

Abstract
Twist1 has been proposed to have oncogenic properties. Although Twist1 was reported to interact with p300/CBP‐associated factor (PCAF) and to inhibit the functions of PCAF, it remains unclear how PCAF affects the functions of Twist1, cell growth, invasive ability, and cellular sensitivity to anticancer agents. We found that PCAF, Twist1, and Y‐box binding protein‐1 (YB‐1) expressions were elevated in cisplatin‐ and doxorubicin‐resistant cancer cells. Luciferase reporter assays revealed that PCAF manipulation modulated YB‐1 transcription in a Twist1‐dependent manner. In addition, PCAF regulated the Twist1 intracellular localization and the Twist1 transcriptional activity through its acetylation function to the Twist1. Suppression of PCAF expression reduced YB‐1 expression in human urothelial cancer KK47 cells. As a result, the cell growth and invasive ability of KK47 cells was retarded by PCAF knockdown, and PCAF knockdown rendered KK47 cells sensitive to cisplatin and doxorubicin, but not to 5‐fluorouracil. The present data suggest that Twist1 and YB‐1 as well as PCAF may be promising molecular therapeutic targets. (Cancer Sci 2010)
In most malignant tumors, high tumor growth rate, invasive and metastatic abilities are well known to correlate with poor prognosis. Successful cancer management will be obtained by controlling these malignant potentials, leading to better prognosis. On the other hand, doxorubicin and cisplatin are widely used in the treatment of many solid tumors including urothelial cancer. However, anticancer drug‐resistance arises before or during chemotherapy through multiple mechanisms, and is an obstacle to successful cancer treatment.( 1 , 2 )
We previously showed that basic helix‐loop‐helix transcription factor Twist1 is involved in both drug resistance and tumor growth through Y‐box binding protein‐1 (YB‐1) expression.( 3 ) Moreover, p53 down‐regulated Twist1 transcriptional activity and YB‐1 expression, and both Twist1 and YB‐1 rescued p53‐mediated growth suppression in human cancer cells.( 4 ) Recently, we reported that programmed cell death protein 4 also interacts with Twist1, down‐regulates YB‐1 expression, and is involved in resistance to cisplatin and paclitaxel.( 5 ) Twist1 has been shown to be involved in several pathways that control tumor cell growth, invasion, metastasis, and resistance to cisplatin, doxorubicin, and paclitaxel.( 3 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 ) On the other hand, YB‐1 is also known to be closely correlated with cell proliferation and to be closely associated with unfavorable clinical outcomes.( 14 , 15 ) Recently, YB‐1 has also been shown to regulate cancer cell invasion.( 16 , 17 ) In addition, YB‐1 is a famous marker for resistance to anticancer treatment.( 15 ) Thus, these previous findings indicate that both Twist1 and YB‐1 are closely implicated in tumor growth, invasion, metastasis, and drug resistance, and suggest that blocking of the Twist1/YB‐1 pathway may be an ideal therapeutic strategy leading to suppression of tumor malignant characteristics.
P300/CBP‐associated factor (PCAF) was the first mammalian histone acetyltransferase (HAT) discovered on the basis of its homology to yeast Gcn5p.( 18 ) P300/CBP‐associated factor (PCAF) is known to acetylate various nuclear proteins in addition to histones.( 19 ) Previously, the N‐terminus region of Twist1, including two nuclear localization signals (NLS), was reported to interact with the C‐terminus of PCAF containing the HAT domain and to inhibit its functions.( 20 ) To clarify a mechanism for controlling Twist1/YB‐1 signaling, elucidating the Twist1‐interaction partner and its function in Twist1/YB‐1 signaling is necessary. However, it remains unclear how PCAF affects the functions of Twist1, and cancer cell phenotypes including cell growth, cellular invasion, and cellular sensitivity to anticancer agents.
In the present study, we investigated the functions of PCAF with regard to Twist1 that are involved in cancer cell proliferation, invasion, and resistance to anticancer drugs. The results revealed that PCAF was able to modulate YB‐1 expression in urothelial cancer cells. Furthermore, knockdown of PCAF expression affected cancer cell growth, invasion, and resistance to cisplatin and doxorubicin, but not to 5‐fluorouracil (5‐FU), resembling the functions of YB‐1.
Materials and Methods
Cell culture. Human urothelial cancer KK47 and T24 cells were cultured in Eagle’s minimal essential medium, which was purchased from Invitrogen (San Diego, CA, USA) and contained 10% fetal bovine serum. KK47/DDP20 and T24/DDP10 cells were established from KK47 and T24 cells, respectively, as described previously.( 21 , 22 ) KK47/ADR and T24/ADR cells were established from KK47 and T24 cells, respectively, as described previously.( 23 ) All cell lines were maintained in a 5% CO2 atmosphere at 37°C.
Antibodies. Anti‐c‐Myc (sc‐40), anti‐pan‐acetylLys (sc‐8663), anti‐PCAF (sc‐13124), and anti‐Twist1 (sc‐81417) antibodies and agarose‐conjugated anti‐c‐Myc antibody (sc‐40 AC) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti‐GFP (AB513) and anti‐YB‐1 antibodies were obtained from Evrogen (Moscow, Russia) and Epitomics (Burlingame, CA, USA), respectively. Anti‐β‐actin and anti‐lamin B1 antibodies were purchased from Sigma (St Louis, MO, USA).
Plasmid construction. The pCMV‐Myc‐PCAF plasmid expressing N‐terminally Myc‐tagged PCAF protein was constructed as described previously.( 24 ) The pCMV‐Myc‐PCAF HAT MT plasmid was constructed as previously described( 25 ) using a KOD Mutagenesis Kit (Toyobo, Osaka, Japan) and the pCMV‐Myc‐PCAF plasmid as a template with the following primer pair: 5′‐GCCGCTAAGAAACAGGGTTTCTCCAAAGAAAGAAATTAAA‐3′ and 5′‐TCCAATTGCATATTCAT‐CTGCATATGTGAGGAAGTTCAGG‐3′. The underlined nucleotides indicate mutated sequences. The Twist1‐GFP plasmid expressing C‐terminally GFP‐tagged Twist1 protein was described previously.( 26 ) Mutations were introduced into the N‐terminus of Twist1 (replacement of lysine residues 73, 76, and 77 with arginine) (Twist1‐GFP K73/76/77R) using a QuikChange Site‐Directed Mutagenesis Kit (Stratagene, Cedar Creek, TX, USA) and the Twist1‐GFP plasmid as a template with the following primer pair: 5′‐CCCAGGGCAGGCGCG‐GCAGGAGGTCTGCGGG‐3′ and 5′‐CCCGCAGACCTCCT‐GCCGCGCCTGCCCTGGG‐3′. The underlined nucleotides indicate mutated sequences.
To construct the reporter plasmid, hYB‐1‐Luc containing the wild‐type human YB‐1 gene (−920 bp–+85 bp), the amplified PCR product using genomic DNA, and the following primer pair: 5′‐AGATCTGTAGACGCTTAAAAAAGAACGAAA‐3′ and 5′‐AAGCTTGGCTGCTCAGGGCTCTCTGGGTC‐3′ was cloned and ligated into the BglII‐HindIII site of the pGL3‐basic vector (Promega, Madison, WI, USA). The hYB‐1‐Luc–357 (−357 bp–+85 bp) and hYB‐1‐Luc–33 (−33 bp–+85 bp) plasmids were constructed from hYB‐1‐Luc by deletion of the SmaI and SmaI‐MscI fragments, respectively. Mutations were introduced into the E‐boxes of hYB‐1‐Luc using a QuikChange Site‐Directed Mutagenesis Kit (Stratagene) with the following primer pairs: 5′‐GGGTGGAGGACTTCAGTAACATACGGTGGCAGCCTC‐3′ and 5′‐GAGGCTGCCACCGTATGTTACTGAAGTCCTCCACCC‐3′ for hYB‐1‐Luc E‐box1 MT; and 5′‐GA‐AACGTGGATAGTAATCCCTCTATACCGTGGCTGTTGCA‐3′ and 5′‐TGCAACAGCCACGGTATAGAGGGATTACTATCCACGTTTC‐3′ for hYB‐1‐Luc E‐box2 MT. An hYB‐1‐Luc E‐box12 MT plasmid was constructed by introducing a mutation into E‐box2 of hYB‐1‐Luc E‐box1 MT. The underlined nucleotides indicate the mutated sequences.
Knockdown analysis using siRNAs. Knockdown analyses using siRNAs were performed as described previously.( 26 , 27 , 28 ) The following double‐stranded RNA 25‐bp oligonucleotides were commercially generated (Invitrogen): 5′‐UUUAGCUCACAUCCCAUUAAAGUGG‐3′ (sense) and 5′‐CCACUUUAAUGGGAUGUGAGCUAAA‐3′ (antisense) for PCAF siRNA #1; and 5′‐AUAUCCUGGAGCUUCUGUUCUCUUC‐3′ (sense) and 5′‐GAAGAGAACAGAAGCUCCAGGAUAU‐3′ (antisense) for PCAF siRNA #2.
Luciferase reporter assay. Luciferase reporter assays were performed as described previously.( 26 , 28 , 29 ) All values are representative of at least three independent experiments.
RNA isolation, reverse transcription, and quantitative real‐time PCR. RNA isolation, reverse transcription, and quantitative real‐time PCR were performed as described previously.( 26 , 27 , 28 ) Briefly, quantitative real‐time PCR was carried out with TaqMan Gene Expression Assays for PCAF (assay identification number Hs00908805_m1), Twist1 (assay identification number Hs00361186_m1), YB‐1 (assay identification number Hs00698625_g1), and GAPDH (assay identification number Hs02758991_g1) (Applied Biosystems, Foster City, CA, USA) and TaqMan Gene Expression Master Mix (Applied Biosystems). The target transcript levels were corrected by the corresponding GAPDH transcript levels. All values are representative of at least three independent experiments.
Immunoprecipitation and western blot analysis. Immuno‐precipitation and western blot analyses were performed as described previously.( 27 , 28 )
Fluorescence microscopy. Fluorescence microscopy was performed as described previously.( 28 ) KK47 cells (1.0 × 105) transfected with various expression plasmids and siRNAs were plated on cover glasses in six‐well plates. After 72 h of incubation, the cells were stained with Hoechst 33342 (Nakalai Tesque, Kyoto, Japan) and observed using a Biozero fluorescence microscope (Keyence, Tokyo, Japan).
Cell proliferation assay. Cell proliferation assays were performed as described previously.( 26 , 27 , 28 ) All values are representative of at least three independent experiments.
Scratch wound assay. KK47 and T24 cells (1 × 105) transfected with 40 nM of various siRNAs were seeded into 12‐well plates and incubated for 72 h until they reached confluency. After creation of a scratch, the KK47 and T24 cells were incubated for 48 or 12 h, respectively. The scratch width in the wounded cells was then determined in three random fields using a Biozero phase‐contrast microscope (Keyence). The invasive abilities of KK47 and T24 cells are represented by the ratios of the decreased scratch width after 48 or 12 h relative to the scratch width at 0 h in the same field, respectively.
Migration assay. KK47 and T24 cells (5 × 104) transfected with 40 nM of various siRNAs for 72 h were suspended in 0.3 mL of medium and placed in the top compartment of a standard 3‐μm pore Boyden chamber (BD Biosciences, San Jose, CA, USA), while 0.8 mL of medium was added to the bottom compartment. Following a 12‐h incubation, non‐migrating cells were scraped from the top compartment and cells that had migrated to the bottom compartment were fixed and stained with DAPI (Vector Laboratories, Burlingame, CA, USA). The membranes were then excised and mounted on standard microscope slides (Curtis Matheson Scientific, Houston, TX, USA). The numbers of migrated cells were determined from three random fields using a Biozero fluorescence microscope (Keyence).
Cytotoxicity analysis. Cytotoxicity analyses were performed as described previously.( 26 , 27 ) All values are representative of at least three independent experiments.
Co‐immunoprecipitation assay. The transient transfection and immunoprecipitation assays were performed as previously described.( 4 , 5 , 29 ) Briefly, KK47 cells (2.5 × 105) were transfected with the indicated amounts of each of the indicated expression plasmids using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions and seeded into six‐well plates. After incubation at 37°C for 48 h with the indicated fresh medium, the cells were lysed in buffer X. The lysates were centrifuged at 21 000 g for 10 min at 4°C and the supernatants (300 μg) were incubated for 2 h at 4°C with agarose‐conjugated anti‐Myc antibody. The immunoprecipitated samples were washed three times with buffer X and the pre‐immunoprecipitated samples (30 μg) were subjected to western blotting analysis with the indicated antibodies.
Results
Exposure to cisplatin and doxorubicin elevates Twist1 and YB‐1 expression and PCAF, Twist1, and YB‐1 are up‐regulated in cisplatin‐ and doxorubicin‐resistant cells. First, KK47 cells were exposed to 1 μM of cisplatin for various times. Quantitative real‐time PCR and western blot analyses showed that the PCAF expression level remained constant during cisplatin exposure for up to 18 h. On the other hand, Twist1 expression was elevated in response to cisplatin after 6 h. Consistent with the finding that Twist1 regulates YB‐1 transcription, the YB‐1 mRNA and protein expression levels were also elevated after cisplatin exposure (Fig. 1a). Similar results were obtained when the cells were exposed to doxorubicin instead of cisplatin (Fig. 1b). We next investigated the expression levels of PCAF, Twist1, and YB‐1 in these cisplatin‐resistant cells, which we previously established.( 21 , 22 ) As shown in Figure 1(c), PCAF, Twist1, and YB‐1 expressions were increased in the cisplatin‐resistant cells compared with those in the parental cells at both the mRNA and protein levels, although Twist1 expression was low in T24 cells. In addition, we compared the expression levels of these proteins in doxorubicin‐resistant cells, which we also previously established.( 23 ) Similar to the findings for the cisplatin‐resistant cells, PCAF, Twist1, and YB‐1 expressions were up‐regulated in the doxorubicin‐resistant cells at both the mRNA and protein levels (Fig. 1d).
Figure 1.

Exposure to cisplatin and doxorubicin elevates Twist1 and Y‐box binding protein‐1 (YB‐1) expressions and p300/CBP‐associated factor (PCAF), Twist1, and YB‐1 are up‐regulated in cisplatin‐ and doxorubicin‐resistant cells. (a) KK47 cells were treated with 1 μM of cisplatin for the indicated durations. Quantitative real‐time PCR was performed for PCAF, Twist1, YB‐1, and GAPDH. Each transcript level from untreated KK47 cells was set as 1. Data represent mean ± SD. Nuclear extracts and whole‐cell lysates were subjected to SDS‐PAGE and western blotting analyses were performed with the indicated antibodies. (b) KK47 cells were treated with 10 nM of doxorubicin. Quantitative real‐time PCR and western blotting analyses were performed as described for (a). (c) Quantitative real‐time PCR using cDNA from the indicated cells was performed for PCAF, Twist1, YB‐1, and GAPDH. Each transcript level from KK47 cells was set as 1. Data represent mean ± SD. Whole‐cell lysates were subjected to SDS‐PAGE and western blotting analyses as described for (a). (d) Quantitative real‐time PCR and western blotting analyses were performed as described for (c).
p300/CBP‐associated factor (PCAF) regulates YB‐1 transcription through an interaction with Twist1. To investigate an effect of PCAF on Twist1/YB‐1 signaling, we constructed a reporter plasmid containing of the wild‐type human YB‐1 gene (−920 bp–+85 bp) (Fig. 2a). Because the 3′ flanking region from the transcription start site is known to play a role in basal transcription, we included this region in the reporter construct. First, KK47 cells were cotransfected with hYB‐1‐Luc, PCAF, and/or Twist1 expression plasmids. As shown in Figure 2(b), Twist1 increased YB‐1 transcription similar to our previous report.( 3 ) Moreover, PCAF overexpression up‐regulated the luciferase activity of hYB‐1‐Luc by about 2‐fold. In addition, Twist1 and PCAF expressions caused cooperative up‐regulation of the luciferase activity by about 8‐fold. Conversely, PCAF knockdown using two kinds of PCAF‐specific siRNAs decreased the luciferase activities of the YB‐1 reporter plasmid in KK47 cells.
Figure 2.
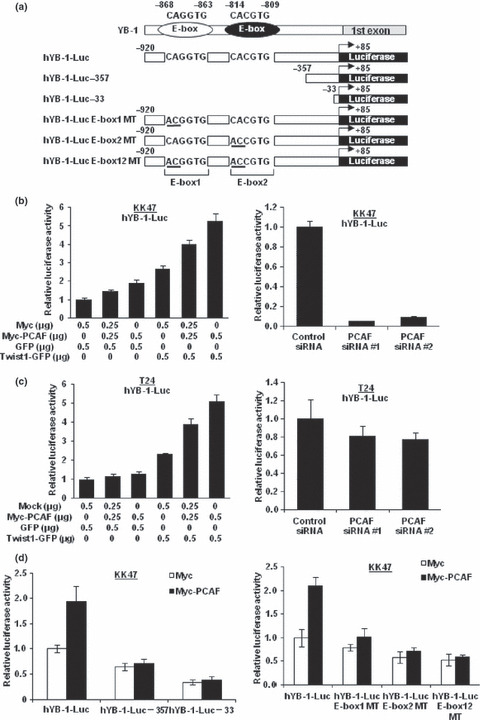
p300/CBP‐associated factor (PCAF) regulates Y‐box binding protein‐1 (YB‐1) transcription through an interaction with Twist1. (a) Schematic representations of the human YB‐1 gene and various YB‐1 reporter plasmids. (b,c) KK47 (b) and T24 (c) cells (1.5 × 105) were transiently co‐transfected with the indicated amounts of expression plasmids or 20 nM of the indicated siRNA in addition to 0.5 μg of hYB‐1‐Luc and 0.05 μg of pRL‐TK. The luciferase activity of hYB‐1‐Luc alone was set as 1. Data represent mean ± SD. (d) KK47 cells (1.5 × 105) were transiently co‐transfected with 1.0 μg of Myc or Myc‐PCAF expression plasmid in addition to 0.5 μg of the indicated reporter plasmid, and 0.05 μg of pRL‐TK. Luciferase reporter assay was performed as described for (b,c).
