A1 to a more polar, inactive glucuronide (SN‐38G). Severe toxicity is attributed, at least in part, to increased exposure to SN‐38 caused by decreased UGT1A1 activity due to genetic polymorphisms. Pharmacogenetic studies of irinotecan toxicity have therefore focused on genetic polymorphisms of the UGT1A1 gene, especially UGT1A1*28, a variant sequence in the promoter region.( 6 , 7 ) Multiple studies have significantly linked this variant to irinotecan‐related toxicity.( 8 ) The United States Food and Drug Administration has required that the package insert of irinotecan states that UGT1A1*28 is associated with an increased risk of toxicity; to decrease this risk, genetic testing is encouraged. The package insert also recommends that the starting dose of irinotecan is reduced by at least one level for patients who are homozygous for UGT1A1*28; however, whether this reduction is sufficient remains unclear. Firm recommendations for patients who are heterozygous for UGT1A1*28 have yet to be established. Such patients are likely to have an intermediate UGT1A1 activity leading to an increased risk of neutropenia, but clinical evidence supporting the use of lower doses of irinotecan is lacking.
The clinical consequences of UGT1A1*6 and UGT1A1*27, single nucleotide polymorphisms in exon 1 of the UGT1A1 gene, also remain poorly understood. These variants are found mainly in Asians.( 9 ) Unlike UGT1A1*28, case‐control studies of Japanese patients have failed to demonstrate a significant relationship between UGT1A1*6 alone and severe irinotecan‐related toxicity.( 10 ) However, the study suggested that the presence of UGT1A1*6 and UGT1A1*27 in addition to UGT1A1*28 might increase susceptibility to irinotecan‐related toxicity considerably. To better understand the clinical significance of these variants, especially the more frequent variant of UGT1A1*6, we examined how the coexistence of UGT1A1*6 or UGT1A1*27 and UGT1A1*28 alters the pharmacokinetics of irinotecan in Japanese patients.
Materials and Methods
Patients. Forty‐six Japanese patients with cancer who received irinotecan monotherapy or various regimens of irinotecan‐based chemotherapy from June 2003 through April 2006 were studied. Some patients with metastatic colorectal cancer were also enrolled in a phase I study of a regimen containing irinotecan, fluorouracil (FU), and leucovorin (LV) (FOLFIRI), carried out at Saitama Medical School (Saitama, Japan). Toxicity was assessed prospectively according to the common terminology criteria for adverse events, version 3.0 (http://ctep.cancer.gov/reporting/ctc_v30.html). Tumor response was not included as a study variable because of various primary diseases and prior therapies. All subjects gave informed consent in writing for their peripheral blood samples and medical information to be used in this study. This study was approved by the Institutional Review Board of Saitama Medical School.
Treatments. Irinotecan as a monotherapy was given weekly at doses of 50, 75 or 100 mg/m2 for the first 3 weeks of a 4‐week cycle or every 2 weeks at a dose of 150 mg/m2 until the onset of disease progression or intolerable toxicity. For combination chemotherapy, a 100‐mg/m2 dose of irinotecan was administered with bolus FU 500 mg/m2 and LV 10 mg/m2 ( l isomer form) weekly for the first 4 weeks of a 6‐week cycle (IFL). The FOLFIRI regimen, administered at 2‐week intervals, comprised irinotecan starting at 150 or 180 mg/m2 and LV 200 mg/m2 administered over the course of 2 h, followed by FU 400 mg/m2 as a bolus injection and FU 2400 mg/m2 as a 46‐h continuous infusion. Regimens containing irinotecan plus cisplatin (IP) were repeated every 4 weeks. Irinotecan was given at a dose of 50–70 mg/m2, followed 2 h later by a 120‐min infusion of cisplatin 80 mg/m2 with adequate hydration on day 1. The same dose of irinotecan was given again on day 15. For all regimens, 50–180 mg/m2 of irinotecan was dissolved in 250 mL of 5% dextrose solution and infused over the course of 90 min
