

Abstract
Endosomal sorting complex required for transport (ESCRT) proteins form a multicomplex sorting machinery that controls multivesicular body (MVB) formation and the sorting of ubiquitinated membrane proteins to the endosomes. Being sorted to the MVB generally results in the lysosome‐dependent degradation of cell‐surface receptors, and defects in this machinery induce dysregulated receptor traffic and turnover. Recent lessons from gene targeting and silencing methodologies have implicated the ESCRT in normal development, cell differentiation, and growth, as well as in the budding of certain enveloped viruses. Furthermore, it is becoming apparent that the dysregulation of ESCRT proteins is involved in the development of various human diseases, including many types of cancers and neurodegenerative disorders. Here, we summarize the roles of ESCRT proteins in MVB sorting processes and the regulation of tumor cells, and we discuss some of their other functions that are unrelated to vesicular transport. (Cancer Sci 2008; 99: 1293–1303)
Endosomal system controls the traffic of membrane‐bound receptors
Tissue homeostasis is orchestrated by a finely tuned balance of signals delivered from one neighboring environment to another. An imbalance of information can cause dysregulated cell growth, differentiation, and cell death. Mammalian cells have several mechanisms for maintaining harmony. One such mechanism is the cells’ control of the lifespan of cell‐surface molecules. Membrane‐bound receptors that are newly generated in the endoplasmic reticulum (ER) are directed to the cell surface via the Golgi. Once at the cell surface, they engage in constant migration between the cell surface and intracellular organelles and, at some point, they are degraded. For example, activated receptors such as epidermal growth factor (EGF) receptors (EGFR) on the cell surface are usually endocytosed and further sorted to various destinations. Although some of the EGFR are recycled to the cell surface, others are transported into the intraluminal vesicles (ILV) of maturing endosomes called multivesicular bodies (MVB).( 1 , 2 ) Following fusion with late endosomes or lysosomes, these membrane‐bound proteins (referred to as cargo) and the ILV containing them within the MVB are degraded by hydrolytic enzymes such as cathepsins, under acidic conditions.( 3 )
When many membrane proteins, such as transferrin receptors and low‐density lipoproteins, bind their respective ligands, they are internalized rapidly into endosomes. From there, they are often sent back to the cell surface via the recycling pathway. This recycling loop is so rapid that the typical half‐life of receptor turnover by recycling is only in the order of minutes. This quick return of the receptors to the cell surface seems to enable their constant reuse for ligand binding and endocytosis of the bound receptor–ligand complex.( 4 ) In addition to these two groups of cell‐surface receptors, other membrane‐bound receptors, such as mannose 6‐phosphate receptor, bind lysosomal proteins in the trans‐Golgi network and are shuttled between the late endosomes or lysosomes and trans‐Golgi network. Thus, different membrane‐bound receptors are reused and recycled by one pathway, or sorted for degradation by another.
Consistent with their central role in intracellular trafficking, endosomes possess a variety of the network's ‘hub’ functions. Endosomes are usually categorized according to their function in cargo processing. For example, two of these compartments, early and late endosomes, are named after the relative time required for the endocytosed cargos to reach the compartments. Although the different endosomal compartments are all part of a functionally linked vesicular system, early endosomes have different characteristics from late endosomes. First, the intravesicular pH is made increasingly acidic from early to late endosomes by vacuolar H+ ATPases; whereas the pH of early endosomes is ~6, the pH of late endosomes and lysosomes is closer to 5. Second, the composition of the compartments changes, including the content of membrane‐bound proteins and lipids. Early endosomes are positive for Rab5 and early endosome antigen‐1, whereas lysosome‐associated membrane proteins‐1, ‐2, and ‐3, Rab7, and lipid lysobisphosphatidic acid are markers for late endosomes and MVB. Third, the endosomal structure changes from the tubulovesicular shape of early endosomes to a multivesicular morphology, and the latter is characterized by the presence of smaller vesicles (40–100 nm in diameter) within it.( 5 ) MVB are formed between the early and late endosomes. They can appear to be hybrid compartments that are part of the early endosome, but in many instances MBV represent vesicles in the transitional state between early and late endosomes. The precise definitions of these compartments sometimes change with the biological situation or as new details are understood; regardless, it is clear that MVB play an important role in cell biology. Indeed, the formation of the MVB from the inward invagination of the endosomal limiting membrane is a key event for many cellular functions that require an alteration in topology to determine the functions of membrane‐bound receptors.
Endosomal sorting complexes required for transport control the MVB sorting of ubiquitinated cargos
Accumulating evidence suggests that cargo sorting and MVB formation require four distinct sorting complexes, called endosomal sorting complexes required for transport (ESCRT), which are essentially conserved throughout the eukaryotes (Table 1).( 1 , 2 , 6 , 7 , 8 ) Prior to the discovery of the ESCRT proteins in mammals, genetic screens in yeast had revealed at least 18 conserved proteins called class E vacuolar protein sorting (Vps) proteins. The mutation of any one of these genes in Saccharomyces cerevisiae results in the formation of aberrant endosomes called ‘class E compartments.’ These class E Vps proteins were shown to be highly conserved between yeast and mammals, and their mutation or deletion in mammals results in the formation of similar aberrant structures. The ESCRT are subdivided into four complexes. ESCRT‐0, which is also called the signal‐transducing adaptor molecule 1 or 2 (STAM)–hepatocyte‐growth factor (HGF)‐regulated growth factor substrate (Hrs) complex, corresponds to the Vps27p–Hse1p complex in S. cerevisiae. The others are ESCRT‐I, ESCRT‐II, and ESCRT‐III (see Table 1). At the cytoplasmic surface of the early endosome, cargo is first captured by ESCRT‐0. At the same time, the cargo is retained in a flat clathrin lattice bound to ESCRT‐0. Cargo is then handed off to ESCRT‐I and subsequently to ESCRT‐II. Close to the final step of sorting, the ESCRT‐III complex, which is composed of at least seven chromatin modifying proteins or charged MVB proteins (CHMP) (CHMP 1–7),( 9 ) concentrates the cargo on the maturing endosomes.( 1 , 10 ) Finally, additional class E Vps and ESCRT‐related AAA‐ATPases, Vps4A and Vps4B, induce dissociation of the ESCRT complex in an ATP‐dependent manner. After this final step, by some unknown mechanism, the cargo and the portion of membrane surrounding them invaginates into the endosome, forming the MVB. Eventually the invaginations separate from the limiting membrane to form the ILV, which carry the cargo proteins on their luminal surface. After the sorting process is complete, the ESCRT and related molecules return to the cytosol, from which they are recruited to the endosomes for reuse in cargo sorting.
Table 1.
Endosomal sorting complex required for transport (ESCRT) and ESCRT‐related proteins
| Complex | Mammalian | Accession | Ub‐binding | Other domains | S. cerevisiae |
|---|---|---|---|---|---|
| ESCRT‐0 | STAM1 | NP_477448 | UIM, VHS | SH3 | Hse1p |
| STAM2 | NM_005843 | UIM, VHS | SH3 | ||
| Hrs | NP_722831 | UIM, VHS | FYVE, coiled‐coil | Vps27p | |
| ESCRT‐I | Tsg101 | NP_006283 | UEV | Coiled‐coil | Vps23p |
| Vps37A | AAH22363 | Coiled‐coil | VpS37p | ||
| Vps37B | NP_078943 | Coiled‐coil | |||
| Vps37C | NP_060436 | Coiled‐coil | |||
| Vps37D | XP_379866 | Coiled‐coil | |||
| Vps28 | NP_057292 | Vps28p | |||
| MVB12A | Mvb12 (YGR206W) | ||||
| MVB12B | |||||
| ESCRT‐II | EAP30 | NP_009172 | Vps22p | ||
| EAP20 | NP_115729 | Vps25p | |||
| EAP45 | NP_057159 | NZF‐C | NZF‐N (GLUE) | Vps36p | |
| ESCRT‐III | CHMP2A | NP_055268 | SNF7, coiled‐coil | Vps2p/Did4p | |
| CHMP2B | NP_940818 | SNF7, coiled‐coil | |||
| CHMP6 | NP_078867 | SNF7, coiled‐coil | VPS20p | ||
| CHMP3 | NP_057163 | SNF7, coiled‐coil | Vps24p | ||
| CHMP4A | NP_789782 | SNF7, coiled‐coil | Vps32p/Snf7p | ||
| CHMP4B | AAH14321 | SNF7, coiled‐coil | |||
| CHMP4C | BC014321 | SNF7, coiled‐coil | |||
| CHMP1A | NM_002768 | SNF7, coiled‐coil | Did2p | ||
| CHMP1B | NP_065145.2 | SNF7, coiled‐coil | |||
| CHMP5 | NP_057494.2 | SNF7, coiled‐coil | Vps60p | ||
| CHMP7 | NM_152272 | SNF7, coiled‐coil | |||
| Others | Vps4A | NP_037377.1 | MIT, AAA‐ATPase | Vps4p | |
| Vps4B | NP_004860.2 | MIT, AAA‐ATPase | |||
| Alix/AIP1 | NP_037506.2 | Bro1, coiled‐coil | Vps31/Bro1p | ||
| LIP5 | NM_016485 | Vta1p |
The four ESCRT complexes are each made up of several molecules. Both the mammalian and S. cerevisiae gene names are listed. Note that some yeast genes have multiple orthologues in mammals. AAA, ATPases associated with a variety of cellular activities; CHMP, chromatin modifying protein, later renamed charged multivesicular body proteins; EAP, ELL‐associated protein; FYVE, Fab, YOTB, Vac1, and EEA‐1; GLUE, GRAM‐like ubiquitin‐binding in EAP45; LIP, LYST‐interacting protein; MIT, microtubule interaction and trafficking; NZF, Npl4‐type zinc finger; NZF‐C, C‐terminal NZF; NZF‐N, N‐terminal NZF; STAM, signal transducing adaptor molecule; SH3, Src homology 3; SNF7, sucrose non‐fermenting 7; Tsg101, tumor‐susceptibility gene 101; Ub, ubiquitin; UEV, ubiquitin E2 variant; UIM, ubiquitin‐interacting motif; VHS, Vps27‐Hrs‐STAM; Vps, vacuolar protein sorting.
