

Abstract
Epstein–Barr virus (EBV)‐associated gastric carcinoma (GC) is a distinct subgroup of GC, comprising 10% of all cases of GC. EBV‐associated carcinoma is the monoclonal growth of EBV‐infected epithelial cells, and it represents a model of virus–host interactions leading to carcinoma. EBV‐infected cells express several latent proteins (latency I program of viral latent gene expression) in EBV‐associated GC. However, latent membrane protein 2A (LMP2A) up‐regulates the cellular survivin gene through the NFkB pathway, conferring resistance to apoptotic stimuli on the neoplastic cells. EBV‐associated GC also shows characteristic abnormality, that is, global and non‐random CpG island methylation of the promoter region of many cancer‐related genes. Since the viral genes are also regulated by promoter methylation in the infected cells, the DNA methylation mechanism specific to EBV‐associated GC may be an exaggeration of the cellular mechanism, which is primarily for defense against foreign DNA. Production of several immunomodulator molecules, inducing tumor‐infiltrating lymphocyte and macrophages, serves to form the characteristic histologic pattern in EBV‐associated GC. The proposed sequence of events within the mucosa is as follows: EBV infection of certain gastric stem cells; expression of viral latent genes; abnormality of signal pathways caused by viral gene products; DNA methylation‐mediated repression of tumor suppressor genes; and monoclonal growth of EBV–infected cells through interaction with other etiologic factors. Potentially useful therapeutic approaches to EBV‐associated GC are those that utilize the virus–host interactions, such as bortezomib‐induced and viral enzyme‐targeted radiotherapy. (Cancer Sci 2008; 99: 1726–1733)
Infectious organisms have been recognized to cause a variety of human neoplasms, but we were surprised to learn that gastric carcinoma (GC) may also be caused by an infectious organism.( 1 ) The skepticism against the role of infectious agents in the cancer research field had originated from the discovery by Fibiger (1926 Nobel Prize honoree) in 1913 that a kind of parasite caused rat stomach cancer, which was later proven to be totally incorrect.( 2 ) However, with acceptance of Helicobacter pylori as a causative agent of GC( 3 , 4 ) Epstein–Barr virus (EBV) has also been regarded to be a GC‐causing infective agent.( 5 , 6 , 7 ) EBV‐associated GC is a distinct subgroup of GC (nearly 10% of all cases of GC), in contrast to the rather ubiquitous relationship of H. pylori with GC, and each agent is uniquely, but not exclusively, involved in carcinogenesis of the stomach (Table 1).
Table 1.
Comparison of Helicobacter pylori and Epstein–Barr virus (EBV) as causative organisms of gastric carcinoma
| Helicobacter pylori | Epstein–Barr virus | |
|---|---|---|
| Carriers | More than 75% of people over age 40. | More than 95% of people over age 20. |
| Involvement in gastric carcinoma | Most GC (intestinal/diffuse types of histology). | 10% of GC, including lymphoepithlioma‐like GC. |
| Higher risk of GC in carriers compared with non‐carriers. | Monoclonal EBV present in all neoplastic cells. | |
| Carcinogenic mechanisms | Hummingbird phenomenon by transfection of the CagA gene into epithelial cells. Activation of ERK mediated by CagA/ SHP2 or Grb2. | Transformation of epithelial cells after transfection of the LMP2A gene. Abnormality in signal transductions mediated by LMP2A. |
| Model animal/Cell line | Mongolian gerbil | GC strain (KT), transplantable to SCID mice. |
| CagA transgenic mouse | GC cell line (SNU‐719). | |
| Other related malignancies | Mucosa associated lymphoid tissue lymphoma | Lymphomas (Burkitt, Hodgkin lymphoma, T/NK cell lymphoma). |
| Nasopharyngeal carcinoma. |
GC: gastric carcinoma.
EBV was the first virus discovered from human neoplastic cells, a Burkitt's lymphoma cell line, in 1964.( 8 ) Subsequent investigations( 9 , 10 ) have identified the virus in nasopharyngeal carcinoma (NPC), Hodgkin's lymphoma, B‐cell lymphomas with or without immunosuppression (e.g. AIDS‐related lymphoma or pyothorax‐associated lymphoma,( 11 ) respectively) and nasal NK/T cell lymphoma. In situ hybridization (ISH) method detecting EBV‐encoded small RNA (EBER) was introduced to the pathology field in 1990, which then led to the unexpected discovery of the presence of EBV in carcinoma tissues of the stomach. Although NPC appears endemic in Southern China, EBV‐associated GC occurs worldwide without regional or racial differences. It is most prevalent among EBV‐associated malignant neoplasms, and more than 90 000 patients worldwide are estimated to develop this type of stomach cancer annually. In this review, we focused on the pathology and pathogenesis of EBV‐associated GC, and the readers can also refer to a recent comprehensive review of this unique type of carcinoma( 7 ) and one specifying its epidemiology.( 12 )
EBV‐associated GC is clinicopathologically a distinct subgroup of GC
EBV‐associated GC is defined by the presence of EBV in neoplastic cells. EBV DNA, or its abundant small RNA, EBER (–107 per infected cell), is always identified in nearly all the neoplastic cells of the tumor tissues when it is present in GC (Fig. 1).
Figure 1.
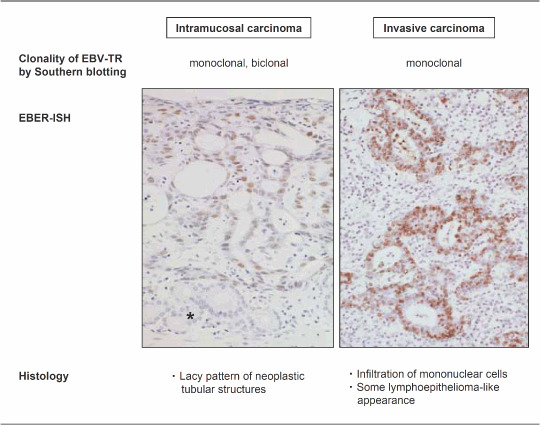
Pathology of Epstein–Barr virus (EBV)‐associated gastric carcinoma. EBV‐encoded small RNA (EBER) in situ hybridization demonstrates that positive signals are present in the nuclei of all the neoplastic cells in intramucosal and advanced carcinomas. The remaining pyloric glands (*) in the intramucosal carcinoma and the infiltrating lymphocytes in the advanced carcinoma are negative for signals.
Clinical features of EBV‐associated GC include male predominance, relatively younger age and preponderant location in the proximal stomach. EBV‐associated GC often takes the form of an ulcerated or saucer‐like tumor accompanied by marked thickening of the gastric wall. These features are well discernible on endoscopic ultrasonograpy and CT scans of the stomach (Maeda E et al. unpublished data). EBV‐associated GC shows a lower rate of lymph‐node involvement, especially during its early stage in the submucosa, and has a relatively favorable prognosis compared with EBV‐negative GC.
Histologically, there are two types of EBV‐associated GC, i.e. lymphoepithelioma‐like and ordinary types of GC. The relative ratio of frequency of the two types is roughly 1:4 in our experience. Lymphoepithelioma‐like GC has a histology typical of poorly differentiated carcinoma with dense infiltration of lymphocytes, which was also reported as ‘GC with lymphoid stroma’ by Watanabe et al. in 1976.( 13 ) More than 80% of lymphoepithelioma‐like GC is infected with EBV.( 14 , 15 ) On the other hand, ordinary‐type GC, comprising 5–10% of all cases of GC, shows features of moderately or poorly differentiated adenocarcinoma with various degrees of lymphocytic infiltration. Both types of carcinomas, however, share the same clinicopathological features. The frequency of EBV‐associated GC is relatively lower in intramucosal and submucosal carcinomas (early GC) compared with that in deeply invasive carcinomas (advanced GC), but there is no statistical difference between them.( 16 ) Some EBV‐associated GC show a ‘lace pattern’( 17 ) at the intramucosal stage, which consists of connection and fusion of neoplastic glands at the intermediate zone of proper mucosa (Fig. 1). Further infiltration of the carcinoma into the submucosa is occasionally accompanied by the infiltration of massive lymphocytes, which consist of CD8‐ and CD4‐positive T‐lymphocytes, and CD68‐positive macrophages, in a ratio of 2:1:1 (Shintani et al. unpublished data). EBV infection is rarely observed in only a very limited number of these infiltrating lymphocytes.
Neoplastic cells of EBV‐associated GC show characteristic profiles of differentiation markers, which might be associated with the cell lineage of the EBV‐infected cells. EBV‐associated GC shows a low level of expression of keratin molecules (CK7, 18, and 19)( 18 ) and has a null/gastric phenotype as determined by the expression pattern of mucin molecules (MUC5AC, MUC6).( 19 ) These findings suggest that the targets of EBV infection and subsequent transformation are the precursor cells with intrinsic differentiation potential toward the gastric cell type but not the intestinal type.
EBV is monoclonal and exists in a latent state in the carcinoma tissue
EBV is a double‐stranded DNA virus (180K kb in size) and takes a linear form in the viral particles. After EBV enters the infected cells, the viral DNA circularizes by fusing terminal repeats (TR, repetitive 500‐bp structures) at both ends( 10 ) and maintains its circular form in the nuclei of the latently infected cells. Southern blot analysis of the specific structure of EBV‐TR provides information about the clonality of EBV, viral integration and the state of viral activation, i.e. replicating (linear configuration) versus latent (episomal circular forms). In EBV‐associated GC, TR analysis has demonstrated that monoclonal EBV is present in an episomal form without integration into the host genome and that the infection is latent with no viral replication. Mono‐ or bi‐clonal EBV was observed in the carcinoma tissues at the intramucosal stage; however, it was always monoclonal at the stage of submucosal invasion( 7 ) and in further advanced carcinoma.( 5 , 6 ) All of the carcinoma cells show a positive signal in EBER‐ISH in all cases of EBV‐associated GC, whether intramucosal or invasive (Fig. 1). Therefore, EBV infection occurs at the initial or a very early stage of carcinoma development.
Latent EBV infection, without replication of viral particles, can take three forms in terms of expression of EBV latent genes: latency I, II and III.( 9 , 10 ) EBV‐associated GC, as well as Burkitt's lymphoma, belongs to latency I, in which the expression of viral latent genes is restricted to EBV‐determined nuclear antigen 1 (EBNA1), EBER, latent membrane protein 2A (LMP2A) and transcripts from the BamHI A region (BARF0 andBARF1).( 5 , 6 , 7 ) Latency II neoplasm includes NPC and Hodgkin's lymphoma, and is characterized by the expression of LMP1, a transmembrane protein with the capacity to transform rodent fibroblasts. EBNA2, 3 A and 3C, essential for immortalizing resting B‐lymphocytes, are additionally expressed owing to the lack of effective immune mechanisms in patients with latency III neoplasms such as AIDS‐ or organ‐transplant‐related lymphomas. Since latency I neoplasms arise without the expression of LMP1 or EBNA2, questions concerning the role of EBV in these neoplasms have been raised. In the case of Burkitt's lymphoma, however, loss of EBV from the EBV‐positive lymphoma cell line, Akata, abolishes colony‐forming capacity in soft agar and tumorigenicity in nude mice, demonstrating that EBV contributes to the tumor growth of latency I‐type neoplasms.( 20 )
Resistance to apoptosis exerted by viral LMP2A
One crucial problem in the investigation of the role of EBV infection in EBV‐associated GC is the lack of an ideal experimental model, such as the Akata cell line in Burkitt's lymphoma as described above. It is well known that EBV is hardly maintained in epithelial cells in culture; therefore, to reiterate the role of EBV infection in EBV‐associated GC in vitro, we have systematically compared the biological characteristics (growth, apoptosis and migration) of GC cell lines (MKN‐1, TMK1, MKN‐74, and MKN‐7) with and without infection of recombinant EBV( 21 ) in which the neomycin resistance gene Neor is inserted into the EBV‐DNA.( 22 ) In this screening, we observed that resistance to serum deprivation‐induced apoptosis is characteristic of EBV‐infected GC cell lines. Subsequent analyses demonstrated that up‐regulation of the cellular survivin gene, by the viral latent protein LMP2A, is responsible for the advantage in survival of EBV‐infected cells. Survivin is the smallest member of a family of proteins, known as inhibitors of apoptosis protein (IAP), which plays a key role in the regulation of apoptosis and cell division. In parallel with these findings in vitro, a lower rate of apoptosis has also been well documented in EBV‐associated GC in vivo. ( 23 ) In immunohistochemical studies, survivin expression was frequently observed in carcinoma tissues of GC, and significantly more expression was found in EBV‐associated GC than in EBV‐negative GC in the advanced stage.( 21 )
EBV uses its latent protein, LMP2A, to activate the NF–κB–survivin pathway to rescue EBV‐infected epithelial cells from serum deprivation in our experimental system.( 21 ) LMP2A (Fig. 2) consists of a long N‐terminal tail, 12 membrane‐spanning domains and a short C‐terminal tail, and forms aggregates in patches on the surface of all forms of latently infected cells, including latency I, II and III.( 9 , 10 , 24 ) There are eight constitutively phosphorylated tyrosine residues in the N‐terminal tail that are critical for interaction with cellular proteins. The sequences around tyrosine 74 and tyrosine 85 form the domains of the immunoreceptor tyrosine‐based activation motif (ITAM), homologous to that found in the immunoglobulin α‐ and β‐signaling subunits of the B‐cell receptor (BCR). LMP2A also has several proline‐rich regions in the N‐terminal tail, which can bind WW domain‐containing Nedd‐4 family ubiquitin–protein ligases. LMP2A is suggested to be a molecular scaffold for the recruitment of both BCR‐associated tyrosine kinases (Syk and Lyn) and E3 protein–ubiquitin ligases. Thus, LMP2A may prevent BCR activation and its induction of lytic replication of EBV in the infected B‐cells. On the other hand, in the in vivo transgenic mouse model, LMP2A activates the phosphatidylinositol 3‐kinase (PI3K)–Akt pathway, and provides developmental and survival signals to BCR‐negative B‐cells.
Figure 2.
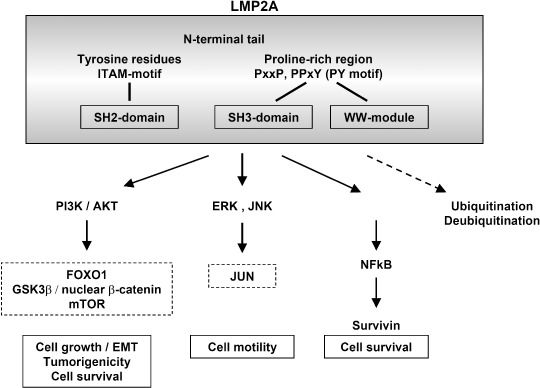
Viral latent membrane protein 2A (LMP2A) in cancer: its structure and downstream signal pathways. The figure summarizes the signal pathways, the activation of which has been reported in epithelial cells and cancer.
The function of LMP2A and its modulation of the intracellular signaling pathway have not yet been fully clarified in epithelial cells, particularly stomach epithelial cells. Although targeting of LMP2A to the epidermis of transgenic mice has no growth‐altering effects,( 25 ) LMP2A greatly affects cell growth and differentiation pathways in a human keratinocyte cell line, HaCaT, in part through activation of the PI3K–Akt pathway.( 26 ) Downstream of the PI3K–Akt pathway, LMP2A is reported to stabilize β‐catenin and activate the Wnt pathway in NPC cell lines.( 27 ) LMP2A inhibits TGFβ1‐induced apoptosis in a GC cell line (HSC‐39)( 28 ) and the constitutive activation of the RAS–PI3K–Akt pathway is necessary for anchorage‐independent cell growth of this cell line.( 29 ) The NF–κB pathway is reported to be necessary for the transformation of rodent cells through inhibition of the death signal activated by oncogenic Ras.( 30 ) Thus, considering our results of the involvement of the NF–κB pathway in EBV‐associated GC, further studies are necessary to clarify the role of LMP2A in the neoplastic growth of stomach epithelial cells. The regulatory mechanism of LMP2A should be also investigated in the EBV‐associated GC. We observed that all of the cases of EBV‐associated GC expressed LMP2A, although the amount of LMP2A transcripts considerably varies in the carcinoma tissues of the surgically resected GC (Hino et al. unpublished data). Since the expression of LMP2A can be regulated by Notch signal in EBV‐infected cells,( 31 ) such an investigation will provide an important insight into the interaction of virus and host cells along the development of the stomach carcinoma.
Aberrant DNA methylation as a key abnormality
CpG‐island methylator phenotype (CIMP) is the most characteristic abnormality among genetic and epigenetic abnormalities that have been investigated in EBV‐associated GC( 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 ) (Table 2). CpG islands are often located in the promoter or 5′‐exon sequences of the coding genes, and their methylation leads to the repression of transcription, inactivating the function of downstream genes. CpG‐island methylation, particularly that of tumor suppressor genes, is now considered as the representative epigenetic abnormality in cancer.( 44 ) The term CIMP was primarily introduced to describe colon carcinomas showing high activity of CpG‐island methylation, and the phenotype was determined by the methylation of two or more MINT (methylation in tumor) genes.( 45 ) To explore the methylated genes specific to GC, Kaneda et al. used methylation‐sensitive representational difference analysis and identified five genes, the promoter regions of which were densely methylated (LOX, HRASLS, FLNC, HAND1 and THBD).( 46 ) By adopting these genes as indicators and using the methylation‐specific polymerase chain method (MSP), we classified GC into three subgroups, CIMP none, intermediate and high (CIMP‐N, I and H), by the numbers of methylated loci, 0, 1–3, and 4 or more, respectively.( 41 ) As a result, nearly all cases (14 of 15) of EBV‐associated GC exhibited CIMP‐H phenotype and concurrently showed frequent CpG‐island methylation among 10 other cancer‐related genes (p14ARF , p15, p16INK4A , p73, TIMP3, E‐cadherin, DAPK, GSTP1, hMLH1 and MGMT). EBV‐associated GC showed significantly higher frequencies of methylation of cancer‐related genes (mean number ± SD = 6.9 ± 1.5) even when compared with EBV‐negative/CIMP‐H GC (3.5 ± 1.8). Thus, in view of quantitative aspects, CpG‐island methylation is characteristically global in EBV‐associated GC. Furthermore, there are several genes, such as p73( 37 ) and Hoxa10,( 43 ) which are specifically methylated in EBV‐associated GC, while the frequency of hMLH1 methylation is similarly low compared with EBV‐negative GC.( 38 , 39 , 40 , 41 ) Thus, these facts suggest that the methylation machinery of EBV‐associated GC is capable of recognizing its own methylation targets.
Table 2.
Genetic and epigenetic abnormalities in Epstein–Barr virus (EBV)‐associated gastric carcinoma (GC)

There are several noteworthy findings regarding methylation. A linkage between promoter methylation and gene silencing is usually inconsistent in EBV‐negative GC, but the repression of gene expression faithfully corresponds to the promoter methylation of p16INK4A ,( 47 ) E‐cadherin,( 48 ) and p73 genes( 37 ) in EBV‐associated GC, respectively. When the methylation state of p14ARF was evaluated with bisulfite sequencing,( 49 ) methylation was observed in all 29 CpG sites of p14ARF and 16 sites of p16INK4A with equally high densities in EBV‐associated GC. On the other hand, in EBV‐negative GC, the methylation profiles differed between the two genes. Promoter methylation was sporadic and variable in p14ARF , and only the last position of CpG in p14ARF was methylated at a high frequency. High‐density methylation in p16INK4A was observed in a subset of GC, but the first position of CpG was never methylated in EBV‐negative GC. These findings reinforce the theory that the epigenetic abnormalities are not stochastic, but are primary defects of EBV‐associated GC.
DNA hypermethylation in the promoter region has also been observed in other EBV‐associated neoplasms, such as NPC,( 50 ) NK/T cell lymphomas,( 51 ) Hodgkin lymphoma,( 52 ) as well as tumors infected with other viruses, such as HTLV1, hepatitis B and C virus (HBV, HCV) and human papilloma virus (HPV).( 53 ) Therefore, CpG‐island methylation of host cells might be a distinct mechanism of survival and propagation in human viruses. Progress in current epigenetic research will eventually reveal how the viruses elicit crosstalk between DNA methylation and histone codes, or between DNA methyltransferases (DNMT) and the Polycomb group (PcG) in stem cells, leading to the development of carcinomas (Fig. 3). One promising clue is up‐regulation of DNMT family proteins by viral gene products.( 54 ) DNMT is the enzyme that adds a methyl group to the 5′ position of the cytosine ring in the CpG dinucleotides. A methyl‐binding protein (MeCP2) forms a complex with histone deacetylase (HDAC) and a transcriptional repressor (Sin‐3), which links CpG methylation and modification of chromatin structure, and subsequent gene silencing. DNMT1 is responsible for copying and maintaining methylation patterns after DNA replication, while DNMT3a and 3b function as de novo methyltransferases. Recently, however, DNMT1 has also been suggested to play an essential role in aberrant de novo methylation in various carcinomas. HPV‐16 E7 stimulates methyltransferase activity of DNMT1 through direct interaction.( 55 ) In addition, hepatitis B virus X protein (HBx) activates a positive circuit mechanism leading to E2F1‐mediated dnmt1 up‐regulation in hepatocellular carcinoma.( 56 ) Tsai et al. demonstrated that the EBV‐LMP1 directly activates dnmt1–P1 promoter via the c‐jun NH2 terminal kinase (JNK)‐activator protein‐1 (AP‐1) pathway in NPC.( 57 ) Chromatin immunoprecipitation assay in LMP1‐inducible cells disclosed that LMP1 induces formation of a transcriptional repression complex (DNMT1 and HDAC) on the E‐cadherin gene promoter. Since DNMT1 over‐expression was also shown in EBV‐associated GC( 58 ) that is devoid of LMP1 expression, further studies are necessary to identify the mechanism specific to EBV‐associated GC.
Figure 3.
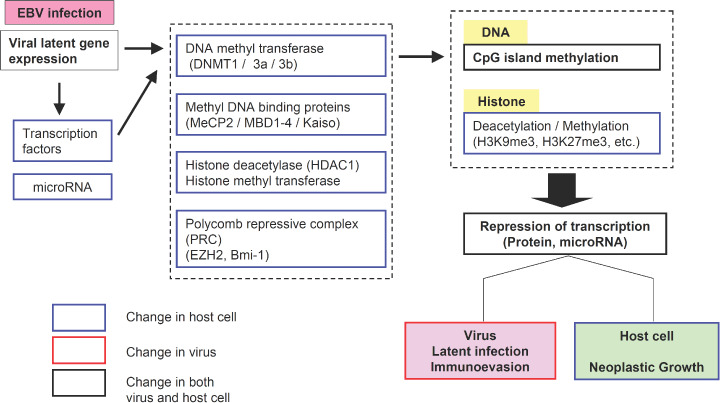
Factors associated with epigenetic abnormality in virus‐associated neoplasms. The possible mechanisms of epigenetic abnormalities that have been reported include direct interaction of viral proteins with DNA methyl transferases (DNMT), up‐regulation of dnmt genes by viral proteins, and increased expression of polycomb group proteins.
DNA methylation is utilized as a host defense mechanism against foreign DNA to suppress the expression of viral genes, but on the other hand, the methylation of the promoter regions of EBV‐latent genes plays a critical role in the transition from the initial infection to the latent infection;( 59 , 60 ) for example, the W‐promoter (Wp) of EBNA2 in EBV‐DNA is active immediately after initial infection. EBNA2 then activates EBV C‐promoter (Cp), and the Wp is soon after silenced by de novo methylation. Cp is also silenced by methylation in the prolonged infection, when the latent genes are driven by the Q‐promoter (Qp). LMP1 is also methylation sensitive and is silenced in a host‐cell‐dependent manner. These epigenetic modifications of EBV‐genes ultimately serve in the escape of the infected cells from cytotoxic T‐cells, and the possible overdriving of defense mechanisms to the cellular genes might cause the transformation of host cells. Further questions regarding methylation in EBV‐associated neoplasms are (i) whether the methylation machineries against viral DNA and host DNA are similar or distinct, and (ii) how the specific DNA sequences are selected.
Other topics that should be investigated
In order to fully understand the developmental process of EBV‐associated GC, further examination of various topics is essential; therefore, in this review we have prepared a list and brief description of those topics.
Interaction with tumor‐infiltrating cells. Recently, global analysis of gene expression profiles has been applied to various carcinomas. In one such study, the expression levels of 326 genes showed a significant association with EBV infection, but a considerable number of them appeared to be related to the lymphoid infiltrate.( 61 ) This fact illustrates the inherent difficulty in investigating the expression profile of carcinomas accompanied with tumor‐associated inflammatory cells. A SCID‐mice transplantable strain of EBV‐associated GC (KT‐strain)( 62 ) lacks these infiltrating cells, and represents the prototype of the gene‐expression profile of neoplastic cells in vivo. According to GeneChip (a high‐density oligonucleotide array) analysis, we identified IL1‐β gene up‐regulation among the cytokine‐related genes and confirmed its characteristic expression by ISH in surgically resected tissues of EBV‐associated GC.( 63 ) IL1‐β has the capacity to inhibit acid secretion from the stomach( 64 ) and to promote the growth of GC cell lines. In addition, IL1‐β plays a role in the recruit of a large number of ineffective T‐lymphocytes, which may counteract the specific cytotoxic T‐lymphocytes against EBV‐infected epithelial cells. Several molecules, such as certain types of chemokine receptors, are also utilized to modulate interaction of neoplastic cells with infiltrating inflammatory cells (Shintani Y et al. unpublished data).