Pharmacokinetic analysis. Blood samples for pharmacokinetic analysis were usually obtained during the first course of treatment with irinotecan. The samples were taken from the arm opposite the infusion site, at the beginning of irinotecan infusion and 0, 0.25, 0.5, 1, 2, 4, 8 and 24 h after the end of the infusion. The blood samples were centrifuged immediately, and the plasma was stored at −80°C until analysis. Total (lactone and carboxylate) plasma concentrations of irinotecan, SN‐38 and SN‐38G were analyzed by a modified reverse‐phase high‐performance liquid chromatographic (HPLC) method.( 11 ) A 150‐µL plasma sample was mixed with 300 µL of methanol, 5% perchloric acid (50:50, v/v) and camptothecin, serving as an internal standard, in a vortex mixer. The mixture was centrifuged at 20 600 g for 10 min, and 200 µL of the supernatant was injected into an HPLC system (Hitachi model 7000 series; Hitachi, Tokyo, Japan), equipped with a TSK‐gel ODS‐120T analytical column (4.6 × 250 mm, 4 µm; TOSOH, Tokyo, Japan). HPLC was carried out at 40°C at a flow rate of 1.0 mL/min. Irinotecan, SN‐38 and SN‐38G were determined fluorometrically (excitation 355 nm; emission 515 nm). The total (lactone and carboxylate) concentrations of irinotecan, SN‐38 and SN‐38G in plasma were quantified. The mobile phase consisted of 75 mM ammonium acetate (pH 4.75) for solvent A, and acetonitrile for solvent B; a 20‐min run was carried out with a linear gradient of 85–65% for solvent A.
The lower limit of quantification was 5 ng/mL (7.4 nM) for irinotecan and 0.5 ng/mL (1.2 nM and 0.88 nM, respectively) for SN‐38 and SN‐38G. The intra‐assay and interassay coefficients of variation were less than 10% for irinotecan and the metabolites.
The area under the plasma time–concentration curve (AUC; µmol·h/L) from the beginning of the infusion of irinotecan to the time of obtaining the last blood sample was calculated by a linear trapezoidal rule. The ratio of the AUC of SN‐38 to that of SN‐38G (AUCSN‐38/AUCSN‐38G) was used as a surrogate for UGT1A1 enzyme activity in vivo.
Genotyping. Genomic DNA was extracted from peripheral blood, which had been stored at −80°C until analysis, with the use of a QIAamp Blood Kit (QIAGEN, Hilden, Germany).
To analyze exon 1, first‐step PCR amplification of a 923‐bp fragment containing exon 1 was carried out as reported previously.( 10 )
For analysis of UGT1A1*6, the second set of polymerase chain reaction (PCR) amplifications was carried out with nested primers designed to amplify a 235‐bp segment. The mismatched forward primer and the reverse primer were 5′‐CTAGCACCTGACGCCTCGTTGTACATCAGAGCC‐3′ (+178 to +210; underlining indicates mismatched site) and 5′‐CCATGAGCTCCTTGTTGTGC‐3′ (+393 to +412), respectively. The forward primer was designed to introduce an MspI (Takara Shuzo Co., Otsu, Japan) restriction site into the wild‐type sequence from +209 to +212. A 1000‐fold diluted product of the first PCR was subjected to nested PCR in a volume of 50 µL containing 0.2 mM of each deoxynucleoside triphosphate, 50 mM KCl, 10 mM Tris‐HCl (pH 8.3), 1.5 mM MgCl2, 0.5 µM of each primer and 1.3 units of Taq polymerase (AmpliTaq Gold; Perkin‐Elmer, Foster City, CA, USA). The PCR conditions were 95°C for 15 min followed by 25 cycles of 94°C for 30 s, 55°C for 40 s and 72°C for 40 s. A 1‐µL PCR product was digested with 4 units of MspI for 1 h at 37°C. DNA from the wild‐type sequence was digested into 203‐ and 32‐bp fragments, whereas DNA from UGT1A1*6 gave an undigested 235‐bp fragment.