The main and best‐studied signal for sorting to the MVB is ubiquitination. Ubiquitin is a highly conserved protein of 76 amino acids that can be covalently conjugated to specific protein substrates by a cascade of ubiquitin‐conjugating systems, including E1, E2, and the catalytic proteins called E3.( 11 ) Cargo is ubiquitinated by the appropriate E3 on cytoplasmic lysine (K) residues. Because ubiquitin itself has lysine residues, the ubiquitin covalently attached to the cargo can itself be ubiquitinated, eventually forming a ubiquitin chain. The best‐studied ubiquitin chains are lysine 48 (K48)‐linked polyubiquitins, which direct the modified protein to be degraded by proteasomes.( 12 , 13 ) More recently, K29‐ and K63‐linked ubiquitin chains were shown to mediate other cellular functions, including DNA repair and endocytosis.( 14 ) Monoubiquitination of target proteins, by contrast, marks them for endocytosis and MVB sorting.( 15 , 16 ) The single ubiquitin moiety is recognized by a variety of ubiquitin‐interacting domains in the ESCRT.( 6 , 17 ) These include the ubiquitin‐interacting motif domain of ESCRT‐0 (STAM and Hrs),( 18 ) the VHS (Vps27‐Hrs‐STAM) domain of STAM,( 19 ) the ubiquitin E2 variant domain of Tsg101 in ESCRT‐I,( 20 ) and the Npl4‐type zinc finger (NZF) or NZF‐C domain of an ESCRT‐II molecule, eleven‐nineteen lysine‐rich (ELL)‐associated protein (EAP) 45.( 21 ) The affinities of these ubiquitin interactions are pretty low (they typically have a K d of 10–500 µmol/L), which in part enables transient cargo binding. Another characteristic of the ubiquitin interaction is that the ubiquitin interaction domains share a binding surface that includes ubiquitin's isoleucine 44 (I44). This finding indicates that ubiquitinated cargo can only be recognized by a single ubiquitin interaction domain at a time.
Apart from its role in recognizing ubiquitinated cargo, the ESCRT‐I molecule Tsg101 binds the proline‐serine‐alanine‐proline (PSAP) motif of the ESCRT‐0 molecule Hrs,( 22 ) and the EAP45 molecule of ESCRT‐II binds Tsg101 though Tsg101's carboxyl terminus NZF (NZF‐C).( 23 ) The double binding mechanisms of the ESCRT‐cargo complex and between the ESCRT complexes seem to assure the sequential transfer of ubiquitinated cargo from upstream to downstream ESCRT. Unlike other ESCRT, however, ESCRT‐III does not possess any ubiquitin‐interacting motifs. Instead, ESCRT‐III is anchored to the endosomal membrane via myristoylation, and it associates with at least two deubiquitinating enzymes (DUB), associated molecule with the SH3 domain of STAM (AMSH) and UBPY.( 24 , 25 ) One possible role for these DUB is to remove ubiquitin from the cargo before its incorporation into the ILV, probably to maintain a sufficient level of free ubiquitin within the cell; however, ubiquitin removal seems not to be essential for the formation of ILV.( 26 )
Roles of individual ESCRT in cellular functions
Endosomal sorting compex required for transport‐0 (STAM–Hrs complex). Hrs and STAM1 or STAM2 (here, ‘STAM’ is used for both molecules) comprise the ESCRT‐0 complex (Fig. 1).( 27 ) The ESCRT‐0 complex has a key function in the initial recognition of ubiquitinated cargo for sorting. It is recruited to the endosomes when cargo is to be sorted, and binds to the flat clathrin lattice.( 18 ) Hrs is anchored via its FYVE domain to the outer surface of the endosome by binding to the endosome‐specific lipid phosphatidylinositol 3‐phosphate (PtdIns3P). Both Hrs and STAM, like Vps27/Hse1 in yeast, bind ubiquitin and are thought to mediate multivalent interactions with ubiquitinated cargo and to transfer the cargo to ESCRT‐I.( 18 , 22 , 28 , 29 ) The crystal structure analysis of ESCRT‐0 shows two intertwined GAT domains, which recruit both ubiquitin ligases and deubiquitinating enzymes.( 30 ) This suggests that ESCRT‐0 has dual functions in terms of ubiquitination and deubiquitination, and is thereby involved in determining whether to sort (ubiquitinate) the cargo or not (i.e. to deubiquitinate it). ESCRT‐0 therefore has a scaffolding role in the MVB‐sorting of ubiquitinated cargos.
Figure 1.
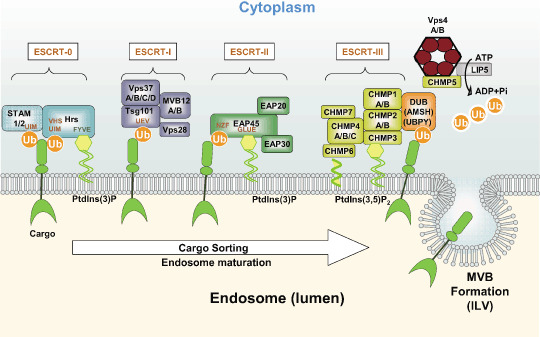
Schematic showing the functions of endosomal sorting complex required for transport (ESCRT) and ESCRT‐related proteins in endosomal sorting of ubiquitinated membrane proteins (cargo). Ubiquitinated cargo is recognized by the ESCRT‐0 complex by binding of the ubiquitin‐interacting motif (UIM) and vacuolar protein sorting (Vps) 27‐hepatocyte‐growth factor‐regulated growth factor substrate (Hrs)‐signal transducing adaptor molecule (STAM) (VHS) domains of STAM and Hrs, respectively. Hrs is anchored to the endosomal surface by the FYVE–PtdIns3P interaction. Cargo is then handed off to the ESCRT‐I complex, which is recruited via Tsg101's ubiquitin E2 variant (UEV) domain. ESCRT‐I (Tsg101) also has an affinity for ESCRT‐0 (Hrs; this interaction is not indicated in the figure). Likewise, ESCRT‐II is recruited by the Vps28–EAP45 interaction, and the Npl4‐type zinc finger (NZF)–ubiquitin (Ub) interaction. The GLUE domain is involved in the membrane binding of ESCRT‐II via PtdIns3P. ESCRT‐III associates with the membrane via the myristoylation of charged multivesicular body protein (CHMP) 6, and the association of CHMP3 with PtdIns3,5P2. ESCRT‐III is thought to be assembled on membranes as a heteromultimer. In the final steps of sorting, ubiquitins are removed from the cargo by the deubiquitinating enzymes (DUB), including association molecule with the SH3 domains of STAM (AMSH) and ubiquitin‐specific peptidase Y (UPBY), and the ESCRT complexes are dissociated by the ATPase activity of Vps4. The Vps4 complex is composed of a Vps4A–Vps4B heteromer that associates with LIP5. Deubiquitinated cargo is carried with the invaginating membrane into the multivesicular body (MVB) lumen and are thereby relocated in intraluminal vesicles (ILV). However, the requirement for deubiquitination at the end of sorting is still controversial. Not all known protein–protein interactions are indicated in the figure.
Multivesicular bodies in Hrs‐depleted cells are greatly enlarged and have few internal vesicles.( 31 ) Hrs seems to be required primarily for the formation of internal vesicles, without affecting the number of MVB per unit cytoplasm. However, the degree of endosome enlargement is greater in Hrs‐depleted cells than in Tsg101 (ESCRT‐I)‐depleted cells. This discrepancy between the depletion of Hrs and of molecules from the other ESCRT suggests that ESCRT‐0 possesses a distinct role in vesicular trafficking. One such role is probably Hrs's ability to inhibit homotypic fusion between endosomes. Hrs prevents the formation of a functional endosomal soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor complex by binding to synaptosome‐associated protein‐25.( 32 ) Another role of Hrs is its involvement in autophagy: Hrs is found on autophagosomes, and facilitates autophagosome–lysosome fusion events.( 33 )
Knocking out Hrs,( 34 , 35 ) or both STAM1 and STAM2, causes embryonic lethality.( 36 ) STAM1 knockout mice develop neurodegeneration.( 37 ) These observations indicate that ESCRT‐0 plays major roles in development and neuronal survival.