Addendum for viral gene products and cellular proteins. According to Lee et al.( 65 ) EBV‐associated GC can be classified into two clusters by the expression profiles of tumor‐associated proteins. Cluster 2, characterized by loss of smad4 and PTEN, showed a more advanced stage and worse prognosis, although further studies are required to confirm their results. The molecules, which affect the behavior of EBV‐associated GC, include variant isoforms of CD44( 66 ) and a growth factor, IGF1,( 67 ) which was shown to be up‐regulated by small RNA molecules of EBV (EBER). Other viral gene products, which have not yet been fully characterized, include BARF0 and BARF1( 68 , 69 ) and microRNA.( 70 )
EBV infection in epithelial cells. It has not been clarified how EBV infection is established in stomach epithelial cells. EBV infections of B‐lymphocytes and epithelial cells occur with different subsets of EBV‐envelope proteins in the process of fusion of the virus with the cell membrane.( 71 ) Since cocultivation of virus producers shows higher efficiency of infection (up to 800‐fold) than cell‐free infection, direct cell‐to‐cell contact is the most likely mode of viral spread to the epithelial cells in vivo. ( 23 ) On the other hand, EBV infection is rarely identified in the non‐neoplastic epithelium of the stomach, as reported in a previous study using EBER‐ISH.( 8 ) There is a possibility of a different form of latent infection (latency 0), in which the latent gene expression is extremely repressed similar to that of circulating EBV‐infected B‐lymphocytes.( 9 , 10 )
A recent case report on EBV‐associated GC in a patient with bone marrow transplantation for the treatment of multiple myeloma( 72 ) has raised controversies regarding EBV mucosal infection. The EBV infection from the donor lymphocytes was likely to occur in the damaged epithelial cells of the stomach, but the carcinoma may have originated from donor‐derived stem cells through reprogramming( 3 ) or cell fusion with the recipient's stem cells.( 73 ) In either case, the incubation time for the development of EBV‐associated GC might be considerably short.
Field cancerization and risk factors. The incidence of multiple carcinomas is higher in EBV‐associated GC than in EBV‐negative carcinomas.( 7 ) Clonal analysis of EBV, as mentioned above, demonstrates different EBV clones in independent foci of the same stomach, indicating that EBV‐associated GC develops at multiple sites independently. These facts suggest that the stomachs bearing EBV‐associated GC may have been conditioned to develop gastric carcinomas by EBV infection (field cancerization). The background gastric mucosa in EBV‐associated GC is characterized by atrophic gastritis and lymphocyte infiltration.( 74 ) Since the proportion of EBV involvement is significantly high after a primary gastrectomy for benign gastric diseases (27–42%),( 75 ) gastritis cystica polyposa, frequently observed in the remnant stomach, might be an additional precursor lesion in the development of EBV‐associated GC.
According to the epigenetic progenitor model( 76 )‘tumor‐progenitor genes’ promote epigenetic disruption of stem/progenitor cells as a first step in the development of cancer. Chronic inflammation expands the pools of epigenetically altered progenitor cells, which are prone to the carcinoma. Although this fact has been confirmed in atrophic gastritis with H. pylori infection, there was no correspondence of quantity and quality of epigenetic burden between CIMP‐H GC (including EBVaGC) and its background mucosa.( 37 , 77 ) Further studies are necessary to identify tumor‐progenitor genes, epigenetic markers of cancer‐prone progenitors and the mechanisms promoting epigenetic abnormalities in the stomach progenitors, e.g. H. pylori's oncoprotein, CagA( 78 ) or host factors such as single‐nucleotide polymorphisms (SNPs) in cytokine genes.( 64 , 79 )
Therapeutic approaches for EBV‐associated GC. It is worthwhile to introduce several therapeutic approaches that utilize virus–host interactions in EBV‐associated neoplasms.( 50 ) Since the viral replication is inhibited by the promoter methylation in the EBV‐DNA, the demethylating agents, such as 5‐aza cytidine, can recover the lytic infection of EBV, leading to the cell lysis of the infected cells. The approach has merit particularly in EBV‐associated GC, in which methylation of the tumor suppressor gene is also a key abnormality. The efficiency of drug delivery and the side‐effects of the demethylating agents are possible drawbacks of this approach. Another promising approach is to activate viral thymidine kinase by a proteosome inhibitor, such as bortezomib.( 80 ) The reactivation coupled with the administration of radio‐labeled substrate would theoretically result in specific targeting of the radioactive end‐product to the EBV‐associated neoplasm.
Concluding remarks
EBV‐associated GC is a distinct subgroup of GC, consisting of monoclonal growth of neoplastic cells with latent infection by EBV. The possible sequence of events within the mucosa (Fig. 4) is EBV infection of certain gastric stem cells, expression of viral latent genes, abnormality of signal pathways caused by LMP2A, DNA methylation‐mediated repression of tumor suppressor genes, growth of the predominant clone and interaction with other etiologic factors (Fig. 4). EBV plays a primary role in these processes and the investigation of this process will lead to resolution of the 100‐year enigma of ‘GC caused by infectious agents’.
Figure 4.
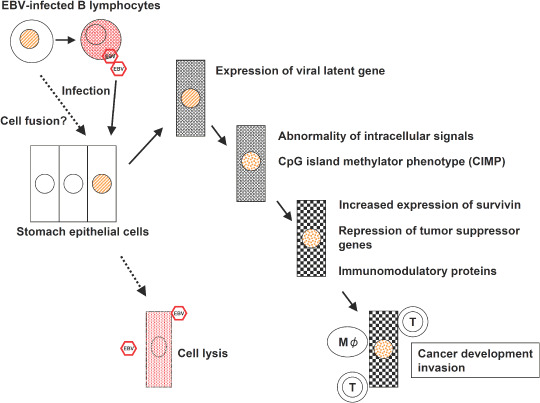
Epstein–Barr virus (EBV)‐associated gastric carcinoma, from EBV infection to the development of carcinoma. A possible sequence of events within the mucosa is illustrated.