For the sequence of UGT1A1*27, another set of the second PCR amplifications was carried out using hemi‐nested primers 5′‐AGTACCTGTCTCTGCCCAC‐3′ (+485 to +503) and 5′‐GTCCCACTCCAATACACAC‐3′ (+865 to +867 and intron 1), designed to amplify a 399‐bp segment. Two BsrI (New England Biolabs, Ipswich, MA, USA) restriction sites exist in UGT1A1*27 (+552 to +556 and +684 to +688), but only one site (+552 to +556) exists in wild type. The method of PCR amplification was identical to that for the MspI restriction fragment length polymorphism, as described above. Digestion of the PCR products with 2.5 units of BsrI for 1 h at 65°C gave 199‐, 132‐ and 68‐bp fragments from UGT1A1*27, or 331‐ and 68‐bp fragments from wild type.
UGT1A1*28 was distinguished from wild type by direct sequencing (−147 to +106) of the 253–255 bp produced by PCR.
A set of PCR amplifications of a 1030‐bp fragment containing UGT1A1*28 and UGT1A1*6 was used to determine the location of these variant sequences on the respective alleles. The reaction mixture was similar to that described previously,( 7 ) except that the concentration of MgCl2 was 2.0 mM, and the amount of Taq polymerase was 1.3 units. The PCR primers were 5′‐AAGTGAACTCCCTGCTACCTT‐3′ (−147 to −127) and 5′‐GTCCCACTCCAATACACAC‐3′ (+865 to +867 and intron 1). The PCR conditions were 95°C for 15 min, followed by 30 cycles of 94°C for 30 s, 58°C for 40 s and 72°C for 40 s. The PCR fragments obtained were subcloned into pT7 Blue T‐Vector (Novagen, Darmstadt, Germany), and sequence analysis was used to examine the gene arrangement of the variants on homologous chromosomes.
Statistical analysis. The statistical significance of differences in the AUCSN‐38/AUCSN‐38G was assessed using the Mann–Whitney U test. This and other statistical analyses were carried out with SPSS for Windows, version 12.0 J (SPSS Japan, Tokyo Japan). Differences were considered statistically significant when the two‐tailed P‐value was less than 0.05.
Results
A total of 46 patients were studied (Table 1). Performance status was generally good; most patients had gastrointestinal tract cancers. Fourteen patients received IP, 20 FOLFIRI, three IFL and nine irinotecan monotherapy. Two of the 14 patients treated with IP, which is one of the experimental arms of an ongoing randomized phase III trial against unresectable gastric cancer in Japan (JCOG9912), were enrolled into JCOG9912, and the other 12 patients treated with IP also fulfilled the recruitment criteria of JCOG9912 except for having no prior chemotherapy.
Table 1.
Patient characteristics (n = 46)
| Characteristic | n |
|---|---|
| Median age (years) | 62 (range: 42–85) |
| Sex | |
| Female | 20 |
| Male | 26 |
| Performance status | |
| 0 | 28 |
| 1 | 16 |
| 2 | 2 |
| Primary organ | |
| Gastric | 14 |
| Colorectal | 25 |
| Other | 7 |
| Previous treatments | |
| Surgery | 31 |
| Radiotherapy | 3 |
| Systemic chemotherapy | 38 |
| 1 regimen | 30 |
| 2 regimens | 8 |
| None | 6 |
Genomic DNA from all patients was genotyped to examine UGT1A1 polymorphisms. No patient was homozygous for UGT1A1*28 or had UGT1A1*27 (Table 2). Two patients were heterozygous for UGT1A1*28. Two patients were homozygous and 15 were heterozygous for UGT1A1*6, all of whom were wild type with respect to UGT1A1*28. Two patients were simultaneously heterozygous for UGT1A1*6 and UGT1A1*28. Sequencing of the subcloned products obtained from the two patients who were heterozygous for both UGT1A1*6 and UGT1A1*28 revealed that the variants existed on different chromosomes (UGT1A1*6/UGT1A1*28 diplotype), which was consistent with the results of Saeki et al.( 11 ) The other 25 patients had none of the variants studied.