Endosomal sorting complex required for transport‐I. ESCRT‐I was originally characterized as a heterotrimer of Tsg101 (Vps23p in yeast), the Vps37 proteins (Vps37A, ‐B, ‐C, and ‐D), and Vps28, but recently new members of the complex, MVB12A and MVB12B, were identified.( 38 , 39 ) Experiments using yeast ESCRT‐I proteins indicate that MVB12A or MVB12B engages ESCRT‐I directly with a nanomolar affinity to form a 1:1:1:1 heterotetramer, but that it has no effect on the affinity of ESCRT‐I for ESCRT‐II. ESCRT‐I mutants in yeast show a typical class E phenotype, except when an MVB12 is the mutant, in which case the phenotype is much milder. Tsg101 depletion in mammalian cells reduces the number of MVB per unit volume of cytoplasm, and causes major morphological changes to EGFR‐containing early endosomes, resulting in tubular clusters and multilamellar cisternae.( 31 , 40 ) The tubular morphology resembles that of recycling compartments, but these tubular clusters are negative for Rab11, a marker for recycling endosomes. This finding suggests that the tubulation caused by Tsg101 depletion does not simply reflect the impaired vesiculation of compartments headed toward the cell surface. Rather, Tsg101 seems to be essential for the formation of stable vacuolar domains within the early endosome, which eventually develop into MVB.
Deletion studies of Tsg101 in primary cell cultures indicate that it is indispensable for normal cell growth and cell survival. Accordingly, Tsg101‐deficient mice die in utero during early development.( 41 ) Detailed studies in Drosophila provide additional interesting findings.( 42 ) Homozygous mutations in erupted (Tsg101) or vps25 (of ESCRT‐II) trigger excess growth of the developing eye imaginal disc, an organ composed primarily of polarized epithelial cells. The proliferative properties of the Tsg101 mutant cells are highly context dependent: in some cases they overproliferate; in others they die by apoptosis. In mosaic flies, Tsg101 mutant cells can also induce ectopic cell proliferation in the surrounding wild‐type cells. With their non‐cell autonomous growth characteristics, Tsg101 and vps25 thus represent a novel class of Drosophila endocytic tumor‐suppressor genes that control developmental patterns of cell proliferation. One underlying mechanism seems to be mediated through the Crumbs apicobasal polarity pathway.( 43 ) In Tsg101 mutant cells, the Crumbs protein mislocalizes to a subapical, cytoplasmic domain, which may alter its ability to influence the pathways that define the apical and basolateral membrane domains. A clear picture of the roles played by ESCRT‐I and ‐II in cell polarity in humans awaits further analysis.
Endosomal sorting complex required for transport‐II. ESCRT‐II is composed of EAP20 (Vps25), EAP30 (Vps22), and EAP45 (Vps36). A heterotetramer consisting of two EAP20 and one each of EAP30 and EAP45 forms the shape of a capital letter Y.( 44 , 45 , 46 , 47 ) Simultaneous and reinforcing interactions between ESCRT‐II and the endosomal membrane (i.e. via PtdIns3P), ESCRT‐I (Vps28), and ubiquitin are critical for the progression of ubiquitinated cargo from early to late endosomes. Although ESCRT‐II mutations in yeast and Drosophila melanogaster induce similar class E compartments, mammalian EAP30 depletion induces abnormally small endosomes, suggesting that ESCRT‐II has different roles in Drosophila and mammals in terms of MVB morphogenesis.( 48 ) Accordingly, mammalian ESCRT‐II proteins seem to be dispensable for some cargo sorting, including the sorting of major histocompatibility complex (MHC)‐class I molecules.( 49 ) One way ESCRT‐II could be ‘skipped’ during sorting is if the ESCRT‐related molecule Alix (AIP1) connects ESCRT‐I and ESCRT‐III, enabling ESCRT‐II to be bypassed. However, as S. cerevisiae encodes similar molecules, Bro1p and Rim20p,( 50 ) a bypass mechanism may not account for the dispensability of ESCRT‐II in mammals. Conversely, the degradation of both CXCR4 and EGFR is significantly reduced by EAP30 depletion in HeLa cells.( 48 ) Therefore, it seems that different cargo have different requirements for ESCRT‐II in their degradation pathway.
Endosomal sorting complex required for transport‐III and related molecules. ESCRT‐III consists of structurally related CHMP family proteins (CHMP 1–7), which have basic N‐ and acidic C‐termini and form higher‐order multimers on membranes. Certain CHMP have different isoforms, such as CHMP4A, ‐B, and ‐C (see Table 1). ESCRT‐III contains two functionally distinct subcomplexes. The CHMP6–CHMP4 (Vps20–Snf7) subcomplex binds to the endosomal membrane via the myristoyl group of CHMP6 (Vps20).( 51 ) The other subcomplex, CHMP2–CHMP3 (Vps2‐Vps24), binds to the lipid PtdIns3,5P2, as well as to the CHMP6–CHMP4 (Vps20–Snf7) complex, and recruits additional cofactors to the site. First, the CHMP2–CHMP3 (Vps2–Vps24) subcomplex recruits Vps4 to endosomes via an interaction between the microtubule‐interacting and trafficking (MIT) domain of Vps4 and the MIT‐interacting motif of CHMP2 (Vps2).( 52 ) Vps4 exists as a dimer in its ADP‐bound state, whereas ATP binding induces its assembly into a homooligomeric complex,( 53 ) apparently a hexamer similar to other AAA‐ATPases.( 54 ) Vps4 also coassembles with LIP5 (Vta1/SBP1), a regulator that also stimulates the ATPase activity of the Vps4 complex.( 55 , 56 , 57 ) CHMP5 (Vps60) further binds to LIP5, and the CHMP5–LIP5 subcomplex seems to be necessary for Vps4 activation.( 50 , 56 , 58 ) Second, the CHMP2–CHMP3 (Vps2–Vps24) subcomplex recruits CHMP1 (Did2). CHMP1 binds Ist1, a positive modulator of MVB sorting, and they form another subcomplex that directs Vps4 to dissociate the ESCRT‐III complex.( 58 ) Although a mammalian orthologue for Ist1 remains unknown, a similar mechanism for recruiting and supporting Vps4 activity is likely to be present in mammals.( 59 ) Finally, ESCRT‐III recruits the DUB to the MVB‐sorting site. CHMP3 binds one of the DUB, AMSH, through the CHMP3 C‐terminal domain, and a newly identified CHMP7 binds another DUB, UBPY, as well as CHMP4B.( 60 , 61 ) Collectively, these findings indicate that ESCRT‐III is a multimolecular complex that sorts and deubiquitinates cargo, and whose function is coupled with active dissociation events.
That ESCRT‐III function is biologically significant is revealed by the depletion of one of its core molecules, CHMP3. CHMP3‐depleted cells show impaired cargo degradation. Similarly, the expression of mutated CHMP2, CHMP4, or CHMP6 in cultured cells causes ubiquitinated cargo to accumulate on enlarged endosomes.( 62 ) CHMP5‐knockout murine cells manifest enlarged endosomes with accumulated ILV containing EGFR and transforming growth factor‐β (TGFβ) receptor type II.( 63 ) The dissociation of the ESCRT machinery by Vps4 is also necessary for the formation of the ILV, and Vps4 inactivation induces the accumulation ESCRT‐III on endosomes. Collectively, these data indicate that ESCRT‐III is not essential for ILV formation, but is required for the degradation of cargo. One reason for its essential role is that ESCRT‐III facilitates MVB–lysosome fusion, which leads to cargo degradation.( 64 )
Endosomal functions of ESCRT in cancer‐related cargo control. One major way that ESCRT contribute to cancer cell control is by downregulating tumor‐related receptors by sorting them into the ILV within the MVB.( 1 , 6 ) Two mechanisms could potentially affect the receptors’ function.( 65 ) First, the sequestration of activated receptors in the ILV could block their ability to transduce signals, and second, the amount of cell‐surface proteins can be modulated via the recycling or secretion machinery.
Most activated growth factor receptors continue to deliver signals after being endocytosed. Early endosomes containing activated receptors are in the ‘on’ state in terms of signaling, and such endosomes are therefore referred to as signaling endosomes. The termination of such signals can be achieved by the inward invagination of the MVB, which changes the location of the receptors from the MVB membrane to the ILV, the vesicles within vesicles. This mechanism permits the ESCRT to modulate the kinetics and amplitude of various signaling pathways from activated receptors.
The mechanism of regulating the presence of the receptors on the cell surface is called cargo breakdown. In this case, cell‐surface molecules that escape from sorting are redirected to the cell surface. By modulating the total amount of cell‐surface proteins, including, for example, adhesion molecules, the malignant phenotypes of cells can be influenced. In the following sections, we describe interactions between the ESCRT and particular cargo that can influence cell transformation.
Epidermal growth factor receptor. EGFR is one of the best‐studied receptor tyrosine kinases, and its excessive signaling is associated with the development of a variety of human cancers.( 66 ) EGF stimulation induces endocytosis of the EGFR as an EGF–EGFR complex,( 67 , 68 , 69 ) but none of the ESCRT is required for its ligand‐mediated internalization.( 70 ) Internalized EGFR is either recycled to the cell surface or sorted to the endosomes, and this decision depends on the strength of the signals. Low doses of EGF induce clathrin‐dependent internalization and recycling, and higher concentrations of EGF induce a clathrin‐independent, lipid raft‐dependent endocytosis that appears to be followed by degradation.( 71 ) In the latter case, EGFR is monoubiquitinated at multiple sites by a RING‐family E3, c‐Cbl, and this modification is essential for the ESCRT‐dependent sorting and degradation, although K63‐linked polyubiquitin chains are also involved (Fig. 2).( 72 , 73 ) However, ubiquitination is not required for EGFR internalization.( 74 )
Figure 2.
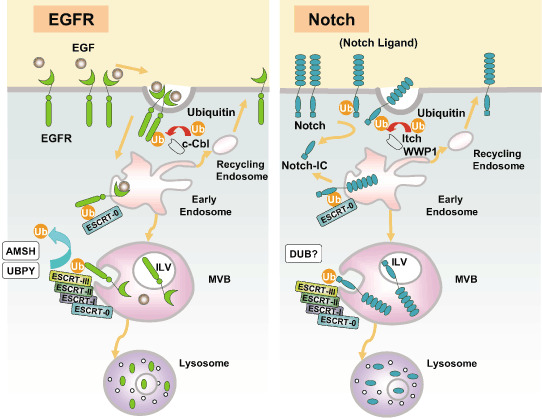
Endocytosis, endosomal sorting, and lysosome‐dependent degradation of epidermal growth factor receptor (EGFR) (left) and Notch (right). (Left) Stimulation of EGFR with epidermal growth factor (EGF) induces the activation and phosphorylation of EGFR. Ubiquitin ligases (E3) including c‐Cbl associate with the intracytoplasmic region of EGFR, which then ubiquitinates EGFR on multiple lysine residues (Ub indicates a single ubiquitin moiety attached to the EGFR). Ubiquitinated EGFR is internalized and transported to the early endosomes. Endosomal sorting complex required for transport (ESCRT)‐0 recognizes the ubiquitinated EGFR and recruits the downstream complex ESCRT‐I. (At this point, EGFR can still deliver a signal, but the signal seems to be terminated by the sequestration of the activated EGFR into the intraluminal vesicles [ILV] of the multivesicular bodies [MVB] later in the process.) The ubiquitinated EGFR is transferred to ESCRT‐II, and then to ESCRT‐III. EGFR is deubiquitinated by association molecule with the SH3 domains of STAM (AMSH) and ubiquitin‐specific peptidase Y (UBPY). The ESCRT complexes are then dissociated, and EGFR is sorted into the ILV within the MVB. Fusion of the MVB with the lysosome results in the degradation of EGFR and other MVB contents in an acid peptidase‐dependent manner. On early endosomes, some of the EGFR is deubiquitinated by UBPY and AMSH (not shown), sorted into the tubular portion of the endosomes, and recycled back to the cell surface via recycling endosomes. (Right) Notch heading for the endosome is ubiquitinated by E3 including Itch (AIP4 in human and Su[dx] in Drosophila) and WWP1, and internalized. ESCRT‐0 captures the ubiquitinated Notch, and it is handed off to ESCRT‐I, ‐II, and ‐III sequentially. Notch that is sorted into the ILV is degraded by the lysosomal acid peptidase. The Notch cellular domain (Notch‐IC) can be produced by proteolysis at the cell surface as well as on the endosomes.
Epidermal growth factor stimulation increases both MVB biogenesis and the inward vesiculation of the EGFR‐containing MVB, with the help of ESCRT‐0 (Hrs and STAM) and ESCRT‐I (Tsg101). EGFR degradation is impaired by the depletion of Hrs or of STAM1 and STAM2,( 75 ) and Hrs‐mutant Drosophila fail to downregulate EGFR. However, in mammals, the effect of depleting these molecules is much milder than that of depleting Tsg101, which causes a severe defect in EGFR degradation. This difference suggests that Tsg101 sorts EGFR in an ESCRT‐0‐independent manner, or that Tsg101 couples with alternative molecules that can substitute for ESCRT‐0, such as Tom1L1, which has both VHS and GAT domains and is capable of binding Tsg101. ESCRT‐II (EAP20) depletion also severely disrupts EGFR degradation.( 31 , 48 ) MVB‐sorting defects result in the activated EGFR staying longer on the limiting membrane of the aberrant endosomes, and ESCRT‐0 and ESCRT‐I inhibition lead to sustained EGFR signaling. On the other hand, although CHMP3 (ESCRT‐III) depletion induces a delay in EGFR degradation,( 70 ) it does not cause sustained signaling, because the receptor signaling is terminated before the engagement of ESCRT‐III.( 64 ) This finding partly reflects the fact that ESCRT‐III deficiency affects MVB–lysosome fusion but not ILV formation. A similar degradation delay with proper signal silencing is reported for ESCRT‐II (EAP30).( 48 ) Conversely, the depletion of Hrs or Tsg101, but not of ESCRT‐II (EAP30) or ESCRT‐III (CHMP3), causes enhanced recycling of endocytosed EGFR, although the mechanism remains unknown. EGFR fate is also controlled by the tyrosine phosphorylation and ubiquitination of Hrs, which regulate Hrs degradation.( 76 ) By modulating Hrs ubiquitination, phosphorylation, and protein levels, c‐Cbl may control the composition of the endosomal sorting machinery and its ability to target EGFR for lysosomal degradation.
Alterations that uncouple EGFR from c‐Cbl‐mediated ubiquitination and thereby from downregulation are tightly associated with the pathogenesis of cancer, and an EGFR mutant lacking only the direct c‐Cbl‐binding site transduces stronger mitogenic signals than the wild‐type receptor.( 77 ) The most common mutation of the EGFR in glioblastomas is the deletion of exons 2–7, known as the EGFRvIII mutation. This mutant receptor is constitutively active irrespective of ligand binding, but the forced overexpression of all three Cbl proteins, c‐Cbl, Cbl‐b, and Cbl‐c, results in its ubiquitination and degradation,( 78 ) and the downregulation of EGFRvIII by Cbl inhibits the transformation of NIH3T3 cells by EGFRvIII. Theoretically, as Cbl downregulates Hrs, a Cbl‐mediated therapeutic strategy could require the simultaneous supplementation of Hrs and other ESCRT. On the other hand, the most oncogenic member of the ErbB family, HER2, couples poorly with c‐Cbl because it lacks a c‐Cbl binding site, and therefore seems unlikely to be regulated by the ESCRT.( 79 ) Nevertheless, several Cbl‐based chimeric ubiquitin ligases have been examined for their potential to downregulate HER2.( 80 ) Chimeras consisting of the Cbl N‐terminal tyrosine kinase‐binding domain, a linker, and the RING domain, with the Cbl SH2 domain replaced with the SH2 from growth factor receptor‐binding protein (Grb) 2, Grb7, p85, or Src not only interacted with HER2 but also enhanced the downregulation of endogenous overexpressed HER2 by effectively ubiquitinating it and marking it for degradation. It is not surprising that this degradation is mediated by ESCRT.
c‐Met. The c‐Met receptor tyrosine kinase regulates a complex array of cellular behaviors collectively known as invasive growth. Although essential for normal development and wound repair, this program is frequently co‐opted by tumors to promote their own growth, motility, and invasion. c‐Met is overexpressed in a variety of human cancers, and this aberrant expression correlates with poor patient prognosis.( 81 ) After HGF stimulation, c‐Met is ubiquitinated, which links c‐Met with the ESCRT machinery. Cellular c‐Met levels are governed in part by c‐Cbl‐mediated ubiquitination and degradation, and uncoupling c‐Met from c‐Cbl‐mediated ubiquitination promotes its transforming activity. To bind with c‐Cbl, c‐Met requires its tyrosine residue Y1001, which is juxtaposed to the cytoplasmic side of the cell membrane, but neither its multifunctional docking site nor any additional C‐terminal tyrosine residues are required.( 82 ) Accordingly, a c‐Met mutant in which the juxtamembrane tyrosine is replaced by phenylalanine is not ubiquitinated, and it has transforming activity in fibroblast and epithelial cells.( 83 ) Furthermore, the ubiquitination‐deficient Met receptor mutant is also tumorigenic in vivo.( 84 ) These results suggest that the coupling of c‐Met with the ubiquitination and ESCRT pathway is essential for cells to avoid Met‐related malignant transformation.
Hrs (ESCRT‐0) was originally identified as a protein that was phosphorylated upon HGF stimulation.( 85 ) Hrs phosphorylation is dramatically reduced by treatment with a proteasome inhibitor. This suggests that proteasome inhibition reduces the intracellular ubiquitin pool, which abolishes the recruitment of Hrs to the ubiquitinated c‐Met. Indeed, c‐Met fails to reach the late endosomes or lysosomes in the presence of a proteasome inhibitor. Conversely, when ubiquitin is present, c‐Met is degraded by acid‐dependent proteases.( 86 ) Accordingly, depletion of Hrs modestly retards c‐Met degradation and markedly prevents the attenuation of c‐Met phosphorylation.( 87 ) Hrs therefore plays a role in signaling longevity from activated c‐Met, by controlling its degradation. Because c‐Met at least partially utilizes the same E3 (c‐Cbl) and sorting machinery as EGFR, it is likely that the MVB sorting of c‐Met also utilizes other ESCRT. Conversely, if a c‐Met mutant that cannot recruit c‐Cbl undergoes normal internalization and colocalization with Hrs, it is unable to induce the phosphorylation of Hrs. The fusion of monoubiquitin to the c‐Met mutant is sufficient for Hrs recruitment and phosphorylation, for decreasing Met receptor stability, and therefore for decreasing in vitro transformation. Therefore, Cbl‐dependent ubiquitination is dispensable for Met internalization, but is critical for targeting the Met receptor to the ESCRT and suppressing its inherent transforming activity.
An additional mechanism for c‐Met sorting was recently identified. In conditional knockout mice for the DUB UBPY (USP8), c‐Met expression is severely reduced.( 88 ) As UBPY binds ESCRT‐0 (STAM1 and STAM2) as well as ESCRT‐III, its role is antagonistic to that of these ESCRT, which are responsible for c‐Met degradation. Taken together, although there is little evidence of a pathogenic linkage between the ESCRT and c‐Met in humans, the c‐Met signal and its transforming activity can be significantly affected by the ESCRT.
Notch. The Notch signaling pathway plays a central role in animal growth and patterning, and its deregulation leads to many human diseases, including cancers.( 89 ) The Notch family of receptors and their ligands are transmembrane proteins, which are degraded after being internalized. Upon engagement by the Notch ligands, the Notch intracellular domain (Notch‐IC) is cleaved and functions as a nuclear coactivator for transcription. Although Notch‐IC is polyubiquitinated and degraded in a proteasome‐dependent manner in the nucleus, a substantial amount of intact Notch is present in endosomes.( 90 ) Studies in D. melanogaster have shown that Notch colocalizes with Rab5 and Rab7, an early and a late endosome marker, respectively.( 91 ) As expected, the endosomal Notch is degraded by the lysosomes, suggesting a possible involvement of the ESCRT sorting machinery in Notch pathways (Fig. 2). The Nedd4‐family HECT‐type E3, including Itch (Drosophila Su[dx]) and WWP1, target Notch for degradation.( 92 , 93 ) Itch and WWP1 associate with the intracellular domain of Notch via the WW domains in the E3 proteins and promote its ubiquitination. Furthermore, Itch interacts genetically with Notch1 in a mouse autoimmune disease model, and Itch is considered to be a negative regulator of Notch signaling.
The intimate relationship between the Nedd4 family E3 and the ESCRT in the sorting of other cargo suggests that a similar interaction controls Notch degradation in the endosome. In fact, a D. melanogaster Vps25 (ESCRT‐II) mutant causes Notch to accumulate on endosomes and upregulates Notch signaling.( 94 , 95 ) The sustained signal is sensitive to a γ‐secretase inhibitor, suggesting that the Notch cleavage machinery is preserved on the Vps25‐depleted endosomes. In another mutant, erupted, a fly equivalent to ESCRT‐I Tsg101, similar Notch accumulation is induced along with sustained signaling. Depletion of the ESCRT‐III component vVps32 (CHMP4) also increases Notch signaling. This relationship between Notch and ESCRT can even be found at the level of ESCRT‐0. Notch colocalizes with Hrs, and Drosophila Su(dx) regulates the postendocytic sorting of Notch within the early endosome to a Hrs‐ and ubiquitin‐enriched subdomain en route to the late endosome. Accordingly, a Hrs mutant induces an increase in Notch signaling,( 96 ) although it is unclear if the Hrs mutant also induces a sustained signal. It is possible that ESCRT‐I and ESCRT‐II recognize Notch irrespective of ESCRT‐0, or an unknown alternative pathway links Notch and ESCRT‐I. The latter possibility could be mediated by VHS and GAT domain‐containing proteins, such as the GGA and TOM1 family proteins, which bind ubiquitinated proteins. An alternative possibility is that the Hrs deficiency augments the recycling process, and the activation of Notch recycling helps to sweep away the accumulated Notch from the endosomes.
Multivesicular body sorting defects of Notch lead to overgrowth phenotypes in vivo, when apoptosis is blocked.( 94 , 97 ) This deregulated growth occurs in a non‐cell‐autonomous manner.( 95 ) When ESCRT are blocked, secretion of unpaired (upd), a target gene of Notch, increases, and induces overgrowth of the surrounding cells via the JAK–STAT pathway. These results suggest that the endocytic sorting of Notch by ESCRT mediates a decision between its activation and downregulation. It seems that the endosomal system provides key points for the modulation of Notch activity by sorting proteins as well as by other signals.
E‐cadherin. A hallmark characteristic of epithelial tumor progression is the loss of the epithelial phenotype and acquisition of a motile or mesenchymal phenotype. Such epithelial‐to‐mesenchymal transitions (EMT) are accompanied by the loss of E‐cadherin function by either transcriptional or post‐transcriptional mechanisms.( 98 , 99 ) Upon the triggering of an EMT, E‐cadherin is internalized and then shuttled to the lysosome, instead of being recycled to the lateral membrane.( 100 ) The modification of E‐cadherin by ubiquitin is essential for its sorting to the lysosome, which occurs through a collaborative process mediated by ESCRT‐0 (Hrs) and the activation of two specific Rab GTPases, Rab5 and Rab7. A c‐Cbl‐like HECT‐family E3, Hakai, monoubiquitinates E‐cadherin and seems to be one of the key regulators for its sorting and degradation.( 101 ) Another E3 ubiquitin ligase, MDM2, interacts in vivo with E‐cadherin on the cell surface and early endosome, resulting in its ubiquitination and degradation.( 102 ) These results suggest the ESCRT are likely to be involved in E‐cadherin degradation, and theoretically in EMT. However, despite much searching, no tumors deficient in Hrs or other ESCRT have been reported. Rather, Hrs expression is significantly increased in a variety of human tumors, compared with normal tissues.( 103 ) It is more likely that Hrs increases, rather than decreases, the malignancy of transformed cells. This hypothesis is supported by a series of experiments showing that Hrs depletion attenuates the proliferation, anchorage‐independent growth, tumorigenesis, and metastatic potential of HeLa cells in vitro and in vivo, and the restoration of Hrs reverses these effects. The upregulation of E‐cadherin and reduced β‐catenin signaling appear to be at least partially responsible for the effects of Hrs depletion. The aberrant accumulation of E‐cadherin on the cell surface most likely results from impaired E‐cadherin degradation in the lysosomes, accompanied by normal or elevated recycling. These results suggest that Hrs may play a critical role in determining the malignancy of cancer cells by regulating E‐cadherin degradation.
Non‐endosomal functions of ESCRT in cancer pathogenesis
Endosomal sorting complexes required for transport control cytokinesis. Much is still unclear about how cells divide into two daughter cells and complete cell division. Cell division in mammals is called cytokinesis, and consists of at least three distinct steps: central spindle assembly, cleavage furrow formation, and the final abscission at the midbody. The final abscission requires breakage of the midbody, a thin membranous stalk connecting the daughter cells. Interestingly, this membrane fission event topologically resembles the pinching‐off step of MVB formation. Carlton et al. recently found that two MVB‐sorting proteins, Tsg101 and Alix, ESCRT‐I‐ and ESCRT‐related, respectively, are recruited to the midbody during cytokinesis by an interaction with centrosome protein 55 (Cep55), a centrosome and midbody protein essential for abscission (Fig. 3).( 104 ) This remarkable finding was extended by Morita et al. who showed that Alix and Tsg101 bind a series of proteins involved in cytokinesis, including Cep55, CD2AP, ROCK1, and IQGAP1.( 105 ) As a result, Tsg101 and Alix, and other ESCRT such as ESCRT‐III and Vps4, concentrate at Flemming bodies during abscission. Depletion of Alix and ESCRT‐I inhibits cytokinesis, and results in an increased number of multinuclear cells. Cytokinesis failure is often accompanied by the generation of cells with an unstable tetraploid content, which predisposes the cells to develop aneuploidy and malignancies. ESCRT may therefore be involved in tumor pathogenesis arising from defective cytokinesis.
Figure 3.

Endosomal sorting complex required for transport (ESCRT) recruitment in multivesicular body (MVB) sorting and cytokinesis. Tsg101 (ESCRT‐I) and Alix are recruited to specialized sites on the membrane by their association with hepatocyte‐growth factor‐regulated growth factor substrate (Hrs) and CEP55, respectively, to carry out similar roles in the terminal membrane fission events of MVB biogenesis and cytokinesis.( 105 ) ESCRT‐III is further recruited by the interactions shown in the figure, and assembled to fulfill its functions by cooperating with vacuolar protein sorting (Vps) A and B.
RNA polymerase control and gene silencing by ESCRT. Mammalian ESCRT‐II was first identified as part of the RNA polymerase II elongation factor ELL complex. For this reason, ESCRT‐II members have been investigated as RNA polymerase modifiers.( 106 ) An ELL‐containing complex that also contains three ESCRT‐II proteins (EAP45, EAP30, EAP20) was named the Holo‐ELL complex, and it is responsible for setting the catalytic rate of transcription elongation by RNA polymerase II.( 107 ) Because ESCRT‐II is not essential for mammalian MVB sorting, at least for several cargo, and ESCRT‐II partially localizes to the nucleus, the nuclear function of ESCRT‐II is of particular interest.( 49 ) A study in D. melanogaster provides interesting insight into one such ESCRT‐II function.( 108 ) The mRNA for bicoid localizes to the anterior of the Drosophila egg, where it is translated to form a morphogenic gradient of Bicoid protein that patterns the head and thorax of the embryo. bicoid mRNA is coupled to the microtubule‐dependent transport pathway by binding Staufen, which forms a complex with three ESCRT‐II proteins, Vps22 (EAP30), Vps25 (EAP20), and Vps36 (EAP45). Accordingly, a mutant form of any of these ESCRT‐II proteins abolishes the bicoid mRNA gradient. The MVB sorting‐independent function of ESCRT‐II in mammals, which is not seen with ESCRT‐I or ‐III, suggests mammalian ESCRT‐II may have a similar RNA‐binding property.
Mammalian ESCRT‐III molecules also seem to have non‐endosomal functions, because they were originally identified as nuclear proteins. It was identified in a screen for conserved partners of the Polycomb‐group (PcG) protein Polycomblike, which represent a diverse set of proteins involved in the maintenance of gene silencing during development.( 9 ) CHMP1 contains a predicted bipartite nuclear localization signal and occurs as distinct forms in the cytoplasm and the nuclear matrix. CHMP1 in the nucleus recruits a PcG protein, BMI1, to these regions of condensed chromatin and can cooperate with coexpressed vertebrate Polycomblike in a Xenopus embryo PcG assay. These observations suggest that CHMP1 plays a role in stable gene silencing in the nucleus. Although the involvement of other ESCRT‐III members in gene silencing is unknown, it is possible that the ESCRT‐III complex has a significant nuclear role.
Exosome formation and secretion. Exosomes are small membrane vesicles of endocytic origin that are secreted by most cells in culture.( 109 ) Accumulating evidence suggests that exosomes play a physiological role in antigen presentation that regulates the immune response against tumors.( 110 ) A recent observation illustrates a pertinent role of the ESCRT, including the Tsg101 protein, in exosome secretion. Exosomes arise from the MVB, and are in fact ILV. Therefore, proteins and lipids contained in (or on) the secreted exosomes are cargo that was sorted into the MVB or are sorting‐associated molecules. The in vivo function of exosomes has been best studied in the field of immunity. Exosomes secreted by the dendritic cells regulate immune activation by combinatorial presentation of MHC class I and class II and the costimulatory molecule CD86, with the result of activating T cells and suppressing tumor growth. Conversely, tumors release exosomes that direct the deletion of reactive lymphocytes. Tumor‐derived exosomes were recently explored as potential vaccines.( 111 ) However, although exosomes express tumor antigens and can be used as tumor vaccines, they also induce apoptosis. In short, the sophisticated and specific use of the exosomal machinery for the reutilization and transfer of cell‐surface molecules, including tumor‐related antigens, needs to be analyzed further.
From a different perspective, exosome formation and secretion is surprisingly similar to the budding of human immunodeficiency virus, in that both utilize ESCRT.( 112 ) Findings such as this from virus studies can be usefully applied to the study of exosomes in the future.
Endosomal sorting complexes required for transport as cell‐cycle‐control and tumor‐maintenance factors. Two of the ESCRT‐I proteins, Tsg101 and Vps37, have been considered to be tumor‐suppressor genes because their genetic loci map to chromosomal regions that are often deleted or mutated in malignancies.( 113 , 114 ) A genetic screen for potential tumor suppressors using insertional mutagenesis in murine NIH3T3 cells also identified Tsg101 as one such candidate. In subsequent studies, genomic deletions and aberrant splice variants of tsg101 were found in sporadic forms of breast cancer and in a variety of other human malignancies.( 115 , 116 , 117 ) However, since then, the observation of genomic deletions of tsg101 has not been verified, and the tsg101 variants all turned out to be alternative splice products generated by exon skipping.( 118 ) The effect of the loss of Tsg101 function in vivo was examined using mice that lack Tsg101 in all cells or specifically in the mammary epithelial cells of adult females.( 41 , 119 ) Tsg101 knockout mice die in utero, but neither haploinsufficiency of tsg101 nor the deletion of both tsg101 alleles in mammary epithelial cells resulted in the development of cancer. Collectively, these studies indicate that Tsg101 is unlikely to be a tumor suppressor.
In contrast, several recent studies suggest that Tsg101 has a tumor‐maintenance function, a possibility that is augmented by the finding that Tsg101, and hence at least ESCRT‐I, plays a role in cell‐cycle control.( 120 ) Two negative regulators of the cell cycle, p53 and p21, accumulate in cells and tissues deficient for Tsg101, and Tsg101 reduces the level of p53 through lysosomal degradation by stimulating its ubiquitination by an E3 ligase, MDM2.( 121 ) These results indicate that Tsg101 is essential for the proliferation and survival of fully neoplastic cells. In support of this idea, tsg101 is upregulated in selected human malignancies, including a subset of invasive human breast cancers.( 122 ) Based on this observation, transgenic mice overexpressing Tsg101 in the developing mammary gland were made and analyzed. Although malignant transformation of mammary epithelia was observed, it was restricted to aging female mice. Judging from the long latency, Tsg101 seems to possess weak oncogenic properties. Like Hrs, which is highly expressed in a variety of cancers,( 103 ) Tsg101 is more likely to be essential for the support of cancer cells than to be an initiator of tumor formation.
Perspectives
With powerful support from yeast genetics, many details of the fascinating machinery for sorting membrane‐bound cargo to the endosomes have been elucidated. Recent progress in cell biology and molecular biology has helped identify the players within the mammalian MVB‐sorting complex, as well as their modes of action. These specialized sorting events are coordinated by protein ubiquitination, and the intimate relationship between this modification and vesicular trafficking is reminiscent of protein phosphorylation and signaling. Although most of the molecules involved in MVB sorting seem to have been identified already, some additional players and unexpected tricks remain. One reason to believe this is that the yeast genetic screens, which were originally used to identify the MVB‐sorting genes, were capable of indicating only mutants that allowed cell survival; therefore, several essential genes may be involved but escaped the screenings.
The essential role of the ESCRT in cytokinesis illustrates a promising avenue of study into the role of the ESCRT in tumorigenesis. Furthermore, many questions remain to be answered. Is ESCRT a tumor suppressor or an inducer? How do the ESCRT control individual key cancer molecules? Which tumors overexpress ESCRT or have lost their expression? The major challenge in elucidating the role of ESCRT in cancer pathogenesis is to define the precise roles of each ESCRT as well as of ESCRT‐related players such as the E3 and DUB. These efforts will require both clinical investigation and basic research using gene‐manipulation technologies. The current enthusiasm in this field is sure to lead soon to a better understanding of ESCRT's biological roles and their functions in cancer.
Acknowledgments
We thank Drs Pejman Soroosh and Noriko Isono for critical reading of the manuscript. This work was supported in part by a Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science, a Grant‐in‐Aid for Scientific Research on Priority Areas, and a Gant‐in Aid from Naito Foundation.
References
- 1. Gruenberg J, Stenmark H. The biogenesis of multivesicular endosomes. Nat Rev Mol Cell Biol 2004; 5: 317–23. [DOI] [PubMed] [Google Scholar]
- 2. Babst M. A protein's final ESCRT. Traffic 2005; 6: 2–9. [DOI] [PubMed] [Google Scholar]
- 3. Luzio JP, Rous BA, Bright NA, Pryor PR, Mullock BM, Piper RC. Lysosome–endosome fusion and lysosome biogenesis. J Cell Sci 2000; 113: 1515–24. [DOI] [PubMed] [Google Scholar]
- 4. Van Der Goot FG, Gruenberg J. Oiling the wheels of the endocytic pathway. Trends Cell Biol 2002; 12: 296–9. [DOI] [PubMed] [Google Scholar]
- 5. Murk JL, Humbel BM, Ziese U et al . Endosomal compartmentalization in three dimensions: implications for membrane fusion. Proc Natl Acad Sci USA 2003; 100: 13 332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Katzmann DJ, Odorizzi G, Emr SD. Receptor downregulation and multivesicular‐body sorting. Nat Rev Mol Cell Biol 2002; 3: 893–905. [DOI] [PubMed] [Google Scholar]
- 7. Hurley JH, Emr SD. The ESCRT complexes: structure and mechanism of a membrane‐trafficking network. Annu Rev Biophys Biomol Struct 2006; 35: 277–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Williams RL, Urbe S. The emerging shape of the ESCRT machinery. Nat Rev Mol Cell Biol 2007; 8: 355–68. [DOI] [PubMed] [Google Scholar]
- 9. Stauffer DR, Howard TL, Nyun T, Hollenberg SM. CHMP1 is a novel nuclear matrix protein affecting chromatin structure and cell‐cycle progression. J Cell Sci 2001; 114: 2383–93. [DOI] [PubMed] [Google Scholar]
- 10. Raiborg C, Rusten TE, Stenmark H. Protein sorting into multivesicular endosomes. Curr Opin Cell Biol 2003; 15: 446–55. [DOI] [PubMed] [Google Scholar]
- 11. Haglund K, Dikic I. Ubiquitylation and cell signaling. EMBO J 2005; 24: 3353–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weissman AM. Regulating protein degradation by ubiquitination. Immunol Today 1997; 18: 189–98. [DOI] [PubMed] [Google Scholar]
- 13. Glickman MH, Maytal V. Regulating the 26S proteasome. Curr Top Microbiol Immunol 2002; 268: 43–72. [DOI] [PubMed] [Google Scholar]
- 14. Hoeller D, Hecker CM, Dikic I. Ubiquitin and ubiquitin‐like proteins in cancer pathogenesis. Nat Rev Cancer 2006; 6: 776–88. [DOI] [PubMed] [Google Scholar]
- 15. Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol 2001; 2: 195–201. [DOI] [PubMed] [Google Scholar]
- 16. Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem 2003; 72: 395–447. [DOI] [PubMed] [Google Scholar]
- 17. Hicke L, Schubert HL, Hill CP. Ubiquitin‐binding domains. Nat Rev Mol Cell Biol 2005; 6: 610–21. [DOI] [PubMed] [Google Scholar]
- 18. Raiborg C, Bache KG, Gillooly DJ, Madshus IH, Stang E, Stenmark H. Hrs sorts ubiquitinated proteins into clathrin‐coated microdomains of early endosomes. Nat Cell Biol 2002; 4: 394–8. [DOI] [PubMed] [Google Scholar]
- 19. Mizuno E, Kawahata K, Kato M, Kitamura N, Komada M. STAM proteins bind ubiquitinated proteins on the early endosome via the VHS domain and ubiquitin‐interacting motif. Mol Biol Cell 2003; 14: 3675–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Teo H, Veprintsev DB, Williams RL. Structural insights into endosomal sorting complex required for transport (ESCRT‐I) recognition of ubiquitinated proteins. J Biol Chem 2004; 279: 28 689–96. [DOI] [PubMed] [Google Scholar]
- 21. Alam SL, Sun J, Payne M et al . Ubiquitin interactions of NZF zinc fingers. EMBO J 2004; 23: 1411–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bache KG, Brech A, Mehlum A, Stenmark H. Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J Cell Biol 2003; 162: 435–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Langelier C, Von Schwedler UK, Fisher RD et al . Human ESCRT‐II complex and its role in human immunodeficiency virus type 1 release. J Virol 2006; 80: 9465–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Clague MJ, Urbe S. Endocytosis: the DUB version. Trends Cell Biol 2006; 16: 551–9. [DOI] [PubMed] [Google Scholar]
- 25. Tanaka N, Kaneko K, Asao H et al . Possible involvement of a novel STAM‐associated molecule ‘AMSH’ in intracellular signal transduction mediated by cytokines. J Biol Chem 1999; 274: 19 129–35. [DOI] [PubMed] [Google Scholar]
- 26. Kyuuma M, Kikuchi K, Kojima K et al . AMSH, an ESCRT‐III associated enzyme, deubiquitinates cargo on MVB/late endosomes. Cell Struct Funct 2007; 31: 159–72. [DOI] [PubMed] [Google Scholar]
- 27. Asao H, Sasaki Y, Arita T et al . Hrs is associated with STAM, a signal‐transducing adaptor molecule: Its suppressive effect on cytokine‐induced cell growth. J Biol Chem 1997; 272: 32 785–91. [DOI] [PubMed] [Google Scholar]
- 28. Bilodeau PS, Winistorfer SC, Kearney WR, Robertson AD, Piper RC. Vps27‐Hse1 and ESCRT‐I complexes cooperate to increase efficiency of sorting ubiquitinated proteins at the endosome. J Cell Biol 2003; 163: 237–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Katzmann DJ, Stefan CJ, Babst M, Emr SD. Vps27 recruits ESCRT machinery to endosomes during MVB sorting. J Cell Biol 2003; 162: 413–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ren J, Kee Y, Huibregtse JM, Piper RC. Hse1, a component of the yeast Hrs‐STAM ubiquitin‐sorting complex, associates with ubiquitin peptidases and a ligase to control sorting efficiency into multivesicular bodies. Mol Biol Cell 2007; 18: 324–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Razi M, Futter CE. Distinct roles for Tsg101 and Hrs in multivesicular body formation and inward vesiculation. Mol Biol Cell 2006; 17: 3469–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun W, Yan Q, Vida TA, Bean AJ. Hrs regulates early endosome fusion by inhibiting formation of an endosomal SNARE complex. J Cell Biol 2003; 162: 125–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tamai K, Tanaka N, Nara A et al . Role of Hrs in maturation of autophagosomes in mammalian cells. Biochem Biophys Res Commun 2007; 360: 721–7. [DOI] [PubMed] [Google Scholar]
- 34. Komada M, Soriano P. Hrs, a FYVE finger protein localized to early endosomes, is implicated in vesicular traffic and required for ventral folding morphogenesis. Genes Dev 1999; 13: 1475–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miura S, Takeshita T, Asao H et al . Hgs (Hrs), a FYVE domain protein, is involved in Smad signaling through cooperation with SARA. Mol Cell Biol 2000; 20: 9346–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yamada M, Ishii N, Asao H et al . Signal‐transducing adaptor molecules STAM1 and STAM2 are required for T‐cell development and survival. Mol Cell Biol 2002; 22: 8648–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yamada M, Takeshita T, Miura S et al . Loss of hippocampal CA3 pyramidal neurons in mice lacking STAM1. Mol Cell Biol 2001; 21: 3807–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Morita E, Sandrin V, Alam SL, Eckert DM, Gygi SP, Sundquist WI. Identification of human MVB12 proteins as ESCRT‐I subunits that function in HIV budding. Cell Host Microbe 2007; 2: 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Audhya A, McLeod IX, Yates JR, Oegema K. MVB‐12, a fourth subunit of metazoan ESCRT‐I, functions in receptor downregulation. PLoS ONE 2007; 2: e956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Doyotte A, Russell MR, Hopkins CR, Woodman PG. Depletion of TSG101 forms a mammalian ‘Class E’ compartment: a multicisternal early endosome with multiple sorting defects. J Cell Sci 2005; 118: 3003–17. [DOI] [PubMed] [Google Scholar]
- 41. Wagner KU, Krempler A, Qi Y et al . Tsg101 is essential for cell growth, proliferation, and cell survival of embryonic and adult tissues. Mol Cell Biol 2003; 23: 150–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gilbert MM, Moberg KH. ESCRTing cell proliferation off the beaten path: lessons from the Drosophila eye. Cell Cycle 2006; 5: 283–7. [DOI] [PubMed] [Google Scholar]
- 43. Bilder D. Epithelial polarity and proliferation control: links from the Drosophila neoplastic tumor suppressors. Genes Dev 2004; 18: 1909–25. [DOI] [PubMed] [Google Scholar]
- 44. Babst M, Katzmann DJ, Snyder WB, Wendland B, Emr SD. Endosome‐associated complex, ESCRT‐II, recruits transport machinery for protein sorting at the multivesicular body. Dev Cell 2002; 3: 283–9. [DOI] [PubMed] [Google Scholar]
- 45. Teo H, Perisic O, Gonzalez B, Williams RL. ESCRT‐II, an endosome‐associated complex required for protein sorting: crystal structure and interactions with ESCRT‐III and membranes. Dev Cell 2004; 7: 559–69. [DOI] [PubMed] [Google Scholar]
- 46. Hierro A, Kim J, Hurley JH. Polycistronic expression and purification of the ESCRT‐II endosomal trafficking complex. Meth Enzymol 2005; 403: 322–32. [DOI] [PubMed] [Google Scholar]
- 47. Kostelansky MS, Sun J, Lee S et al . Structural and functional organization of the ESCRT‐I trafficking complex. Cell 2006; 125: 113–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Malerod L, Stuffers S, Brech A, Stenmark H. Vps22/EAP30 in ESCRT‐II mediates endosomal sorting of growth factor and chemokine receptors destined for lysosomal degradation. Traffic 2007; 8: 1617–29. [DOI] [PubMed] [Google Scholar]
- 49. Bowers K, Piper SC, Edeling MA et al . Degradation of endocytosed epidermal growth factor and virally ubiquitinated major histocompatibility complex class I is independent of mammalian ESCRTII. J Biol Chem 2006; 281: 5094–105. [DOI] [PubMed] [Google Scholar]
- 50. Bowers K, Lottridge J, Helliwell SB, Goldthwaite LM, Luzio JP, Stevens TH. Protein–protein interactions of ESCRT complexes in the yeast Saccharomyces cerevisiae . Traffic 2004; 5: 194–210. [DOI] [PubMed] [Google Scholar]
- 51. Babst M, Katzmann DJ, Estepa‐Sabal EJ, Meerloo T, Emr SD. Escrt‐III: an endosome‐associated heterooligomeric protein complex required for MVB sorting. Dev Cell 2002; 3: 271–82. [DOI] [PubMed] [Google Scholar]
- 52. Obita T, Saksena S, Ghazi‐Tabatabai S et al . Structural basis for selective recognition of ESCRT‐III by the AAA ATPase Vps4. Nature 2007; 449: 735–9. [DOI] [PubMed] [Google Scholar]
- 53. Babst M, Wendland B, Estepa EJ, Emr SD. The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J 1998; 17: 2982–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol 2005; 6: 519–29. [DOI] [PubMed] [Google Scholar]
- 55. Scott A, Chung HY, Gonciarz‐Swiatek M et al . Structural and mechanistic studies of VPS4 proteins. EMBO J 2005; 24: 3658–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Azmi I, Davies B, Dimaano C et al . Recycling of ESCRT by the AAA‐ATPase Vps4 is regulated by a conserved VSL region in Vta1. J Cell Biol 2006; 172: 705–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lottridge JM, Flannery AR, Vincelli JL, Stevens TH. Vta1p and Vps46p regulate the membrane association and ATPase activity of Vps4p at the yeast multivesicular body. Proc Natl Acad Sci USA 2006; 103: 6202–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rue SM, Mattei S, Saksena S, Emr SD. Novel Ist1–Did2 complex functions at a late step in MVB sorting. Mol Biol Cell 2008; 19: 475–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dimaano C, Jones CB, Hanono A, Curtiss M, Babst M. Ist1 Regulates Vps4 localization and assembly. Mol Biol Cell 2008; 19: 465–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Horii M, Shibata H, Kobayashi R et al . CHMP7, a novel ESCRT‐III‐related protein, associates with CHMP4b and functions in the endosomal sorting pathway. Biochem J 2006; 400: 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Row PE, Liu H, Hayes S et al . The MIT domain of UBPY constitutes a CHMP binding and endosomal localization signal required for efficient epidermal growth factor receptor degradation. J Biol Chem 2007; 282: 30 929–37. [DOI] [PubMed] [Google Scholar]
- 62. Shim S, Kimpler LA, Hanson PI. Structure/function analysis of four core ESCRT‐III proteins reveals common regulatory role for extreme C‐terminal domain. Traffic 2007; 8: 1068–79. [DOI] [PubMed] [Google Scholar]
- 63. Shim JH, Xiao C, Hayden MS et al . CHMP5 is essential for late endosome function and downregulation of receptor signaling during mouse embryogenesis. J Cell Biol 2006; 172: 1045–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bache KG, Stuffers S, Malerod L et al . The ESCRT‐III subunit hVps24 is required for degradation but not silencing of the epidermal growth factor receptor. Mol Biol Cell 2006; 17: 2513–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bache KG, Slagsvold T, Stenmark H. Defective downregulation of receptor tyrosine kinases in cancer. EMBO J 2004; 23: 2707–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schlessinger J. Ligand‐induced, receptor‐mediated dimerization and activation of EGF receptor. Cell 2002; 110: 669–72. [DOI] [PubMed] [Google Scholar]
- 67. Honegger AM, Schmidt A, Ullrich A, Schlessinger J. Separate endocytic pathways of kinase‐defective and ‐active EGF receptor mutants expressed in same cells. J Cell Biol 1990; 110: 1541–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Barbieri MA, Roberts RL, Gumusboga A et al . Epidermal growth factor and membrane trafficking: EGF receptor activation of endocytosis requires Rab5a. J Cell Biol 2000; 151: 539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Burke P, Schooler K, Wiley HS. Regulation of epidermal growth factor receptor signaling by endocytosis and intracellular trafficking. Mol Biol Cell 2001; 12: 1897–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Raiborg C, Malerod L, Pedersen NM, Stenmark H. Differential functions of Hrs and ESCRT proteins in endocytic membrane trafficking. Exp Cell Res 2008; 314: 801–13. [DOI] [PubMed] [Google Scholar]
- 71. Sigismund S, Woelk T, Puri C et al . Clathrin‐independent endocytosis of ubiquitinated cargos. Proc Natl Acad Sci USA 2005; 102: 2760–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol 2003; 5: 461–6. [DOI] [PubMed] [Google Scholar]
- 73. Huang F, Kirkpatrick D, Jiang X, Gygi S, Sorkin A. Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol Cell 2006; 21: 737–48. [DOI] [PubMed] [Google Scholar]
- 74. Huang F, Goh LK, Sorkin A. EGF receptor ubiquitination is not necessary for its internalization. Proc Natl Acad Sci USA 2007; 104: 16 904–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kanazawa C, Morita E, Yamada M et al . Effects of deficiencies of STAMs and Hrs, mammalian class E Vps proteins, on receptor downregulation. Biochem Biophys Res Commun 2003; 309: 848–56. [DOI] [PubMed] [Google Scholar]
- 76. Stern KA, Visser Smit GD, Place TL, Winistorfer S, Piper RC, Lill NL. Epidermal growth factor receptor fate is controlled by Hrs tyrosine phosphorylation sites that regulate Hrs degradation. Mol Cell Biol 2007; 27: 888–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Waterman H, Katz M, Rubin C et al . A mutant EGF‐receptor defective in ubiquitylation and endocytosis unveils a role for Grb2 in negative signaling. EMBO J 2002; 21: 303–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ryan PE, Davies GC, Nau MM, Lipkowitz S. Regulating the regulator: negative regulation of Cbl ubiquitin ligases. Trends Biochem Sci 2006; 31: 79–88. [DOI] [PubMed] [Google Scholar]
- 79. Levkowitz G, Klapper LN, Tzahar E, Freywald A, Sela M, Yarden Y. Coupling of the c‐Cbl protooncogene product to ErbB‐1/EGF‐receptor but not to other ErbB proteins. Oncogene 1996; 12: 1117–25. [PubMed] [Google Scholar]
- 80. Li X, Shen L, Zhang J et al . Degradation of HER2 by Cbl‐based chimeric ubiquitin ligases. Cancer Res 2007; 67: 8716–24. [DOI] [PubMed] [Google Scholar]
- 81. Haddad R, Lipson KE, Webb CP. Hepatocyte growth factor expression in human cancer and therapy with specific inhibitors. Anticancer Res 2001; 21: 4243–52. [PubMed] [Google Scholar]
- 82. Taher TE, Tjin EP, Beuling EA, Borst J, Spaargaren M, Pals ST. c‐Cbl is involved in Met signaling in B cells and mediates hepatocyte growth factor‐induced receptor ubiquitination. J Immunol 2002; 169: 3793–800. [DOI] [PubMed] [Google Scholar]
- 83. Peschard P, Fournier TM, Lamorte L et al . Mutation of the c‐Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol Cell 2001; 8: 995–1004. [DOI] [PubMed] [Google Scholar]
- 84. Abella JV, Peschard P, Naujokas MA et al . Met/hepatocyte growth factor receptor ubiquitination suppresses transformation and is required for Hrs phosphorylation. Mol Cell Biol 2005; 25: 9632–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Komada M, Kitamura N. Growth factor‐induced tyrosine phosphorylation of Hrs, a novel 115‐kilodalton protein with a structurally conserved putative zinc finger domain. Mol Cell Biol 1995; 15: 6213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hammond DE, Urbe S, Vande Woude GF, Clague MJ. Down‐regulation of MET, the receptor for hepatocyte growth factor. Oncogene 2001; 20: 2761–70. [DOI] [PubMed] [Google Scholar]
- 87. Hammond DE, Carter S, McCullough J, Urbe S, Vande Woude G, Clague MJ. Endosomal dynamics of Met determine signaling output. Mol Biol Cell 2003; 14: 1346–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Niendorf S, Oksche A, Kisser A et al . Essential role of ubiquitin‐specific protease 8 for receptor tyrosine kinase stability and endocytic trafficking in vivo . Mol Cell Biol 2007; 27: 5029–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Radtke F, Raj K. The role of Notch in tumorigenesis: oncogene or tumour suppressor? Nat Rev Cancer 2003; 3: 756–67. [DOI] [PubMed] [Google Scholar]
- 90. Lai EC. Protein degradation: four E3s for the notch pathway. Curr Biol 2002; 12: R74–8. [DOI] [PubMed] [Google Scholar]
- 91. Hutterer A, Knoblich JA. Numb and α‐Adaptin regulate Sanpodo endocytosis to specify cell fate in Drosophila external sensory organs. EMBO Report 2005; 6: 836–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Qiu L, Joazeiro C, Fang N et al . Recognition and ubiquitination of Notch by Itch, a hect‐type E3 ubiquitin ligase. J Biol Chem 2000; 275: 35 734–7. [DOI] [PubMed] [Google Scholar]
- 93. Jennings MD, Blankley RT, Baron M, Golovanov AP, Avis JM. Specificity and autoregulation of Notch binding by tandem WW domains in suppressor of Deltex. J Biol Chem 2007; 282: 29 032–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Vaccari T, Bilder D. The Drosophila tumor suppressor vps25 prevents nonautonomous overproliferation by regulating notch trafficking. Dev Cell 2005; 9: 687–98. [DOI] [PubMed] [Google Scholar]
- 95. Moberg KH, Schelble S, Burdick SK, Hariharan IK. Mutations in erupted, the Drosophila ortholog of mammalian tumor susceptibility gene 101, elicit non‐cell‐autonomous overgrowth. Dev Cell 2005; 9: 699–710. [DOI] [PubMed] [Google Scholar]
- 96. Jekely G, Rorth P. Hrs mediates downregulation of multiple signalling receptors in Drosophila. EMBO Report 2003; 4: 1163–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Thompson BJ, Mathieu J, Sung HH, Loeser E, Rorth P, Cohen SM. Tumor suppressor properties of the ESCRT‐II complex component Vps25 in Drosophila . Dev Cell 2005; 9: 711–20. [DOI] [PubMed] [Google Scholar]
- 98. Thiery JP. Epithelial‐mesenchymal transitions in tumour progression. Nat Rev Cancer 2002; 2: 442–54. [DOI] [PubMed] [Google Scholar]
- 99. Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol 2003; 4: 657–65. [DOI] [PubMed] [Google Scholar]
- 100. Palacios F, Tushir JS, Fujita Y, D'Souza‐Schorey C. Lysosomal targeting of E‐cadherin: a unique mechanism for the down‐regulation of cell–cell adhesion during epithelial to mesenchymal transitions. Mol Cell Biol 2005; 25: 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Fujita Y, