

Abstract
Cisplatin, a commonly used chemotherapeutic agent, causes tumor cell death by producing DNA damage and generating reactive oxygen intermediates, which have been reported to activate the early growth response‐1 (Egr‐1) promoter through specific cis‐acting sequences, termed CArG elements. The aim of this study was to construct an adenoviral vector containing CArG elements cloned upstream of the cDNA for human wt‐p53, and to observe the effect of this vector on human non‐small cell lung cancer (NSCLC) xenografts in athymic nude mice when combined with cisplatin treatment. The adenoviral vector AdEgr–p53 was generated by inserting CArG elements upstream of human wt‐p53 cDNA. Two human NSCLC cell lines of varying p53 gene status, A549 (containing wild‐type p53) and H358 (containing an internal homozygous deletion of the p53 gene) were used for in vitro and in vivo experiments. Wt‐p53 production in cultured tumor cells and xenografts treated with the combination of AdEgr–p53 and cisplatin were detected by enzyme‐linked immunosorbent assays. The antitumor responses in nude mice with the A549 or H358 xenografts following treatment with AdEgr–p53 and cisplatin were observed. We found that p53 was produced in tumor cells and xenografts treated with a combination of AdEgr–p53 and cisplatin. Furthermore, the Egr‐1 promoter is induced by cisplatin, and this induction is mediated in part through the CArG elements. There was an enhanced antitumor response without an increase in toxicity following treatment with AdEgr–p53 and cisplatin, compared with either agent alone. Cisplatin‐inducible p53 gene therapy may provide a means to control transgene expression while enhancing the effectiveness of commonly used chemotherapeutic agents. This is a novel treatment for human NSCLC. (Cancer Sci 2005; 96: 706 – 712)
Abbreviations:
- Egr‐1
early growth response‐1
- IR
ionizing radiation
- i.t.
intratumoral
- LA
luciferase activity
- NS
normal saline
- ROI
reactive oxygen intermediate.
Lung cancer is a common cause of cancer deaths worldwide.( 1 ) Conventional treatments are not adequate for the majority of lung cancer patients. Attempts to overcome drug resistance with higher doses of radiation and chemotherapeutic agents inevitably result in an unacceptable degree of toxicity and bystander damage to normal tissues.( 2 , 3 ) Novel strategies are needed to further improve the treatment outcome for lung cancer patients.
Radio‐inducible gene therapy is a novel strategy for cancer treatment, in which an ionizing radiation‐inducible regulatory sequence is linked with an adjuvant tumor‐therapeutic gene, and transfected into tumor cells. The expression of the therapeutic gene, therefore, will be induced by radiotherapy. The transfected cancer cells will be destroyed by both radiation and the radiation‐inducible gene. Radio‐inducible CArG [CC(A/T)6GG] DNA elements of the early growth response‐1 (Egr‐1) promoter are widely used as ionizing radiation (IR)‐inducible sequences in radio‐genetic therapy.( 4 , 5 , 6 , 7 , 8 , 9 , 10 )
Mechanistic studies of Egr‐1 induction by IR have demonstrated a role for free radical activation of the CArG elements of the Egr‐1 promoter. The role of reactive oxygen intermediates (ROI) was confirmed by the finding that activation of the Egr‐1 promoter by H2O2 is quantitatively and temporally similar to that obtained with IR. Moreover, treatment with N‐acetyl‐L‐cysteine, a free radical scavenger, decreased Egr‐1 induction by IR or H2O2.( 11 , 12 , 13 ) These findings suggest that activation of the Egr‐1 promoter is mediated by both DNA damage and ROI production.
Cisplatin is a commonly used chemotherapeutic agent that can stimulate ROI generation in cells. In the present study, cisplatin is used to induce the production of p53 in human non‐small cell lung cancer (NSCLC) cells infected with an adenoviral vector encoding the CArG elements of the Egr‐1 promoter ligated upstream to a cDNA encoding wt‐p53. Importantly, significant synergistic antitumor effects between wt‐p53 and cisplatin were observed in these experimental tumors. These findings provide support for a novel approach that combines cisplatin treatment with the temporal and spatial control of gene therapy.
Materials and Methods
Cells and cell culture
Two human NSCLC cell lines, A549 and H358, with varying p53 gene status were used for in vitro and in vivo experiments. The A549 line, which contains wild‐type p53, was maintained in Ham's F12 medium supplemented with 10% fetal calf serum (FCS). The H358 line, which has an internal homozygous deletion of the p53 gene, was maintained in RPMI‐1640 supplemented with 10% FCS and 5% glutamine. All of the cells were incubated in a humidified incubator supplied with 5% carbon dioxide. All of the cell cultures were tested regularly for the presence of Mycoplasma.
Animals
Six‐week‐old female athymic nude mice (Experimental Animal Research Center, Sichuan University, China) received food and water ad libitum. Experiments were in accordance with the guidelines of the Institutional Animal Care and Use Committee of the University of Sichuan.
Plasmids and recombinant adenovirus construction
Plasmid DNA was purified using Qiagen tip‐100 columns (Qiagen, USA). Isolation of DNA fragments was performed with a DNA isolation kit (Qiagen). The wild type promoter of the human Egr‐1 gene was isolated as a 614‐bp fragment by HindIII–XbaI restriction from the plasmid p‐600 (kindly provided by Dr KM Sakamoto, Department of Pediatrics, UCLA School of Medicine). This fragment was subcloned into pUC19 and then inserted into the vector, a BglII–NcoI‐deletion derivative of pGL3‐C (Promega, Berlin, Germany) lacking the original SV40‐promoter. Subsequent intramolecular re‐ligation resulted in the plasmid pEL, in which luc reporter expression is driven only by the activity of the Egr‐1 promoter. The recombinant adenovirus (AV) AdEgr–p53, which carries a wt‐p53 gene under the control of the Egr‐1 promoter was constructed using the AdEasy system (Qbiogene, USA). Briefly, the Egr‐1 promoter was isolated as a 625‐bp HindIII–KpnI fragment, subcloned into a promoterless derivative of the vector pwtp53 (kindly provided by Dr Y‐S He, University of Sichuan), and finally cloned together with wt‐p53 as a 3328‐bp HindIII–AflII (blunt) fragment into the plasmid pShuttle to generate pEp53. Recombinant AV genomes were generated by cotransformation of pEp53 linearized by PmeI, and pAdEasy1, an adenoviral backbone vector, into the Escherichia coli recBC mutant JB5183 with subsequent selection for kanamycin resistant (KanR) clones. After selection and isolation of a correct recombinant AV plasmid (pAdEgr–p53), it was transformed into the E. coli strain DH5 to obtain large amounts of intact pAdEgr–p53 DNA. Recombinant AV was generated by transfecting HEK293 cells with PacI‐linearized pAdEgr–p53 followed by incubation for a further 8–12 days. The isolated recombinant AV (AdEgr–p53) was propagated and concentrated to titers of approximately 1 × 109 p.f.u./mL by subsequently passaging in the same E1A‐transcomplementing competent cell line. As a control vector, AdEasy1–p53 was generated by inserting the wt‐p53 fragment into the AdEasy1 vector. The titer of the concentrated lysates was determined by a plaque‐forming assay.
In vitro measurement of p53 protein
Cells were plated at 105 cells per well in 12‐well plates (Becton Dickinson, Bedford, MA, USA), grown overnight, and infected with AdEgr–p53, AdEasy1–p53 or AdEasy1 at a multiplicity of infection of 100 in serum‐free medium for 2–3 h. Cells in the IR group were exposed to 5 Gy in complete medium using a Pantak PCM 1000 X‐ray generator (Pantak, East Haven, CT, USA). Cells in the cisplatin group were exposed to 5 µM cisplatin in complete medium. Cells and supernatants were harvested at 1, 3, 8, 12 and 24 h by scraping, and human p53 production was quantified using a Quantikine enzyme‐linked immunosorbent assay (elisa) kit (R&D Systems, Minneapolis, MN, USA) following three freeze–thaw lysis cycles. These experiments were performed in triplicate. Duplicate treatment plates were used to adjust for the cytotoxicity of IR and cisplatin. Cells were harvested using versene (0.02% ethylenediamine tetraacetic acid [EDTA] in Hanks’ balanced salt solution) and trypsin–EDTA (0.25% trypsin, 1 mM EDTA 4Na; Invitrogen Life Technologies), and cells were counted using a hemocytometer with trypan blue (0.4%) exclusion (Invitrogen Life Technologies). Protein assays were performed to normalize protein concentration using the Bio‐Rad dye reagent (Bio‐Rad Laboratories, Hercules, CA, USA). Data are expressed as mean ± SEM of experiments performed in triplicate.
Western blot analysis of p53
Approximately 1 × 107 cells were washed twice with phosphate‐buffered saline (PBS) and lysed in 0.65 mL ice‐cold lysis buffer (1 × PBS, 1% NP40, 0.5% sodium deoxycholate, and 0.1% sodium dodecylsulfate [SDS]). The cell lysates were prepared by treating plated cell monolayers with sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) sample buffer. The protein content of the lysates was then determined by Bio‐Rad protein assay. Next, each lane on a SDS‐polyacrylamide (12%) gel was loaded with 60 µg of cell lysate and electrophoresed to separate proteins under reducing conditions for the protein of interest. After being electrophoresed at 120 V for 2 h, the proteins were transferred to high bond‐enhanced chemiluminescence (ECL) membranes (Amersham, Arlington Heights, IL, USA). The membranes were than incubated with the primary and secondary antibodies, and developed according to the Amersham ECL protocol. Actin was used as a control. Antibody to actin (monoclonal) was obtained from Sigma‐Aldrich (St. Louis, MO, USA). Antibody to p53 (monoclonal) was obtained from Dako (Carpinteria, CA, USA).
In vitro luciferase reporter assay
The Egr‐1 constructs pEL (600 bp, containing all CArG elements and no AP‐1 sites) and pE660 (the minimal Egr‐1 promoter of 115 bp, containing no CArG elements)( 12 ) were evaluated following sequence confirmation and insertion of the PCR product into the pGL3 basic firefly luciferase reporter plasmid construct (Promega, Madison, WI, USA) by enzyme restriction and ligation. JM109‐competent cells (Stratagene, La Jolla, CA, USA) were transformed with these plasmids, and endotoxin‐free maxipreps (Qiagen, Valencia, CA, USA) were prepared. Product confirmation was performed by PCR, sequencing, enzyme restriction, and gel electrophoresis. Cells were plated at 105 cells per well in 12‐well plates, and were transfected with the firefly luciferase reporter plasmid constructs (pGL3 basic [promoterless, negative control], pE660 [minimal Egr‐1 promoter], or pEL [Egr‐1 promoter containing all CArG elements]) using the TransFast transfection reagent (Promega). All groups were cotransfected with the Renilla luciferase reporter plasmid construct pRL‐TK (herpes simplex virus thymidine kinase promoter) to normalize transfection efficiency. Forty‐eight hours later, cells were exposed to IR (10 Gy) or cisplatin (25 µM). Cells were harvested 6 h later, and luciferase activity (LA) was measured using the Dual‐Luciferase reporter assay system (Promega).
In vivo measurement of p53 protein
Lung cancer cells (5 × 106 per 0.1 mL) were injected subcutaneously into the right hind limbs of nude mice. Tumor‐bearing mice were randomized to one of six groups: intratumoral (i.t.) AdEasy1 (2 × 108 particle units p.f.u./10 µL) with i.p. normal saline (NS) or cisplatin (3 mg/kg), i.t. AdEasy1–p53 (2 × 108 p.f.u./10 µL) with i.p. NS or cisplatin and i.t. AdEgr–p53 (2 × 108 p.f.u./10 µL) with i.p. NS or cisplatin. i.p. NS or cisplatin treatments were administered 20 h after transfection with the i.t. vector, and two consecutive i.t. and i.p. injections were given. Animals were killed, and xenografts were harvested 48 h after the second i.p. injection. Xenografts were snap‐frozen in liquid nitrogen and homogenized in RIPA buffer (150 mM NaCl, 10 mM Tris at pH 7.5, 5 mM EDTA at pH 7.5, 100 mM PMSF, 1 µg/mL leupeptin, and 2 µg/mL aprotinin) using a Brinkman Polytron homogenizer (Kinematica AG, Lucerne, Switzerland). After three freeze–thaw lysis cycles, the homogenate was centrifuged at 780 g in a Sorvall RC‐5C SS34 rotor (Kendro Laboratory Products, Newtown, CT, USA) for 10 min at 4°C. p53 levels in the supernatants were measured as described above.
Tumor samples were washed twice with cold PBS, and 100–200 mg of tumor tissues were homogenized on ice in a lysis buffer. After centrifugation at 600 g for 10 min at 4°C, the supernatants containing the cellular proteins were used for analysis. Western blotting was performed as described earlier.
In vivo evaluation of tumor growth
Lung cancer cells (5 × 106 per 0.1 mL) were injected subcutaneously into the right hind limbs of nude mice. In the preliminary experiment, to work out the optimal doses, various dosages of virus and cisplatin were used in the treatment of tumor‐bearing mice (data not shown). The dosages as follows were the optimal virus and drug doses for suppressing tumor growth. Tumor‐bearing mice were assigned to one of six groups: i.t. AdEasy1 (2 × 108 p.f.u./10 µL) with i.p. NS or cisplatin (3 mg/kg), i.t. AdEasy1–p53 (2 × 108 p.f.u./10 µL) with i.p. NS or cisplatin and i.t. AdEgr–p53 (2 × 108 p.f.u./10 µL) with i.p. NS or cisplatin. Animals were injected i.p. with NS or cisplatin 20 h after the i.t. vector injection. i.t. and i.p. injections were given for five consecutive days. Xenografts were measured every 2 days using calipers, and tumor volume was calculated as (length × width × thickness)/2. Fractional tumor volumes (V/V 0 where V 0 = volume on day 0) were calculated and plotted. Day 0 is the first day of treatment (i.t. injection vector) and the day that the mice were distributed into treatment groups. Tumor volumes represented on graphs begin on day 0. The performance status and survival rates of mice in six groups were observed over the entire experimental course.
Statistical analysis
Statistical significance was determined using the two‐tailed Student's t‐test.