Table 2.
UGT1A1 genetic profiles and plasma concentration–time curve (AUC) ratios
| UGT1A1*6 | UGT1A1*28 | n † | AUCSN‐38/AUCSN‐38G |
|---|---|---|---|
| –/– | –/– | 24 | 0.43 (0.08–1.39) ‡ |
| +/– | –/– | 15 | 0.61 (0.26–1.42) ‡ |
| +/+ | –/– | 2 | 1.40, 1.10 |
| –/– | +/– | 2 | 0.48, 0.44 |
| +/– | –/+ | 2 | 1.04, 2.16 |
Total n = 45. One patient was excluded because of an incomplete set of blood samples;
‡ median (range).
Complete sets of blood samples were obtained in 45 of the 46 patients. In the remaining one patient who received FOLFIRI, a blood sample could not be obtained for technical reasons at the end of the infusion. Pharmacokinetic profiling showed that SN‐38G levels were higher than SN‐38 levels; AUCSN‐38/AUCSN‐38G was therefore usually under 1 (Table 2; Fig. 1A). There was no apparent relationship between AUCSN‐38/AUCSN‐38G and the dose of irinotecan (data not shown). In the patient who was excluded from AUC analysis because of the missing blood sample, SN‐38G levels were higher than SN‐38 levels at all other time points. This patient had none of the variants studied.
Figure 1.
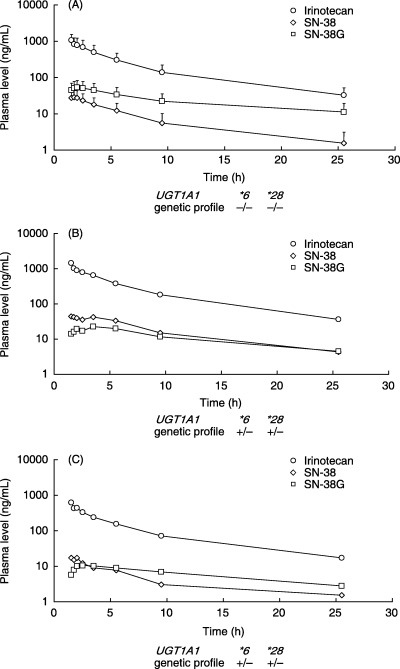
Pharmacokinetic profiles of patients who received irinotecan. (A) The profile of 24 patients without UGT1A1*28, UGT1A1*6 and UGT1A1*27 showed that SN‐38 glucuronide (SN‐38G) levels were higher than the SN‐38 levels reported in previous studies. Each data point indicates mean SD. (B) The patient with colon cancer received irinotecan at a dose of 150 mg/m2 in FOLFIRI. (C) The patient with gastric cancer received irinotecan at a dose of 70 mg/m2 in IP. SN‐38G levels were lower than SN‐38 levels in these two patients who were heterozygous for UGT1A1*6 and UGT1A1*28.
In the two patients who had the heterozygous UGT1A1*6/UGT1A1*28 diplotype, SN‐38G levels were lower than or approximated SN‐38 levels. Consequently, their AUCSN‐38/AUCSN‐38G ratios were higher than those of the patients who had no variants or who were heterozygous for either UGT1A1*6 or UGT1A1*28 alone (Table 2; Figs 1B,C, 2). One of these patients who received the FOLFIRI regimen at an irinotecan dose of 150 mg/m2 was studied again, after obtaining additional informed consent, to confirm reproducibility of the pharmacokinetic profile. The dose of irinotecan was reduced from 150 mg/m2 in the first course to 100 mg/m2 in the second course because of grade 4 neutropenia during the first course. The patient had grade 2 neutropenia in the second course. The pharmacokinetic profiles were similar for the first and second courses of treatment; the AUCSN‐38/AUCSN‐38G was 2.16 in the first course and 1.56 in the second. The other patient who had the heterozygous UGT1A1*6/UGT1A1*28 diplotype received IP safely, with no apparent toxicity.