To confirm the above findings in KK47 cells, T24 cells were cotransfected with hYB‐1‐Luc, PCAF, and/or Twist1 expression plasmids. As expected, the luciferase activity of hYB‐1‐Luc was elevated by Twist1 overexpression, whereas PCAF overexpression without Twist1 expression did not increase the activity in T24 cells expressing little Twist1. When Twist1 was overexpressed in T24 cells, PCAF overexpression led to an increase in the luciferase activity. Next, we investigated the effect of PCAF knockdown on YB‐1 transcription in T24 cells. PCAF knockdown did not decrease the luciferase activity of hYB‐1‐Luc in T24 cells (Fig. 2c).
We also constructed deletion mutants of hYB‐1‐Luc as shown in Figure 2(a), and conducted luciferase reporter assays. When the E‐boxes located in the YB‐1 promoter region were deleted, the basal luciferase activity of YB‐1 was reduced and the increase in the luciferase activity induced by PCAF overexpression was abolished (Fig. 2d). These findings suggest that the E‐boxes located within the YB‐1 promoter region play a critical role and that the effect of PCAF on YB‐1 transcription is dependent on these E‐boxes. To confirm the above results, we constructed YB‐1 reporter plasmids with introduced mutations in the E‐boxes responsible for YB‐1 transcription by Twist1 as shown in Figure 2(a). The introduction of these mutations into the E‐boxes abolished the up‐regulation of the luciferase activities by Twist1 in KK47 cells, as we previously reported.( 3 ) As shown in Figure 2(d), the various mutated YB‐1 reporter plasmids exhibited dramatically lower luciferase activities than the wild‐type YB‐1 reporter plasmid. In addition, PCAF overexpression induced an up‐regulation of the luciferase activity of wild‐type hYB‐1‐Luc, but had little effect on the activities of the E‐box‐mutated YB‐1 reporter plasmids, suggesting that the up‐regulation of YB‐1 transcription by PCAF overexpression is dependent on the E‐boxes located within the promoter region of YB‐1.
p300/CBP‐associated factor (PCAF) acetylates Twist1 and disruptions of Twist1 acetylation decrease YB‐1 transcription. Next, we investigated whether the introduction of mutations into the HAT domain of PCAF could affect this interaction. Luciferase reporter assays using a HAT activity‐negative PCAF expression plasmid (pCMV‐Myc‐PCAF HAT MT) revealed that the HAT activity was indispensable for the transactivation of YB‐1 by PCAF (Fig. 3a). Then, we investigated whether PCAF could acetylate Twist1. As expected, the acetylation level of Twist1‐GFP was reduced when PCAF expression was silenced (Fig. 3b). To confirm the above result, we constructed a Twist1‐GFP expression plasmid with introduced mutations that converted lysine residues (amino acids 73, 76 and 77) within the N‐terminus of Twist1, which is known to be an interactive domain with PCAF( 20 ) and to be compatible with acetylation consensus motif KX1–2(X/K)K,( 30 ) to non‐acetylation‐mimicking arginine residues (Twist1‐GFP K73/76/77R) (Fig. 3c). The acetylation level of mutated Twist1‐GFP was reduced compared with that of wild‐type Twist1‐GFP (Fig. 3d). The Twist1 expression levels were approximately equal among the Twist1‐GFP WT and Twist1‐GFP K73/76/77R expression plasmids. The YB‐1 transcription measured by luciferase reporter assays using hYB‐1‐Luc revealed that the non‐acetylation‐mimicking mutations could reduce the transactivating activity of Twist1 (Fig. 3e).
Figure 3.
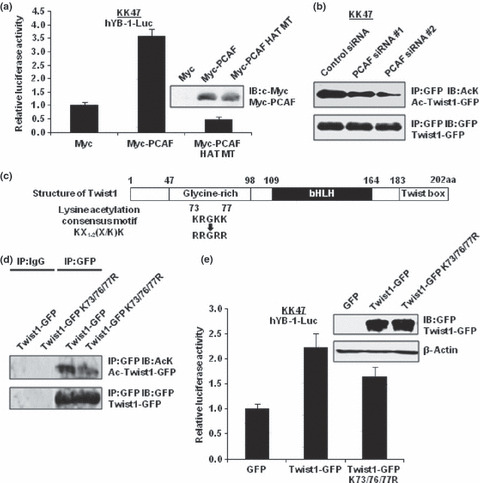
p300/CBP‐associated factor (PCAF) acetylates Twist1 and disruptions of Twist1 acetylation decrease Y‐box binding protein‐1 (YB‐1) transcription. (a) KK47 cells (1.5 × 105) were transfected with 1.0 μg of the indicated expression plasmids. After 48 h of incubation, whole‐cell lysates were subjected to SDS‐PAGE, and western blotting was performed using an anti‐Myc antibody. KK47 cells (1.5 × 105) were transiently cotransfected with 0.5 μg of the indicated expression plasmids in addition to 0.5 μg of hYB‐1‐Luc and 0.05 μg of pRL‐TK. The luciferase activity of hYB‐1‐Luc alone was set as 1. Data represent mean ± SD. (b) KK47 cells (2.5 × 105) were transfected with 1.0 μg of Twist1‐GFP expression plasmid and 20 nM of the indicated siRNA. After 72 h of incubation, whole‐cell extracts (500 μg) were immunoprecipitated with 2.0 μg of mouse IgG or anti‐GFP antibody, and 20 μL of protein A/G agarose. The immunoprecipitants were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies. (c) Schematic representation of the Twist1 gene structure showing the positions of the mutated lysine residues. (d) KK47 cells (2.5 × 105) were transfected with 2.0 μg of Twist1‐GFP and Twist1‐GFP K73/76/77R expression plasmids. After 48 h of incubation, immunoprecipitation and western blotting were performed as described for (b). (e) KK47 cells (1.5 × 105) were transfected with 1.0 μg of the indicated Twist1‐GFP and Twist1‐GFP K73/76/77R expression plasmids. After 48 h of incubation, whole‐cell lysates were subjected to SDS‐PAGE, and western blotting was performed using an anti‐GFP antibody. KK47 cells (1.5 × 105) were transiently co‐transfected with 0.5 μg of wild‐type or mutated Twist1‐GFP expression plasmid in addition to 0.5 μg of hYB‐1‐Luc and 0.05 μg of pRL‐TK. The luciferase activity of hYB‐1‐Luc alone was set as 1. Data represent mean ± SD.