Results
In vitro induction of p53 in human lung cancer cells following infection with AdEgr–p53 and exposure to cisplatin
No p53 protein was detectable in H358 cell pellets or supernatants from cultures infected with the null vector (AdEasy1) and treated with IR or cisplatin (data not shown). In contrast, significantly increased levels of p53 protein were detected at 1, 3, 8, 12 and 24 h in cultures of H358 cells infected with the AdEgr–p53 vector and exposed to IR (5 Gy) compared with cells infected with vector alone (P < 0.001). Combined treatment with AdEgr–p53 and IR resulted in 1.5‐, 6.3‐, 3.1‐, 1.4‐ and 1.1‐fold increases in p53 production, respectively (rounded to the nearest 0.1). A similar induction of p53 protein was detected in H358 cells infected with the AdEgr–p53 vector and exposed to 5 µM cisplatin (compared with vector alone) for 1, 3, 8, 12 and 24 h (P < 0.001). Combined treatment with AdEgr–p53 and cisplatin thus resulted in 1.4‐, 5.3‐, 7.4‐, 6.3‐ and 3.2‐fold increases in p53 production, respectively. No induction of p53 protein was detected in cells infected with the control vector AdEasyl–p53 when exposed to IR or cisplatin (Fig. 1).
Figure 1.

In vitro measurement of p53 protein. p53 production by AdEgr–p53‐infected cells exposed to IR (5 Gy) or cisplatin (5 µM) was measured using enzyme‐linked immunosorbent assay and western blotting. Significant increases in the levels of p53 protein were detected at 1, 3, 8, 12 and 24 h following exposure to AdEgr–p53 plus IR (P < 0.001) and AdEgr–p53 plus cisplatin (P < 0.001) compared with vector alone in H358 cultures (a,b) and A549 cultures (a,c). Data are reported as the mean ± SEM of three independent experiments.
Comparable experiments were conducted with A549 cell cultures. No p53 protein was detectable in A549 cell pellets or supernatants from cultures infected with the null vector (AdEasy1), and minimal levels of p53 protein were detectable in cells treated with IR or cisplatin (data not shown). Similar results of p53 protein induction were found in A549 cells infected with the AdEgr–p53 or AdEasyl–p53 vector and exposed to IR (5 Gy) or cisplatin (5 µM) (Fig. 1). These findings from the H358 and A549 cell lines demonstrate that IR and cisplatin induce p53 expression.
CArG elements of the Egr‐1 promoter mediate induction of p53 by cisplatin
Minimal LA (expressed as relative luminescence) was detectable in A549 cells transfected with the pGL3 basic plasmid construct (LA = 0.22–0.35) or with the pE660 plasmid construct (LA = 0.46–0.77). However, A549 cells transfected with the pEL plasmid construct exhibited a 2.8‐fold increase (P = 0.005) in relative LA (to 18.27) following exposure to IR (10 Gy) compared with the untreated control (LA = 6.53), and a 2.0‐fold increase (P = 0.005) in LA (to 13.06) following exposure to cisplatin (25 µM) compared with the untreated control (Fig. 2).
Figure 2.
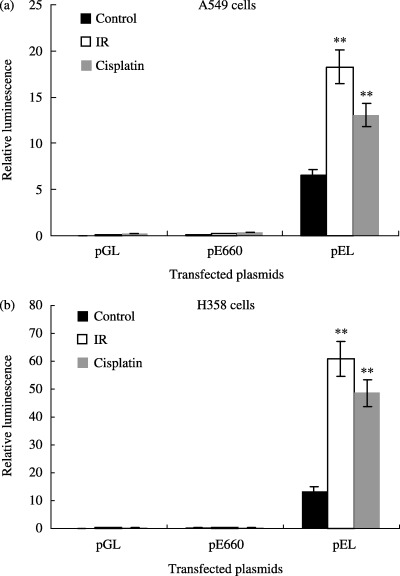
In vitro reporter assays. Luciferase reporter constructs were used to evaluate induction of the Egr‐1 promoter by IR or cisplatin. Minimal LA was detectable following transfection with either the pGL3 basic (negative control) or the pE660 plasmid (minimal Egr‐1 promoter) constructs. (a) In A549 cells transfected with pEL, a 2.8‐fold increase (P = 0.005) in relative LA was observed following exposure to IR (10 Gy), and a 2.0‐fold increase (P = 0.005) was seen following exposure to cisplatin (25 µM). (b) In H358 cells transfected with pEL, there was a 4.5‐fold increase (P = 0.004) in relative LA following exposure to IR (10 Gy), and a 3.6‐fold increase (P = 0.01) following exposure to cisplatin (25 µM). Data are reported as mean ± SEM. **P < 0.01 versus control.
Similar results were obtained with the H358 cell line. Minimal LA was detectable in H358 cells transfected with the pGL3 basic plasmid construct (LA = 0.25–0.35) or with the pE660 plasmid construct (LA = 0.63–1.25). H358 cells transfected with the pEL plasmid construct exhibited a 4.5‐fold increase (P = 0.004) in LA (to 60.84) following exposure to IR (10 Gy) compared with the untreated control (LA = 13.52), and a 3.6‐fold increase (P = 0.01) in LA (to 48.67) following exposure to cisplatin (25 µM) compared with the untreated control (Fig. 2). These data demonstrate that CArG elements of the Egr‐1 promoter are inducible by cisplatin and mediate the transcriptional activation of the chimeric Egr–p53 gene.