Figure 2.
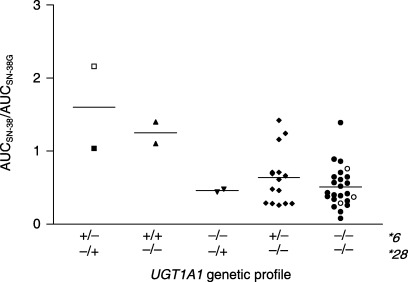
Ratio of the area under the plasma concentration–time curve (AUC) for SN‐38 to the AUC for SN‐38 glucuronide (SN‐38G), used as a surrogate for UGT1A1 activity (AUCSN‐38/AUCSN‐38G). Results are shown according to UGT1A1 genetic profile for 45 Japanese patients who received various regimens of irinotecan chemotherapy. Open symbols in the figure represent the patients suffering from grade 4 neutropenia. Lines indicate median values. One patient was excluded because of an incomplete set of blood samples.
Two patients were homozygous for UGT1A1*6. Their AUCSN‐38/AUCSN‐38G ratios were relatively higher than those of the other patients (Table 2; Fig. 2). One received irinotecan at a dose of 180 mg/m2 in the FOLFIRI regimen and had grade 2 neutropenia in the first course of treatment. Pharmacokinetic studies were repeated in this patient with the same dose of irinotecan after obtaining additional informed consent. The patient had grade 3 neutropenia in the second course. Similar pharmacokinetic profiles were obtained; the AUCSN‐38/AUCSN‐38G ratio was 1.40 in the first course and 2.73 in the second. The other patient received irinotecan monotherapy at a dose of 75 mg/m2 and had grade 3 nausea and grade 1 neutropenia. AUCSN‐38/AUCSN‐38G in this patient was 1.10. The AUCSN‐38/AUCSN‐38G ratios in the four patients who were heterozygous for UGT1A1*28/UGT1A1*6 or homozygous for UGT1A1*6 were significantly higher than those in the other 41 patients (P = 0.0039).
Among the 45 patients, four patients suffered from grade 4 neutropenia. Two of them received FOLFIRI regimen. One of the patients with UGT1A1*28/UGT1A1*6 received a FOLFIRI regimen including irinotecan 150 mg/m2, and the absolute AUC for SN‐38 was the highest (1.06 µmol·h/L) among all patients who received FOLFIRI (median 0.56 µmol·h/L; range 0.49–1.06 µmol·h/L; irinotecan dose 150 or 180 mg/m2; n = 19). One who had wild‐type UGT1A1 alleles received FOLFIRI with irinotecan 180 mg/m2, and the absolute AUC for SN‐38 was not high (0.66 µmol·h/L). Another two who harbored wild‐type UGT1A1 alleles received IP regimens with irinotecan 60 or 70 mg/m2. The AUCs for SN‐38 were, respectively, 0.39 µmol·h/L and 0.26 µmol·h/L, similar to the values in the other patients given IP (median 0.30 µmol·h/L; range 0.20–0.68 µmol·h/L; n = 12). No patient had grade 3 or higher diarrhea in this study.
Discussion
The present study demonstrated that patients who were heterozygous for both UGT1A1*6 and UGT1A1*28 (UGT1A1*6/UGT1A1*28 diplotype) had markedly lower SN‐38 glucuronidation activity than those who were heterozygous for either of these variants, let alone patients who had neither variant. Previous pharmacokinetic studies of irinotecan have reported that UGT1A1*6 decreases SN‐38 glucuronidation, despite the effects being somewhat lower than UGT1A1*28 does.( 12 , 13 ) Heterozygous UGT1A1*28 status is considered to result in UGT activity intermediate between that of wild type and that of homozygous UGT1A1*28. Thus, the presence of both UGT1A1*6 and UGT1A1*28, even when heterozygous, apparently lowers SN‐38 glucuronidation activity additively, resulting in a phenotypic effect similar to that associated with homozygous UGT1A1*28. The mechanisms responsible for this add‐on effect are unclear. UGT1A1*6 and UGT1A1*28 decrease UGT1A1 activity via different mechanisms; the former variant lessens protein function directly, whereas the latter reduces transcriptional activity of the promoter. Genetic effects on UGT activity compensate for each other when these variants exist separately. In contrast, compensation might be hindered by the coexistence of UGT1A1*6 and UGT1A1*28, resulting in remarkably lower UGT activity.