Acknowledgments
This work was supported by Grant‐in aid for Scientific Research on Priority Areas (18390110 and 17790229) from the Ministry of Education, Culture, Science, Sports, and Technology of Japan.
References
- 1. IRC monograph on the evaluation of carcinogenic risks to humans. Schistosomes, Liver Flukes and Helicobactor Pylori . Lyon: IRC, 1994; 177–240. [PMC free article] [PubMed] [Google Scholar]
- 2. Weinberg RA. The Biology of Cancer. New York: Garland Science, 2007; 61. [Google Scholar]
- 3. Correa P, Houghton J. Carcinogenesis of Helicobactor pylori . Gastroenterology 2007; 133: 659–72. [DOI] [PubMed] [Google Scholar]
- 4. Hatakeyama M. Helicobacter pylori CagA – a bacterial intruder conspiring gastric carcinogenesis. Int J Cancer 2006; 119: 1217–23. [DOI] [PubMed] [Google Scholar]
- 5. Imai S, Koizumi S, Sugiura M et al . Gastric carcinoma: monoclonal epithelial malignant cells expressing Epstein–Barr virus latent infection protein. Proc Natl Acad Sci USA 1994; 91: 9131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fukayama M, Hayashi Y, Iwasaki Y et al . Epstein–Barr virus‐associated gastric carcinoma and Epstein–Barr virus infection of the stomach. Laboratory Invest 1994; 71: 73–81. [PubMed] [Google Scholar]
- 7. Uozaki H, Fukayama M. Epstein–Barr virus associated gastric carcinoma‐viral carcinogenesis through epigenetic mechanisms. Int J Clin Exp Pathol 2008; 1: 198–216. [PMC free article] [PubMed] [Google Scholar]
- 8. Epstein MA, Barr YM, Achong BG. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet 1964; 15: 702–3. [DOI] [PubMed] [Google Scholar]
- 9. Young LS, Rickinson AB. Epstein–Barr virus: 40 years on. Nat Rev Cancer 2004; 4: 757–68. [DOI] [PubMed] [Google Scholar]
- 10. Kutok JL, Wang F. Spectrum of Epstein–Barr virus‐associated diseases. Annu Rev Pathol Mech Dis 2006; 1: 375–404. [DOI] [PubMed] [Google Scholar]
- 11. Fukayama M, Ibuka T, Hayashi Y, Ooba T, Koike M, Mizutani S. Epstein–Barr virus in pyothorax‐associated pleural lymphoma. Am J Pathol 1993; 143: 1044–9. [PMC free article] [PubMed] [Google Scholar]
- 12. Akiba S, Koriyama C, Herrera‐Goepfert R, Eizuru Y. Epstein–Barr virus associated gastric carcinoma: epidemiological and clinicopathological features. Cancer Sci 2008; 99: 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Watanabe H, Enjoji M, Imai T. Gastric carcinoma with lymphoid stroma. Its morphologic characteristics and prognostic correlations. Cancer 1976; 38: 232–43. [DOI] [PubMed] [Google Scholar]
- 14. Nakamura S, Ueki T, Yao T, Ueyama T, Tsuneyoshi M. Epstein–Barr virus in gastric carcinoma with lymphoid stroma. Special reference to its detection by the polymerase chain reaction and in situ hybridization in 99 tumors, including a morphologic analysis. Cancer 1994; 73: 2239–49. [DOI] [PubMed] [Google Scholar]
- 15. Matsunou H, Konishi F, Hori H et al . Characteristics of Epstein–Barr virus‐associated gastric carcinoma with lymphoid stroma in Japan. Cancer 1996; 77: 1998–2004. [DOI] [PubMed] [Google Scholar]
- 16. Tokunaga M, Land CE, Uemura Y, Tokudome T, Tanaka S, Sato E. Epstein–Barr virus in gastric carcinoma. Am J Pathol 1993; 143: 1250–4. [PMC free article] [PubMed] [Google Scholar]
- 17. Arikawa J, Tokunaga M, Satoh E, Tanaka S, Land CE. Morphological characteristics of Epstein–Barr virus‐related early gastric carcinoma: A case‐control study. Pathol Int 1997; 47: 360–7. [DOI] [PubMed] [Google Scholar]
- 18. Uozaki H, Chong JM, Fujimoto E et al . Soft and hard keratin expression in Epstein–Barr‐virus‐associated gastric carcinoma. Anticancer Res 2005; 25: 3183–90. [PubMed] [Google Scholar]
- 19. Barua RR, Uozaki H, Chong JM et al . Phenotype analysis by MUC2, MUC5AC, MUC6, and CD10 expression in Epstein–Barr virus‐associated gastric carcinoma. J Gastroenterol 2006; 41: 733–9. [DOI] [PubMed] [Google Scholar]
- 20. Shimizu N, Tanabe‐Tochikura A, Kuroiwa Y, Takada K. Isolation of Epstein–Barr virus (EBV)‐negative cell clones from the EBV‐positive Burkitt's lymphoma (BL) line Akata. J Virol 1994; 68: 6069–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hino R, Uozaki H, Inoue Y et al . Survival advantage of EBV‐associated gastric carcinoma: survivin up‐regulation by viral latent membrane protein 2A. Cancer Res 2008; 68: 1427–35. [DOI] [PubMed] [Google Scholar]
- 22. Imai S, Nishikawa J, Takada K. Cell‐to‐cell contact as an efficient mode of Epstein–Barr virus infection of diverse human epithelial cells. J Virol 1998; 72: 4371–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ohfuji S, Osaki M, Tsujitani S, Ikeguchi M, Sairenji T, Ito H. Low frequency of apoptosis in Epstein–Barr virus‐associated gastric carcinoma with lymphoid stroma. Int J Cancer 1996; 68: 710–15. [DOI] [PubMed] [Google Scholar]
- 24. Brinkmann MM, Schuz TF. Regulation of intracellular signaling by terminal membrane proteins of members of the. Gammaherpesvirinae J General Virol 2006; 87: 1047–74. [DOI] [PubMed] [Google Scholar]
- 25. Longan L, Longnecker R. Epstein–Barr virus latent membrane protein 2A has no growth‐altering effects when expressed in differentiating epithelia. J General Virol 2000; 81: 2245–52. [DOI] [PubMed] [Google Scholar]