Induction of p53 in human lung cancer xenografts following treatment with AdEgr–p53 and cisplatin
No p53 protein was detected in H358 tumor homogenates following injection of the AdEasy1 vector and systemic treatment with either NS or cisplatin (data not shown). A significant increase (3.8‐fold) in i.t. p53 protein was observed following combined treatment with AdEgr–p53 and cisplatin (1385.0 ± 252.7 pg/mg) compared with vector treatment alone (364.5 ± 51.3 pg/mg; P < 0.05; Fig. 3). Minimal p53 protein was detected in A549 tumor homogenates following injection with AdEasy1 vector and systemic treatment with either NS or cisplatin (data not shown). However, a significant increase (3.1‐fold) in i.t. p53 protein was observed following combined treatment with AdEgr–p53 and cisplatin (979.5 ± 53.5 pg/mg) compared with vector treatment alone (315.8 ± 22.7 pg/mg; P < 0.001; Fig. 3). No induction of p53 protein was detected in xenografts treated with the control vector AdEasyl–p53 and cisplatin. These findings demonstrate the in vivo induction of p53 protein by cisplatin and verify that the p53 protein is a product of the AdEgr–p53 vector rather than of the tumor tissue.
Figure 3.
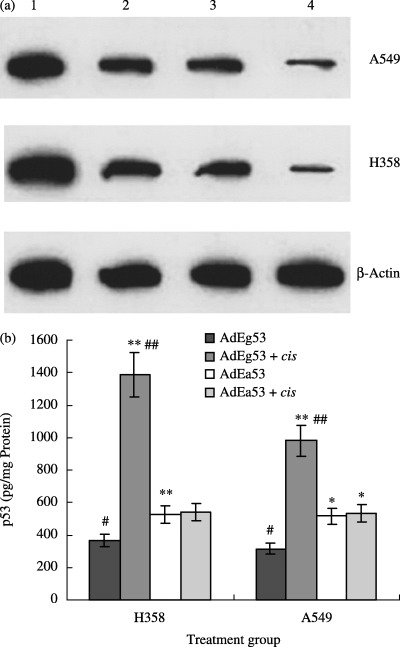
In vivo measurement of p53 protein. p53 production by AdEgr–p53‐injected xenografts was measured by (a) western blotting and (b) enzyme‐linked immunosorbent assay. A significant increase in i.t. p53 protein concentration was observed following combined treatment with AdEgr–p53 and cisplatin compared with treatment with AdEgr–p53 vector alone in H358 (3.8‐fold increase; P < 0.05) and A549 xenografts (3.1‐fold increase; P < 0.001). Data are reported as mean ± SEM. In western blot analysis: 1, AdEg53 + cis; 2, AdEa53 + cis; 3, AdEa53; 4, AdEg53. Versus AdEg53: *P < 0.05, **P < 0.01.
Cisplatin‐inducible AdEgr–p53 enhances treatment of human NSCLC xenografts
In the A549 studies, mean tumor volume on day 0 (initiation of treatment) was 364.8 ± 12.6 mm3 (n = 15 mice per group in each of six treatment groups). Xenografts were injected i.t. with either AdEasy1, AdEasyl–p53 or AdEgr–p53. Mice were injected i.p. with either NS or cisplatin. Control tumors (treated with AdEasy1 plus NS) doubled in size by day 4 and exhibited a 5.4‐fold increase in mean tumor volume by day 14. A similar growth pattern was observed in tumors treated with the AdEgr–p53 vector and NS (a 2.0‐fold increase by day 4 and a 5.1‐fold increase in mean volume by day 14). Significant inhibition of tumor growth was observed in the tumors receiving combined treatment with AdEgr–p53 and cisplatin compared with tumors treated with the null vector and cisplatin on days 4 (P = 0.045), 6 (P < 0.005), 8 (P < 0.002), 10 (P < 0.001), 12 (P < 0.004), and 14 (P < 0.021) after the initiation of treatment. Enhanced tumor growth inhibition was also found in the AdEasyl–p53 plus cisplatin treatment group, but to a lesser extent than for the AdEgr–p53 plus cisplatin group (Fig. 4).
Figure 4.
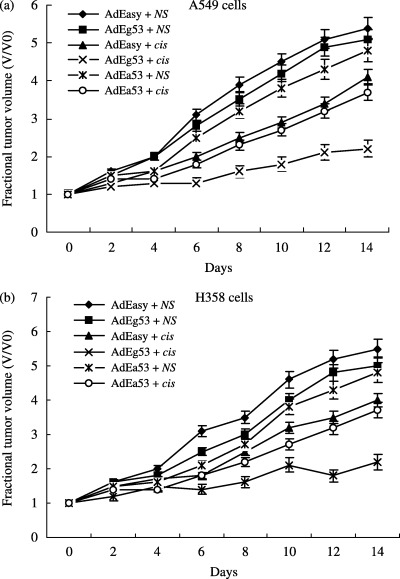
In vivo evaluation of tumor growth. The effect of combined treatment with AdEgr–p53 and cisplatin was evaluated by measuring the volume of xenografts injected with AdEasy1, AdEasy1–p53 or AdEgr–p53 with or without cisplatin. Day 0 represents the first day of treatment. (a) In A549 xenografts, combined treatment with AdEgr–p53 and cisplatin produced significant tumor regression compared with tumors treated with AdEasy1 and cisplatin. Obvious tumor inhibition was also found in the AdEasy1–p53 and cisplatin treatment group, but to a lesser extent than that found in the AdEgr–p53 and cisplatin group. (b) In H358 xenografts, similar tumor growth inhibition was found as for the A549 xenografts. Data are reported as mean ± SEM.