Our results suggest that patients who have the UGT1A1*6/UGT1A1*28 diplotype are at elevated risk for severe irinotecan‐related toxicity due to increased exposure to SN‐38. This notion is supported by the fact that one of the four patients with severe toxicity had the UGT1A1*6/UGT1A1*28 diplotype. The AUC for SN‐38 in this patient was higher than that of any other patients who received the same FOLFIRI regimen. We therefore believe that patients who have the UGT1A1*6/UGT1A1*28 diplotype should be treated carefully, similar to those who are homozygous for UGT1A1*28. Furthermore, 2.5–4.6% of the Japanese population is estimated to have both UGT1A1*6 and UGT1A1*28,( 13 , 14 , 15 ) which is similar to, or slightly higher than, the percentage homozygous for UGT1A1*28. These findings suggest that pharmacogenetic testing for irinotecan toxicity should include not only UGT1A1*28, but also the coding region variant UGT1A1*6, at least in Japanese patients.
Although the patients who were homozygous for UGT1A1*6 had lower SN‐38 glucuronidation activity than the others in this study, UGT1A1*6 alone appears to have only a limited effect on irinotecan‐related toxicity. Indeed, UGT1A1*6 was not associated with severe toxicity, and even patients who were homozygous for UGT1A1*6 could be treated safely in a previous study.( 10 ) Thus, patients who are heterozygous or even homozygous for UGT1A1*6 alone can most likely tolerate conventional starting doses of irinotecan, unless they have other conditions associated with an increased risk of toxicity. In in vitro expression studies, however, homozygosity and heterozygosity for UGT1A1*6 were associated with approximately 30 and 60% reductions in UGT activity, respectively.( 16 ) Otherwise, UGT1A1*6 could be partially linked to other genetic polymorphisms that decrease overall UGT activity. We have recently identified genetic linkages of UGT1A7 and UGT1A9 polymorphisms to UGT1A1*6, including those related to lower catalytic or transcriptional activities of UGT enzymes (K. Fujita et al., unpublished data, 2006). In addition, a previous pharmacokinetic study showed that the patients who had UGT1A1*6 alone showed slightly but not significantly decreased SN‐38 glucuronidation activity,( 13 ) consistent with the results of our study. As for hyperbilirubinemia, UGT1A1*6 is a significant contributory factor to unconjugated hyperbilirubinemia, including Gilbert's syndrome, especially among Japanese neonates.( 14 , 15 ) These findings suggest that patients who have UGT1A1*6, especially when homozygous, are at increased risk for irinotecan‐related toxicity. Although definitive evidence of this risk is lacking, we believe that toxicity should be monitored rigorously during irinotecan chemotherapy in patients who have UGT1A1*6 alone.