- 26. Scholle F, Bendt KM, Raab Traub N. Epstein–Barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt. J Virol 2000; 74: 10681–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morrison JA, Klingelhutz AJ, Raab Traub N. Epstein–Barr virus latent membrane protein 2A activates beta‐catenin signaling in epithelial cells. J Virol 2003; 77: 12276–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fukuda M, Longnecker R. Latent membrane protein 2A inhibits transforming growth factor‐beta 1‐induced apoptosis through the phosphatidylinositol 3‐kinase/Akt pathway. J Virol 2004; 78: 1697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fukuda M, Longnecker R. Epstein–Barr virus latent membrane protein 2A mediates transformation through constitutive activation of the Ras/PI3‐K/Akt Pathway. J Virol 2007; 81: 9299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jo H, Zhang R, Zhang H et al . NF–κB is required for H‐ras oncogene induced abnormal cell proliferation and tumorigenesis. Oncogene 2000; 19: 841–9. [DOI] [PubMed] [Google Scholar]
- 31. Anderson LJ, Longnecker R. An auto‐regulatory loop for EBV LMP2A involves activation of Notch. Virology 2008; 371: 257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zur Hausen A, Van Grieken NC, Meijer GA et al . Distinct chromosomal aberrations in Epstein–Barr virus‐carrying gastric carcinomas tested by comparative genomic hybridization. Gastroenterology 2001; 121: 612–18. [DOI] [PubMed] [Google Scholar]
- 33. Chan WY, Liu Y, Li CY et al . Recurrent genomic aberrations in gastric carcinomas associated with Helicobacter pylori and Epstein–Barr virus. Diagn Mol Pathol 2002; 11: 127–34. [DOI] [PubMed] [Google Scholar]
- 34. Chong JM, Fukayama M, Hayashi Y et al . Microsatellite instability in the progression of gastric carcinoma. Cancer Res 1994; 54: 4595–7. [PubMed] [Google Scholar]
- 35. Van Rees BP, Caspers E, Zur Hausen A et al . Different pattern of allelic loss in Epstein–Barr virus‐positive gastric cancer with emphasis on the p53 tumor suppressor pathway. Am J Pathol 2002; 161: 1207–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chang MS, Lee HS, Kim HS et al . Epstein–Barr virus and microsatellite instability in gastric carcinogenesis. J Pathol 2003; 199: 447–52. [DOI] [PubMed] [Google Scholar]
- 37. Ushiku T, Chong JM, Uozaki H et al . p73 gene promoter methylation in Epstein–Barr virus‐associated gastric carcinoma. Int J Cancer 2007; 120: 60–6. [DOI] [PubMed] [Google Scholar]
- 38. Kusano M, Toyota M, Suzuki H et al . Genetic, epigenetic, and clinicopathologic features of gastric carcinomas with the CpG island methylator phenotype and an association with Epstein–Barr virus. Cancer 2006; 106: 1467–79. [DOI] [PubMed] [Google Scholar]
- 39. Kang GH, Lee S, Kim WH et al . Epstein–Barr virus‐positive gastric carcinoma demonstrates frequent aberrant methylation of multiple genes and constitutes CpG island methylator phenotype‐positive gastric carcinoma. Am J Pathol 2002; 160: 787–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chang MS, Uozaki H, Chong JM et al . CpG island methylation status in gastric carcinoma with and without infection of Epstein–Barr virus. Clin Cancer Res 2006; 12: 2995–3002. [DOI] [PubMed] [Google Scholar]
- 41. Enomoto S, Maekita T, Tsukamoto T et al . Lack of association between CpG island methylator phenotype in human gastric cancers and methylation in their background non‐cancerous gastric mucosae. Cancer Sci 2007; 98: 1853–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chong JM, Sakuma K, Sudo M et al . Global and non‐random CpG‐island methylation in gastric carcinoma associated with Epstein–Barr virus. Cancer Sci 2003; 94: 76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kang GH, Lee S, Cho NY et al . DNA methylation profiles of gastric carcinoma characterized by quantitative DNA methylation analysis. Lab Invest 2008; 88: 161–70. [DOI] [PubMed] [Google Scholar]
- 44. Esteller M. Epigenetics in cancer. N Engl J Med 2008; 358: 1148–59. [DOI] [PubMed] [Google Scholar]
- 45. Toyota M, Ahuja N, Ohe Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 1999; 96: 8681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kaneda A, Kaminishi M, Yanagihara K, Sugimura T, Ushijima T. Identification of silencing of nine genes in human gastric cancers. Cancer Res 2002; 62: 6645–50. [PubMed] [Google Scholar]
- 47. Osawa T, Chong JM, Sudo M et al . Reduced expression and promoter methylation of p16 gene in Epstein–Barr virus‐associated gastric carcinoma. Jap J Cancer Res 2002; 93: 1195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sudo M, Chong JM, Sakuma K et al . Promoter hypermethylation of E‐cadherin and its abnormal expression in Epstein–Barr virus‐associated gastric carcinoma. Int J Cancer 2004; 109: 194–9. [DOI] [PubMed] [Google Scholar]
- 49. Sakuma K, Chong JM, Sudo M et al . High‐density methylation of p14ARF and p16INK4A in Epstein–Barr virus‐associated gastric carcinoma. Int J Cancer 2004; 112: 273–8. [DOI] [PubMed] [Google Scholar]
- 50. Tao Q, Chan AT. Nasopharyngeal carcinoma: molecular pathogenesis and therapeutic developments. Expert Rev Mol Med 2007; 9: 1–24. [DOI] [PubMed] [Google Scholar]
- 51. Siu LL, Chan JK, Wong KF, Kwong YL. Specific patterns of gene methylation in natural killer cell lymphomas: p73 is consistently involved. Am J Pathol 2002; 160: 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dutton A, Woodman CB, Chukwuma MB et al . Bmi‐1 is induced by the Epstein–Barr virus oncogene LMP1 and regulates the expression of viral target genes in Hodgkin lymphoma cells. Blood 2007; 109: 2597–603. [DOI] [PubMed] [Google Scholar]
- 53. Flanagan JM. Host epigenic modifications by oncogenic viruses. Br J Cancer 2007; 96: 183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kanai Y, Hirohashi S. Alterations of DNA methylation associated with abnormalities of DNA methyltransferases in human cancers during transition from a precancerous to a malignant state. Carcinogenesis 2007; 28: 2434–42. [DOI] [PubMed] [Google Scholar]