In the H358 studies, mean tumor volume on day 0 was 285.9 ± 8.3 mm3 (n = 12 mice per group in each of six treatment groups). Control tumors (treated with AdEasy1 plus NS) grew steadily, doubling in size by day 4, exhibiting a 5.5‐fold increase in mean tumor volume by day 14. A similar growth pattern was observed in tumors treated with the AdEgr–p53 vector and NS (1.8‐fold increase by day 4 and 5.0‐fold increase in mean volume by day 14). Significant tumor regression was observed in the tumors receiving combined treatment with AdEgr–p53 and cisplatin compared with tumors treated with the null vector and cisplatin on days 4 (P = 0.045), 6 (P < 0.001), 8 (P = 0.048), 10 (P < 0.001), 12 (P < 0.001), and 14 (P = 0.002) (Fig. 4b). Similar results were found in the AdEasyl–p53 and cisplatin treatment groups and the A549 studies.
As shown in Table 1, the survival rates of mice bearing lung cancer xenografts treated with AdEg–p53 and cisplatin were much higher than those of mice treated with AdEg–p53 and NS. Increased survival rates were also found in the AdEg–p53 and cisplatin treatment group compared with mice treated with the AdEasy1 vector or AdEasyl–p53 and cisplatin. Taken together, these data support an antitumor interaction between AdEgr–p53 and cisplatin in xenografts of human lung cancer. These findings are consistent with, and supported by, p53 induction by cisplatin observed in the in vitro and in vivo experiments. Although toxicity was observed after treatment with cisplatin, no additional toxicity was observed following combined treatment with cisplatin and AdEgr–p53.
Table 1.
Survival rates of mice bearing lung cancer xenografts treated with various viral vectors combined with NS or cisplatin (%, mean ± SEM)
| Cell line | AdEasy + NS | AdEasy +cis | AdEa53 + NS | AdEa53 + cis | AdEg53 + NS | AdEg53 + cis |
|---|---|---|---|---|---|---|
| A549 | 33.7 ± 3.5 | 41.2 ± 3.7* | 39.9 ± 5.2 | 46.9 ± 5.5**, † | 35.4 ± 3.6 | 79.1 ± 6.8**, †† |
| H358 | 35.5 ± 4.1 | 44.8 ± 4.3* | 40.5 ± 4.6 | 48.3 ± 5.7**, † | 36.8 ± 4.8 | 84.5 ± 9.2**, †† |
Versus AdEasy + NS: *P < 0.05, **P < 0.01; versus AdEasy +cis: † P < 0.05, †† P < 0.01.
Discussion
The 5′‐CArG sequences are known to mediate the induction of Egr‐1 following exposure to agents that induce intracellular ROI, as is the case with IR. Therefore, we hypothesized that the use of cisplatin, a commonly used chemotherapeutic agent that alters intracellular radical oxygen formation and damages DNA, might be useful in inducing the p53 gene under control of the DNA‐damaging and ROI‐inducible CArG elements of the Egr‐1 promoter.
We used an E1/E3/E4‐deleted replication‐incompetent adenoviral vector containing the chimeric promoter–effector construct Egr–p53 (AdEgr–p53) to deliver the cDNA construct to human NSCLC cell lines. We report that the CArG sequences are activated by cisplatin in vitro when ligated to p53 or to the luciferase reporter gene. Additionally, induction of p53 by cisplatin was noted in tumor xenografts in vivo. Most importantly, cisplatin induction of p53 demonstrates significantly enhanced tumor growth inhibition compared with either agent alone. Moreover, although toxicity was observed following treatment with cisplatin, no additional toxicity was observed with the combination of cisplatin and p53.
In a recent study, Yamini et al. investigated the combined use of Ad.Egr‐TNF, a replication‐defective adenoviral vector encoding the cDNA for tumor necrosis factor (TNF)‐α under the control of the Egr‐1 gene promoter, and i.p. temozolomide in an intracranial human malignant glioma model. The Ad.Egr‐TNF and temozolomide combination leads to a synergistic decrease in U87 cell viability at 72 h compared with either treatment alone. Median survival for animals treated with Ad.Egr‐TNF alone, temozolomide alone, and Ad.Egr‐TNF/temozolomide was 21, 28, and 74 days, respectively.( 14 ) In another study, Lopez et al. reported that resistance of PC‐3 human prostate carcinoma and PROb rat colon carcinoma tumors to doxorubicin in vivo was reversed by combining doxorubicin with Ad.Egr‐TNF and resulted in significant antitumor effects.( 15 ) In our study, using wt‐p53 as the therapeutic gene combined with cisplatin, similar antitumor efficacy was gained in the treatment of NSCLC as was achieved in the cited reports using TNF‐α combined with cisplatin.
The use of an inducible promoter in viral gene therapy for cancer has broad potential applicability in oncology practice, as demonstrated in a recently completed phase I trial evaluating the use of Ad.Egr.TNF.11D with radiotherapy.( 16 ) This study included patients with locally advanced/radioresistant melanoma and tumors of the pancreas, head and neck, and breast. A 60% complete response–partial response rate and a 30% stable disease rate was achieved, with no added toxicity compared with radiotherapy alone.
Control of gene expression is an important issue in gene therapy.( 17 ) Our studies demonstrate a potential clinical utility for inducible gene therapy using a genotoxic agent currently used in cancer therapy (cisplatin) and a viral vector containing a promoter with known inducible properties based on DNA damage.( 11 , 12 , 13 ) The studies reported herein reinforce the importance and relevance of transcriptional targeting with potentially toxic therapeutic agents under circumstances where tight transcriptional control of gene expression is essential to achieve a high therapeutic index.
For many common human neoplasms, grossly visible tumors are not effectively treated with most standard chemotherapeutic agents. The transcriptional targeting strategy of Egr–p53 and cisplatin may be useful when it is possible to infuse or directly inject macroscopic tumors, even in the presence of micrometastases, since the vector/cisplatin combination is effective against primary tumors and cisplatin is effective against micrometastatic disease. The direct injection of tumors should be improved with the recent advances in radiographic imaging analysis of tumors (e.g. positron emission tomography [PET] scans) combined with computed tomography (CT) image reconstruction.( 18 , 19 ) Additionally, recent developments in the targeting of viral vectors to tumors may provide additional specificity to chemo‐inducible gene therapy of metastatic cancer.( 20 , 21 ) The use of cisplatin in a strategy for targeting a cisplatin‐inducible vector has potentially important implications for improvements in clinical outcome, by employing currently used chemotherapies that damage DNA or mediate gene transcription through ROI.
The status of the p53 tumor suppressor gene in tumor cells has been shown to be a strong determinant of cellular response to treatment with either radiation or chemotherapy; the vulnerability of tumor cells to radiation or chemotherapy is greatly reduced by mutations that abolish p53‐dependent apoptosis.( 22 , 23 , 24 , 25 , 26 ) Existing studies suggest that the inactivation of p53 might produce treatment‐resistance of tumor cells to cisplatin chemotherapy and radiotherapy. However, restoration of p53 function in p53‐deficient cells or overexpression of exogenous p53 in p53‐wild type tumor cells might overcome cellular resistance and enhance cellular response to either chemotherapy or radiotherapy via a mechanism leading to p53‐dependent apoptosis.( 27 , 28 , 29 ) Consequently, our results strongly suggest that the combined‐modality therapy studied here, with cisplatin chemotherapy and AdEgr–p53 gene therapy, might be an effective therapeutic option for patients with advanced NSCLC as well as other types of cancers.
Acknowledgments
This work was supported by a grant from the National Natural Science Fund of China grant (no. 30300097). We thank KM Sakamoto for kindly donating the Egr‐1 promoter, and Dr Y‐S He for the wt‐p53 cDNA.
References
- 1. Jemal A, Murray T, Ward E et al. Cancer statistics, 2005. CA Cancer J Clin 2005; 55: 10–30. [DOI] [PubMed] [Google Scholar]
- 2. Spiro SG, Silvestri GA. The treatment of advanced non‐small cell lung cancer. Curr Opin Pulm Med 2005; 11: 287–91. [DOI] [PubMed] [Google Scholar]
- 3. Farray D, Mirkovic N, Albain KS. Multimodality therapy for stage III non‐small‐cell lung cancer. J Clin Oncol 2005; 23: 3257–69. [DOI] [PubMed] [Google Scholar]
- 4. Jin GH, Jin SZ, Liu Y et al. Therapeutic effect of gene‐therapy in combination with local X‐irradiation in a mouse malignant melanoma model. Biochem Biophys Res Commun 2005; 330: 975–81. [DOI] [PubMed] [Google Scholar]
- 5. Anton M, Gomaa IE, von Lukowicz T et al. Optimization of radiation controlled gene expression by adenoviral vectors in vitro. Cancer Gene Ther 2005; 12: 640–6. [DOI] [PubMed] [Google Scholar]