The genetic basis for irinotecan‐related toxicity apparently differs among distinct ethnic populations. That is to say, the same genetic variants affect toxicity differently. UGT1A1 genetic polymorphisms differ considerably among genetically distinct populations; the allele frequency of UGT1A1*28 is several times higher in whites (0.3–0.4) than in Asians (around 0.15).( 17 , 18 ) UGT1A1*6 and UGT1A1*27 have been identified only in Asians (0.11–0.23 for UGT1A1*6 and 0.01–0.03 for UGT1A1*27).( 6 , 9 , 12 , 14 ) As for hyperbilirubinemia, bilirubin levels are generally higher in Asians than in whites, at least among infants, suggesting that the genetic basis for hyperbilirubinemia differs between these two ethnic groups.( 19 , 20 , 21 ) The neonatal hyperbilirubinemia found in Japanese is significantly related to UGT1A1*6, not UGT1A1*28,( 14 , 15 ) whereas that occurring in whites during the first 2 days of life is associated with homozygosity for UGT1A1*28.( 22 ) In the present study, the total bilirubin levels observed in the patients who were homozygous for UGT1A1*6 and heterozygous for both of UGT1A1*6 and UGT1A1*28 were not significantly higher than those observed in the other patients. The complex genetic basis for UGT1A1 activity makes it difficult to compare irinotecan‐related toxicity among different ethnic groups. In addition, other factors potentially related to irinotecan‐associated toxicity include patients’ age, organ functions, prior treatments, dosing schedule and concurrently administered drugs. However, because genotype‐based individualized chemotherapy with irinotecan is now practiced widely, studies comparing irinotecan toxicity among the different ethnic populations appear to be warranted.
In conclusion, our study showed that the coexistence of two UGT1A1 variants, the promoter variant UGT1A1*28 and the coding region variant UGT1A1*6, remarkably altered the disposition of irinotecan, potentially increasing susceptibility to toxicity, even when these variants were heterozygous. Patients who had the UGT1A1*6/UGT1A1*28 diplotype should therefore be treated similarly to those who are homozygous for UGT1A1*28. Genotyping for UGT1A1*6 in addition to UGT1A1*28 is necessary to predict the risk of irinotecan‐related toxicity, at least in Asian patients. Genomic information is hereafter expected to play an increasingly important role in optimizing the use of irinotecan therapy.
Acknowledgments
We thank Ms Yuko Akiyama for technical help in the genotype analysis as well as Ms Kaori Kawara for serving as a research nurse. This study was supported in part by a Grant‐in‐Aid from the Ministry of Health and Welfare of Japan (13‐10). This study was presented in part at the 42nd Annual Meeting of American Society of Clinical Oncology, Atlanta, GA, 2–6 June 2006.
References
- 1. Meyerhardt JA, Mayer RJ. Systemic therapy for colorectal cancer. N Engl J Med 2005; 352: 476–87. [DOI] [PubMed] [Google Scholar]
- 2. Noda K, Nishiwaki Y, Kawahara M et al. Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small‐cell lung cancer. N Engl J Med 2002; 346: 85–91. [DOI] [PubMed] [Google Scholar]
- 3. Tadokoro J, Hasegawa H, Hayakawa K. Post‐marketing surveillance (PMS) of all patients treated with irinotecan in Japan: clinical experience and ADR profile of 13 935 patients. Proc Am Soc Clin Oncol 2002; 21: 259. [DOI] [PubMed] [Google Scholar]
- 4. Kubota K, Nishiwaki Y, Ohashi Y. The Four‐Arm Cooperative Study (FACS) for advanced non‐small‐cell lung cancer (NSCLC). J Clin Oncol 2004; 22: 618. [Google Scholar]
- 5. Mathijssen RH, Van Alphen RJ, Verweij J et al. Clinical pharmacokinetics and metabolism of irinotecan (CPT‐11). Clin Cancer Res 2001; 7: 2182–94. [PubMed] [Google Scholar]