Disruptions of Twist1 acetylation inhibit nuclear local‐ization. Next, the mechanism of the decrease in Twist1 transcriptional activity by disruption of Twist1 acetylation was investigated. Since amino acids 73, 76, and 77 are known to be part of the nuclear localization signal (NLS),( 20 ) we speculated that acetylation of these sites may affect the intracellular localization of Twist1. First, we investigated the intracellular localizations of wild‐type Twist1‐GFP and Twist1‐GFP K73/76/77R. Western blotting and fluorescence microscopy revealed that almost all the wild‐type Twist1‐GFP was localized in the nuclei of KK47 cells whereas Twist1‐GFP K73/76/77R was localized in both the nuclei and the cytoplasm, which was consistent with the result shown in Figure 3(d) wherein mutated Twist1 was reduced, but still retained its transcriptional ability (Fig. 4a,b). When PCAF expression was suppressed, the nuclear localizations of Twist1‐GFP and endogenous Twist1 were reduced (Fig. 4c,d). In addition, the Twist1 acetylation level in cytoplasm was reduced compared with that in the nucleus when PCAF expression was silenced (Fig. 4e).
Figure 4.

Disruptions of Twist1 acetylation inhibit nuclear localization. (a) KK47 cells (3.0 × 105) were transfected with 2.0 μg of the indicated expression plasmids. After 48 h of incubation, nuclear and cytoplasmic extracts were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies. (b) KK47 cells were transfected with 2.0 μg of the indicated expression plasmids. After 48 h of incubation, the cells were stained with Hoechst33342 and observed by fluorescence microscopy. Representative images of high‐magnification fields are shown. (c) KK47 cells (3.0 × 105) were transfected with 1.0 μg of Twist1‐GFP expression plasmid and 20 nM of the indicated siRNAs. After 72 h of incubation, western blotting was performed as described for (a). (d) KK47 cells were transfected with 1.0 μg of Twist1‐GFP expression plasmid and 20 nM of the indicated siRNAs. After 72 h of incubation, the cells were stained with Hoechst33342 and observed by fluorescence microscopy. Representative images of high‐magnification fields are shown. (e) KK47 cells (2.5 × 105) were transfected with 1.0 μg of Twist1‐GFP expression plasmid and 20 nM of the indicated siRNA. After 72 h of incubation, nuclear and cytoplasmic extracts (100 μg and 300 μg, respectively) were immunoprecipitated with 2.0 μg of mouse IgG or anti‐GFP antibody, and 20 μL of protein A/G agarose. The immunoprecipitants were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies.
Knockdown of PCAF decreases YB‐1 expression, cancer cell growth, invasion, and resistance to cisplatin and doxorubicin in a Twist1‐dependent manner. When PCAF was silenced, both the mRNA and protein expression levels of PCAF were almost completely abolished in KK47 cells. Although the Twist1 expression level was not affected by PCAF knockdown, the mRNA and protein expression levels of YB‐1 were decreased to about 20–30% in KK47 cells (Fig. 5a). However, PCAF knockdown induced little reduction of the YB‐1 mRNA and protein expression levels in T24 cells expressing little Twist1 (Fig. 5b).
Figure 5.

Knockdown of p300/CBP‐associated factor (PCAF) decreases Y‐box binding protein‐1 (YB‐1) expression and retards cancer cell growth in a Twist1‐dependent manner. (a,b) KK47 (a) and T24 (b) cells (3.0 × 105) were transiently transfected with 40 nM of the indicated siRNA. At 72 h after transfection, quantitative real‐time PCR was performed using specific primers and probes for PCAF, Twist1, YB‐1, and GAPDH. Each transcript level from cells transfected with the control siRNA was set as 1. Data represent mean ± SD. Whole‐cell lysates were subjected to SDS‐PAGE, and western blotting analysis was performed using the indicated antibodies. (c) KK47 and T24 cells were transfected with 40 nM of control siRNA, PCAF siRNA #1, or PCAF siRNA #2. At the indicated time points, the cell numbers were counted. The results were normalized by the cell numbers at 0 h. Data represent means ± SD.
To investigate an effect of PCAF on cancer cell growth, we performed a cell proliferation assay after PCAF knockdown. As shown in Figure 5(c), KK47 cell growth was retarded by PCAF knockdown. However, T24 cell growth was hardly affected although YB‐1 knockdown retarded the cell growth of both KK47 and T24 cells (data not shown).
Subsequently, we investigated the invasive ability of cancer cells after PCAF knockdown. When PCAF expression was reduced in KK47 cells, their invasive ability was reduced. On the other hand, the invasive ability was hardly affected when PCAF was knocked down in T24 cells (Fig. 6a), although YB‐1 knockdown reduced the invasive abilities of both KK47 and T24 cells (data not shown). Similarly, the motility ability judged by Boyden‐chamber assays was reduced in KK47 cells after PCAF knockdown, whereas the motility ability in T24 cells was hardly affected (Fig. 6b). Y‐box binding protein‐1 (YB‐1) suppression also decreased the motility abilities in both KK47 and T24 cells (data not shown).
Figure 6.
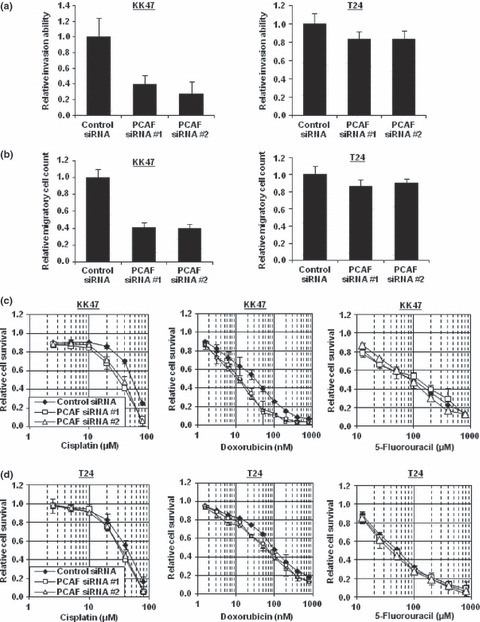
Knockdown of p300/CBP‐associated factor (PCAF) decreases cancer cell invasion, motility, and resistance to cisplatin and doxorubicin, but not to 5‐fluorouracil (5‐FU), in a Twist1‐dependent manner. (a) KK47 and T24 cells were transfected with 40 nM of the indicated siRNA. At 72 h after transfection, the cells were scratched. After 48 h (KK47 cells) or 12 h (T24 cells) of incubation, the scratch widths were measured. Data represent mean ± SD. (b) KK47 and T24 cells were transfected with 40 nM of the indicated siRNA. At 48 h after transfection, the cells were applied to Boyden chambers. After 6 h of incubation, the cells were fixed and stained with DAPI. The numbers of mobilized cells were counted. Data represent mean ± SD. (c,d) KK47 (c) and T24 (d) cells were transfected with 40 nM of the indicated siRNA. On the following day, various concentrations of cisplatin, doxorubicin, or 5‐FU were applied. After 48 h, the cell survival rates were analyzed by cytotoxicity analyses. Cell survival in the absence of cisplatin, doxorubicin, or 5‐FU was set as 1. Data represent mean ± SD.
Finally, we examined the cell survival rates after treatment with cisplatin, doxorubicin, and 5‐FU when PCAF expression was silenced. As shown in Figure 6(c), PCAF knockdown sensitized KK47 cells to cisplatin and doxorubicin, but not to 5‐FU. On the other hand, PCAF knockdown hardly sensitized T24 cells to these anticancer drugs (Fig. 6d).
Discussion
p300/CBP‐associated factor (PCAF) belongs to the HAT family and is known to acetylate and modulate its target proteins. Previously, it was demonstrated that p53 interacts directly with PCAF and that acetylation of the C‐terminal domain of p53 by PCAF is critical for its regulation.( 31 , 32 , 33 , 34 ) The acetylated forms of p53 leading to an increase of its sequence‐specific DNA‐binding ability have been observed, as have the levels of acetylation increase in response to DNA damage, including that caused by ultraviolet light and ionizing radiation.( 33 , 35 ) Moreover, the function of the cell cycle‐related activator E2F is also regulated by acetylation.( 36 ) One of the E2F family members, E2F1, was previously shown to be acetylated by PCAF, and this acetylation has several positive effects on E2F function.( 37 ) Two types of HAT mutations in PCAF have previously been reported, comprising deletion mutations and point mutations.( 25 , 38 ) Since deletion mutations may alter the interaction between PCAF and Twist1, we employed a point mutation construct, and showed that mutation into the HAT domain abolished PCAF’s effect on Twist1 transcriptional ability. In addition, introduction of mutations into lysine sites located in an interactive domain of Twist1 with PCAF affected the Twist1 transcriptional ability and intracellular localization of Twist1, although an interacting strength between mutated Twist1 and PCAF was similar to that between wild‐type Twist1 and PCAF (Fig. S1a). In KK47 cells, PCAF localized in the nucleus as well as in cytoplasm, in which Twist1 appeared to be acetylated by PCAF and translocate into nucleus (Fig. S1b). These findings suggest that PCAF exerts its effects on the intracellular distribution of Twist1 and on the Twist1 transcriptional activity through its acetylating activity toward the Twist1, although further analysis analyzing the acetylation status of individual amino acid using equipment such as mass spectrometry is needed to determine whether these residues were acetylated by PCAF.
As stated above, PCAF can modulate several transcription factors other than Twist1, including p53 and E2F1. p300/CBP‐associated factor (PCAF) suppression led to cell growth retardation and the suppression of cancer invasion in KK47 cells, but not in T24 cells. This discrepancy may be accounted for by the differences in the basal Twist1 expression levels and the YB‐1 expression levels after PCAF knockdown between KK47 and T24 cells. Although it remains unclear what caused these findings, they seemed to result from YB‐1 suppression through PCAF knockdown, and not from modulation of p53, because KK47 and T24 cells both express mutated p53. In addition, they could not have resulted from E2F1 modulation because the cell proliferation of T24 cells in which YB‐1 expression was hardly affected by PCAF knockdown, but in which E2F1 was adequately expressed (data not shown), was not modulated.
In the present study, cisplatin‐ and doxorubicin‐resistant cells expressed PCAF at more than 2‐fold higher levels compared with their parental cells, although the mechanism for the PCAF expression remains unknown. Genetic or epigenetic changes in drug‐resistant cells may be responsible for alterations in the PCAF expression level. As a PCAF‐interacting partner, Twist1 was dramatically induced by stimulations with anticancer agents and overexpressed in anticancer drug‐resistant cells. Therefore, PCAF and Twist1 may coordinately confer resistance to cisplatin and doxorubicin by modulating the expression levels of their target genes. Y‐box binding protein‐1 (YB‐1) has been shown to be one of the target genes for Twist1.( 3 ) The present data have revealed that PCAF also regulated the expression of YB‐1 in KK47 cells, which express abundant Twist1. However, PCAF did not have this effect in T24 cells that express little Twist1. These findings indicate that the modulation of YB‐1 expression by PCAF is dependent on the Twist1 expression level. Previously, both Twist1 and YB‐1 were demonstrated to be involved in cisplatin resistance. Therefore, PCAF also appears to be involved in cisplatin resistance. In addition, Twist1 is known to be related to doxorubicin resistance. The finding that the Twist1 target gene YB‐1 is also involved in doxorubicin resistance suggests that Twist1 confers doxorubicin resistance, at least in part, through YB‐1 expression. Similarly, PCAF also conferred doxorubicin resistance on Twist1‐expressing cancer cells. Since PCAF interacts with a number of proteins that may affect drug sensitivity, PCAF may be involved in drug resistance through molecules other than YB‐1. However, PCAF appears to regulate drug sensitivity through YB‐1 expression because the drug sensitivity of T24 cells, in which the YB‐1 expression was barely affected by PCAF knockdown, was hardly affected by PCAF suppression.