- 55. Burgers WA, Blanchon L, Pradhan S, De Launoit Y, Kouzarides T, Fuks F. Viral oncoproteins target the DNA methyltransferases. Oncogene 2007; 26: 1650–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jung JK, Arora P, Pagano JS, Jang KL. Expression of DNA methyltransferase 1 is activated by hepatitis B virus X protein via a regulatory circuit involving the p16INK4a‐cyclin D1‐CDK 4/6‐pRb‐E2F1 pathway. Cancer Res 2007; 67: 5771–8. [DOI] [PubMed] [Google Scholar]
- 57. Tsai CL, Li HP, Lu YJ et al . Activation of DNA methyltransferase 1 by EBV LMP1 Involves c‐Jun NH2‐terminal kinase signaling. Cancer Res 2006; 66: 11668–76. [DOI] [PubMed] [Google Scholar]
- 58. Etoh T, Kanai Y, Ushijima S et al . Increased DNA methyltransferase 1 (DNMT1) protein expression correlates significantly with poorer tumor differentiation and frequent DNA hypermethylation of multiple CpG islands in gastric cancers. Am J Pathol 2004; 164: 689–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li H, Minarovits J. Host cell‐dependent expression of latent Epstein–Barr virus genomes: regulation by DNA methylation. Adv Cancer Res 2003; 89: 133–56. [DOI] [PubMed] [Google Scholar]
- 60. Lieberman PM. Chromatin organization and virus gene expression. J Cell Physiol 2008; 216: 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chen X, Leung SY, Yuen ST et al . Variation in gene expression patterns in human gastric cancers. Mol Biol Cell 2003; 14: 3208–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Iwasaki Y, Chong JM, Hayashi Y et al . Establishment and characterization of a human Epstein–Barr virus‐associated gastric carcinoma in SCID mice. J Virol 1998; 72: 8321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chong JM, Sakuma K, Sudo M et al . Interleukin‐1beta expression in human gastric carcinoma with Epstein–Barr virus infection. J Virol 2002; 76: 6825–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. El‐Omar EM, Carrington M, Chow WH et al . Interleukin‐1 polymorphisms associated with increased risk of gastric cancer. Nature 2000; 404: 398–402. [Erratum in: Nature 2001; 412: 99]. [DOI] [PubMed] [Google Scholar]
- 65. Lee HS, Chang MS, Yang HK, Lee BL, Kim WH. Epstein–Barr virus‐positive gastric carcinoma has a distinct protein expression profile in comparison with Epstein–Barr virus‐negative carcinoma. Clin Cancer Res 2004; 10: 1698–705. [DOI] [PubMed] [Google Scholar]
- 66. Chong JM, Fukayama M, Hayashi Y et al . Expression of CD44 variants in gastric carcinoma with or without Epstein–Barr virus. Int J Cancer 1997; 74: 450–4. [DOI] [PubMed] [Google Scholar]
- 67. Iwakiri D, Eizuru Y, Tokunaga M, Takada K. Autocrine growth of Epstein–Barr virus‐positive gastric carcinoma cells mediated by an Epstein–Barr virus‐encoded small RNA. Cancer Res 2003; 63: 7062–7. [PubMed] [Google Scholar]
- 68. Zur Hausen A, Brink AA, Craanen ME, Middeldorp JM, Meijer CJ, Van Den Brule AJ. Unique transcription pattern of Epstein–Barr virus (EBV) in EBV‐carrying gastric adenocarcinomas: expression of the transforming BARF1 gene. Cancer Res 2000; 60: 2745–8. [PubMed] [Google Scholar]
- 69. Seto E, Yang L, Middeldorp J et al . Epstein–Barr virus (EBV)‐encoded BARF1 gene is expressed in nasopharyngeal carcinoma and EBV‐associated gastric carcinoma tissues in the absence of lytic gene expression. J Med Virol 2005; 76: 82–8. [DOI] [PubMed] [Google Scholar]
- 70. Kim DN, Chae HS, Oh ST et al . Expression of viral microRNAs in Epstein–Barr virus‐associated gastric carcinoma. J Virol 2007; 81: 1033–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Borza CM, Hutt Fletcher LM. Alternate replication in B cells and epithelial cells switches tropism of Epstein–Barr virus. Nat Med 2002; 8: 594–9. [DOI] [PubMed] [Google Scholar]
- 72. Au WY, Pang A, Chan EC, Chu KM, Shek TW, Kwong YL. Epstein–Barr virus‐related gastric adenocarcinoma: an early secondary cancer post hemopoietic stem cell transplantation. Gastroenterology 2005; 129: 2058–63. [DOI] [PubMed] [Google Scholar]
- 73. Duelli D, Lazebnik Y. Cell‐to‐cell fusion as a link between viruses and cancer. Nat Rev Cancer 2007; 7: 968–76. [DOI] [PubMed] [Google Scholar]
- 74. Kaizaki Y, Sakurai S, Chong JM, Fukayama M. Atrophic gastritis, Epstein–Barr virus infection, and Epstein–Barr virus‐associated gastric carcinoma. Gastric Cancer 1999; 2: 101–8. [DOI] [PubMed] [Google Scholar]
- 75. Kaizaki Y, Hosokawa O, Sakurai S, Fukayama M. Epstein–Barr virus‐associated gastric carcinoma in the remnant stomach: de novo and metachronous gastric remnant carcinoma. J Gastroenterol 2005; 40: 570–7. [DOI] [PubMed] [Google Scholar]
- 76. Ushijima T. Epigenetic field for cancerization. J Biochem Mol Biol 2007; 40: 142–50. [DOI] [PubMed] [Google Scholar]
- 77. Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet 2006; 7: 21–33. [DOI] [PubMed] [Google Scholar]
- 78. Ohnishi N, Yuasa H, Tanaka S et al . Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci USA 2008; 105: 1003–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sakuma K, Uozaki H, Chong JM et al . Cancer risk to the gastric corpus in Japanese, its correlation with interleukin‐1beta gene polymorphism (+3953*T) and Epstein–Barr virus infection. Int J Cancer 2005; 115: 93–7. [DOI] [PubMed] [Google Scholar]
- 80. Fu DX, Tanhehco YC, Chen J et al . Bortezomib‐induced enzyme‐targeted radiotherapy in herpesvirus‐associated tumors. Nat Med in press. [DOI] [PMC free article] [PubMed] [Google Scholar]