- 6. Bosma PJ, Chowdhury JR, Bakker C et al. The genetic basis of the reduced expression of bilirubin UDP‐glucuronosyltransferase 1 in Gilbert's syndrome. N Engl J Med 1995; 333: 1171–5. [DOI] [PubMed] [Google Scholar]
- 7. Monaghan G, Ryan M, Seddon R, Hume R, Burchell B. Genetic variation in bilirubin UDP‐glucuronosyltransferase gene promoter and Gilbert's syndrome. Lancet 1996; 347: 578–81. [DOI] [PubMed] [Google Scholar]
- 8. Ando Y, Hasegawa Y. Clinical pharmacogenetics of irinotecan (CPT‐11). Drug Metab Rev 2005; 37: 565–74. [DOI] [PubMed] [Google Scholar]
- 9. Akaba K, Kimura T, Sasaki A et al. Neonatal hyperbilirubinemia and mutation of the bilirubin uridine diphosphate‐glucuronosyltransferase gene: a common missense mutation among Japanese, Koreans and Chinese. Biochem Mol Biol Int 1998; 46: 21–6. [DOI] [PubMed] [Google Scholar]
- 10. Ando Y, Saka H, Ando M et al. Polymorphisms of UDP‐glucuronosyltransferase gene and irinotecan adverse reactions: a pharmacogenetic analysis. Cancer Res 2000; 60: 6921–6. [PubMed] [Google Scholar]
- 11. Saeki M, Saito Y, Jinno H et al. Haplotype structures of the UGT1A gene complex in a Japanese population. Pharmacogenomics J 2006; 6: 63–75. [DOI] [PubMed] [Google Scholar]
- 12. Ando Y, Ueoka H, Sugiyama T, Ichiki M, Shimokata K, Hasegawa Y. Polymorphisms of UDP‐glucuronosyltransferase and pharmacokinetics of irinotecan. Ther Drug Monit 2002; 24: 111–16. [DOI] [PubMed] [Google Scholar]
- 13. Sai K, Saeki M, Saito Y et al. UGT1A1 haplotypes associated with reduced glucuronidation and increased serum bilirubin in irinotecan‐administered Japanese patients with cancer. Clin Pharmacol Ther 2004; 75: 501–15. [DOI] [PubMed] [Google Scholar]
- 14. Akaba K, Kimura T, Sasaki A et al. Neonatal hyperbilirubinemia and a common mutation of the bilirubin uridine diphosphate‐glucuronosyltransferase gene in Japanese. J Hum Genet 1999; 44: 22–5. [DOI] [PubMed] [Google Scholar]
- 15. Maruo Y, Nishizawa K, Sato H, Doida Y, Shimada M. Association of neonatal hyperbilirubinemia with bilirubin UDP‐glucuronosyltransferase polymorphism. Pediatrics 1999; 103: 1224–7. [DOI] [PubMed] [Google Scholar]
- 16. Yamamoto K, Sato H, Fujiyama Y, Doida Y, Bamba T. Contribution of two missense mutations (G71R and Y486D) of the bilirubin UDP glycosyltransferase (UGT1A1) gene to phenotypes of Gilbert's syndrome and Crigler–Najjar syndrome type II. Biochim Biophys Acta 1998; 1406: 267–73. [DOI] [PubMed] [Google Scholar]
- 17. Beutler E, Gelbart T, Demina A. Racial variability in the UDP‐glucuronosyltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism? Proc Natl Acad Sci USA 1998; 95: 8170–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ando Y, Chida M, Nakayama K, Saka H, Kamataki T. The UGT1A1*28 allele is relatively rare in a Japanese population. Pharmacogenetics 1998; 8: 357–60. [DOI] [PubMed] [Google Scholar]
- 19. Horiguchi T, Bauer C. Ethnic differences in neonatal jaundice: comparison of Japanese and Caucasian newborn infants. Am J Obstet Gynecol 1975; 121: 71–4. [DOI] [PubMed] [Google Scholar]
- 20. Fischer AF, Nakamura H, Uetani Y, Vreman HJ, Stevenson DK. Comparison of bilirubin production in Japanese and Caucasian infants. J Pediatr Gastroenterol Nutr 1988; 7: 27–9. [DOI] [PubMed] [Google Scholar]
- 21. Yamauchi Y, Yamanouchi I. Transcutaneous bilirubinometry in normal Japanese infants. Acta Paediatr Jpn 1989; 31: 65–72. [DOI] [PubMed] [Google Scholar]
- 22. Bancroft JD, Kreamer B, Gourley GR. Gilbert syndrome accelerates development of neonatal jaundice. J Pediatr 1998; 132: 656–60. [DOI] [PubMed] [Google Scholar]