- 6. Xia J, Xia K, Feng Y et al. The combination of suicide gene therapy and radiation enhances the killing of nasopharyngeal carcinoma xenographs. J Radiat Res (Tokyo) 2004; 45: 281–9. [DOI] [PubMed] [Google Scholar]
- 7. Kufe D, Weichselbaum R. Radiation therapy: activation for gene transcription and the development of genetic radiotherapy‐therapeutic strategies in oncology. Cancer Biol Ther 2003; 2: 326–9. [DOI] [PubMed] [Google Scholar]
- 8. Schmidt M, Heimberger T, Gruensfelder P et al. Inducible promoters for gene therapy of head and neck cancer: an in vitro study. Eur Arch Otorhinolaryngol 2004; 261: 208–15. [DOI] [PubMed] [Google Scholar]
- 9. Hsu H, Rainov NG, Quinones A et al. Combined radiation and cytochrome CYP4B1/4‐ipomeanol gene therapy using the EGR1 promoter. Anticancer Res 2003; 23: 2723–8. [PubMed] [Google Scholar]
- 10. Wang WD, Chen ZT, Li DZ et al. Experimental study on lung carcinoma‐targeted suicide gene therapy induced by irradiation. Zhonghua Jie He He Hu Xi Za Zhi 2003; 26: 84–7. [PubMed] [Google Scholar]
- 11. Hallahan DE, Mauceri HJ, Senug LP et al. Spatial and temporal control of gene therapy using ionizing radiation. Nat Med 1995; 1: 786–91. [DOI] [PubMed] [Google Scholar]
- 12. Datta R, Taneja N, Sukhatme VP et al. Reactive oxygen intermediates target CC (A/T) 6GG sequences to mediate activation of the early growth response 1 transcription factor gene by ionizing radiation. Proc Nat Acad Sci USA 1993; 90: 2419–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pines A, Bivi N, Romanello M et al. Cross‐regulation between Egr‐1 and APE/Ref‐1 during early response to oxidative stress in the human osteoblastic HOBIT cell line: evidence for an autoregulatory loop. Free Radic Res 2005; 39: 269–81. [DOI] [PubMed] [Google Scholar]
- 14. Yamini B, Yu X, Gillespie GY et al. Transcriptional targeting of adenovirally delivered tumor necrosis factor by temozolomide in experimental glioblastoma. Cancer Res 2004; 64: 6381–4. [DOI] [PubMed] [Google Scholar]
- 15. Lopez CA, Kimchi ET, Mauceri HJ et al. Chemoinducible gene therapy: a strategy to enhance doxorubicin antitumor activity. Mol Cancer Ther 2004; 3: 1167–75. [PubMed] [Google Scholar]
- 16. Senzer N, Mani S, Rosemurgy A et al. TNFerade biologic, an adenovector with a radiation‐inducible promoter, carrying the human tumor necrosis factor alpha gene: a phase I study in patients with solid tumors. J Clin Oncol 2004; 22: 592–601. [DOI] [PubMed] [Google Scholar]
- 17. Crystal RG. Transfer of genes to humans: early lessons and obstacles to success. Science 1995; 270: 404–10. [DOI] [PubMed] [Google Scholar]
- 18. Hany TF, Steinert HC, Goerres GW et al. PET diagnostic accuracy: improvement with in‐line PET‐CT system: initial results. Radiology 2002; 225: 575–81. [DOI] [PubMed] [Google Scholar]
- 19. Rizzo G, Cattaneo GM, Castellone P et al. Multi‐modal medical image integration to optimize radiotherapy planning in lung cancer treatment. Ann Biomed Eng 2004; 32: 1399–408. [DOI] [PubMed] [Google Scholar]
- 20. Demeneix B, Hassani Z, Behr JP. Towards multifunctional synthetic vectors. Curr Gene Ther 2004; 4: 445–55. [DOI] [PubMed] [Google Scholar]
- 21. Jia W, Zhou Q. Viral vectors for cancer gene therapy: viral dissemination and tumor targeting. Curr Gene Ther 2005; 5: 133–42. [DOI] [PubMed] [Google Scholar]
- 22. Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin 2005; 55: 178–94. [DOI] [PubMed] [Google Scholar]
- 23. Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer 2005; 5: 231–7. [DOI] [PubMed] [Google Scholar]
- 24. Ei‐Aneed A. Current strategies in cancer gene therapy. Eur J Pharmacol 2004; 498: 1–8. [DOI] [PubMed] [Google Scholar]
- 25. Fang B, Roth JA. The role of gene therapy in combined modality treatment strategies for cancer. Curr Opin Mol Ther 2003; 5: 475–82. [PubMed] [Google Scholar]
- 26. Lebedeva IV, Su ZZ, Sarkar D, Fisher PB. Restoring apoptosis as a strategy for cancer gene therapy: focus on p53 and mda‐7. Semin Cancer Biol 2003; 13: 169–78. [DOI] [PubMed] [Google Scholar]
- 27. Torigoe T, Izumi H, Ishiguchi H et al. Cisplatin resistance and transcription factors. Curr Med Chem Anti-Canc Agents 2005; 5: 15–27. [DOI] [PubMed] [Google Scholar]
- 28. Pommier Y, Sordet O, Antony S et al. Apoptosis defects and chemotherapy resistance: molecular interaction maps and networks. Oncogene 2004; 23: 2934–49. [DOI] [PubMed] [Google Scholar]
- 29. Irwin MS. Family feud in chemosensitvity: p73 and mutant p53. Cell Cycle 2004; 3: 319–23. [PubMed] [Google Scholar]