In summary, this study has revealed that PCAF promotes Twist1 localization into the nucleus and activates Twist1 transcriptional ability through its HAT activity. Consequently, PCAF was implicated in cancer cell growth, invasion, and drug resistance. Therefore, Twist1 and YB‐1, as well as PCAF, may be promising molecular therapeutic targets through a modulation of YB‐1 expression by an interaction with Twist1. Intensive search for a clinically useful HAT inhibitor of PCAF is important. In future, a HAT inhibitor may be a clinically useful modality, especially combined with chemotherapy including cisplatin and doxorubicin.
Supporting information
Fig. S1. (a) KK47 cells (2.5 × 105) were transfected with 1.0 μg of each of the indicated expression plasmids. After 48 h of incubation, whole‐cell lysates were immunoprecipitated with 20 μL of agarose‐conjugated anti‐Myc antibody. The pre‐immunoprecipitants as input and the immunoprecipitants were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies. (b) KK47 cells were divided into nuclear and cytoplasmic fractions. Equal amounts of nuclear and cytoplasmic extracts were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
This work was supported in part by Health Sciences Research Grants for Clinical Research for Evidenced‐Based Medicine and Grants‐in‐Aid for Cancer Research (016) from the Ministry of Health, Labor and Welfare of Japan. The authors would like to acknowledge the technical expertise of the Support Center for Education and Research, Kyushu University. We would like to thank Dr Dongchon Kang (Kyushu University, Fukuoka, Japan) for help with the quantitative real‐time PCR, Edanz Group Japan for editorial assistance, and Ms Noriko Hakoda and Ms Seiko Kamori for their technical assistance.
References
- 1. Naito S, Yokomizo A, Koga H. Mechanisms of drug resistance in chemotherapy for urogenital carcinoma. Int J Urol 1999; 6: 427–39. [DOI] [PubMed] [Google Scholar]
- 2. Naito S, Koga H, Yokomizo A et al. Molecular analysis of mechanisms regulating drug sensitivity and the development of new chemotherapy strategies for genitourinary carcinomas. World J Surg 2000; 24: 1183–6. [DOI] [PubMed] [Google Scholar]
- 3. Shiota M, Izumi H, Onitsuka T et al. Twist promotes tumor cell growth through YB‐1 expression. Cancer Res 2008; 68: 98–105. [DOI] [PubMed] [Google Scholar]
- 4. Shiota M, Izumi H, Onitsuka T et al. Twist and p53 reciprocally regulate target genes via direct interaction. Oncogene 2008; 27: 5543–53. [DOI] [PubMed] [Google Scholar]
- 5. Shiota M, Izumi H, Tanimoto A et al. Programmed cell death protein 4 down‐regulates Y‐box binding protein‐1 expression via a direct interaction with Twist1 to suppress cancer cell growth. Cancer Res 2009; 69: 3148–56. [DOI] [PubMed] [Google Scholar]
- 6. Maestro R, Dei Tos AP, Hamamori Y et al. Twist is a potential oncogene that inhibits apoptosis. Genes Dev 1999; 13: 2207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Funato N, Ohtani K, Ohyama K, Kuroda T, Nakamura M. Common regulation of growth arrest and differentiation of osteoblasts by helix‐loop‐helix factors. Mol Cell Biol 2001; 21: 7416–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Valsesia‐Wittmann S, Magdeleine M, Dupasquier S et al. Oncogenic cooperation between H‐Twist and N‐Myc overrides failsafe programs in cancer cells. Cancer Cell 2004; 6: 625–30. [DOI] [PubMed] [Google Scholar]
- 9. Wang X, Ling MT, Guan XY et al. Identification of a novel function of TWIST1, bHLH protein, in the development of acquired taxol resistance in human cancer cells. Oncogene 2004; 23: 474–82. [DOI] [PubMed] [Google Scholar]
- 10. Yang J, Mani SA, Donaher JL et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004; 117: 927–39. [DOI] [PubMed] [Google Scholar]
- 11. Alexander NR, Tran NL, Rekapally H, Summers CE, Glackin C, Heimark RL. N‐cadherin gene expression in prostate carcinoma is modulated by integrin‐dependent nuclear translocation of Twist1. Cancer Res 2006; 66: 3365–9. [DOI] [PubMed] [Google Scholar]
- 12. Li J, Wood WH, Becker KG, Weeraratna AT, Morin PJ. Gene expression response to cisplatin treatment in drug‐sensitive and drug‐resistant ovarian cancer cells. Oncogene 2007; 26: 2860–72. [DOI] [PubMed] [Google Scholar]
- 13. Zhou WL, Wang Y, Zhou XL, Zhang YS, Chen ZT. Short interfering RNA directed against TWIST, a novel zinc finger transcription factor, increases A549 cell sensitivity to cisplatin via MAPK/mitochondrial pathway. Biochem Biophys Res Commun 2008; 369: 1098–102. [DOI] [PubMed] [Google Scholar]
- 14. Kohno K, Izumi H, Uchiumi T, Ashizuka M, Kuwano M. The pleiotrophic functions of the Y‐box‐binding protein, YB‐1. Bioessays, 2003; 25: 691–8. [DOI] [PubMed] [Google Scholar]
- 15. Kuwano M, Oda Y, Izumi H et al. The role of nuclear Y‐box binding protein 1 as a global marker in drug resistance. Mol Cancer Ther 2004; 3: 1485–92. [PubMed] [Google Scholar]
- 16. Astanehe A, Finkbeiner MR, Hojabrpour P et al. The transcriptional induction of PIK3CA in tumor cells is dependent on the oncoprotein Y‐box binding protein‐1. Oncogene 2009; 28: 2406–18. [DOI] [PubMed] [Google Scholar]
- 17. Evdokimova V, Tognon C, Ng T et al. Translational activation of snail1 and other developmentally regulated transcription factors by YB‐1 promotes an epithelial‐mesenchymal transition. Cancer Cell 2009; 15: 402–15. [DOI] [PubMed] [Google Scholar]
- 18. Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y. A p300/CBP‐associated factor that competes with the adenoviral oncoprotein E1A. Nature 1996; 382: 319–24. [DOI] [PubMed] [Google Scholar]
- 19. Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev 2000; 14: 1553–77. [PubMed] [Google Scholar]
- 20. Hamamori Y, Sartorelli V, Ogryzko V et al. Regulation of histone acetyltransferases p300 and PCAF by the bHLH protein Twist and adenoviral oncoprotein E1A. Cell, 1999; 96: 405–13. [DOI] [PubMed] [Google Scholar]
- 21. Kotoh S, Naito S, Yokomizo A et al. Increased expression of DNA topoisomerase I gene and collateral sensitivity to camptothecin in human cisplatin‐resistant bladder cancer cells. Cancer Res 1994; 54: 3248–52. [PubMed] [Google Scholar]
- 22. Kotoh S, Naito S, Yokomizo A, Kohno K, Kuwano M, Kumazawa J. Enhanced expression of gamma‐glutamylcysteine synthetase and glutathione S‐transferase genes in cisplatin‐resistant bladder cancer cells with multidrug resistance phenotype. J Urol 1997; 157: 1054–8. [PubMed] [Google Scholar]
- 23. Hasegawa S, Abe T, Naito S et al. Expression of multidrug resistance‐associated protein (MRP), MDR1 and DNA topoisomerase II in human multidrug‐resistant bladder cancer cell lines. Br J Cancer 1995; 71: 907–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Masatsugu T, Yamamoto K. Multiple lysine methylation of PCAF by Set9 methyltransferase. Biochem Biophys Res Commun 2009; 381: 22–6. [DOI] [PubMed] [Google Scholar]
- 25. Korzus E, Torchia J, Rose DW et al. Transcription factor‐specific requirements for coactivators and their acetyltransferase functions. Science 1998; 279: 703–7. [DOI] [PubMed] [Google Scholar]
- 26. Shiota M, Yokomizo A, Tada Y et al. Castration resistance of prostate cancer cells caused by castration‐induced oxidative stress through Twist1 and androgen receptor overexpression. Oncogene 2010; 24: 114–27. [DOI] [PubMed] [Google Scholar]
- 27. Shiota M, Yokomizo A, Kashiwagi E et al. Foxo3a expression and acetylation regulate cancer cell growth and sensitivity to cisplatin. Cancer Sci 2010; doi: 10.1111/j.1349-7006.2010.01503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shiota M, Yokomizo A, Masubuchi D et al. Tip60 promotes prostate cancer cell proliferation by translocation of androgen receptor into the nucleus. Prostate 2010; 70: 540–54. [DOI] [PubMed] [Google Scholar]
- 29. Shiota M, Izumi H, Miyamoto N et al. Ets regulates peroxiredoxin1 and 5 expressions through their interaction with the high mobility group protein B1. Cancer Sci 2008; 99: 1950–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang XJ. Lysine acetylation and the bromodomain: a new partnership for signaling. Bioessays 2004; 26: 1076–87. [DOI] [PubMed] [Google Scholar]
- 31. Lill NL, Grossman SR, Ginsberg D, DeCaprio J, Livingston DM. Binding and modulation of p53 by p300/CBP coactivators. Nature 1997; 387: 823–7. [DOI] [PubMed] [Google Scholar]
- 32. Scolnick DM, Chehab NH, Stavridi ES et al. CREB‐binding protein and p300/CBP‐associated factor are transcriptional coactivators of the p53 tumor suppressor protein. Cancer Res 1997; 57: 3693–6. [PubMed] [Google Scholar]
- 33. Liu L, Scolnick DM, Trievel RC et al. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol Cell Biol 1999; 19: 1202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gu W, Roeder RG. Activation of p53 sequence‐specific DNA binding by acetylation of the p53 C‐terminal domain. Cell 1997; 90: 595–606. [DOI] [PubMed] [Google Scholar]
- 35. Sakaguchi K, Herrera JE, Saito S et al. DNA damage activates p53 through a phosphorylation‐acetylation cascade. Genes Dev 1998; 12: 2831–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Adams PD, Kaelin WG Jr. Transcriptional control by E2F. Semin Cancer Biol 1995; 6: 99–108. [DOI] [PubMed] [Google Scholar]
- 37. Martínez‐Balbás MA, Bauer UM, Nielsen SJ, Brehm A, Kouzarides T. Regulation of E2F1 activity by acetylation. EMBO J 2000; 19: 662–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang H, Lu H, Schiltz RL et al. PCAF interacts with tax and stimulates tax transactivation in a histone acetyltransferase‐independent manner. Mol Cell Biol 1999; 19: 8136–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. (a) KK47 cells (2.5 × 105) were transfected with 1.0 μg of each of the indicated expression plasmids. After 48 h of incubation, whole‐cell lysates were immunoprecipitated with 20 μL of agarose‐conjugated anti‐Myc antibody. The pre‐immunoprecipitants as input and the immunoprecipitants were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies. (b) KK47 cells were divided into nuclear and cytoplasmic fractions. Equal amounts of nuclear and cytoplasmic extracts were subjected to SDS‐PAGE, and western blotting was performed using the indicated antibodies.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item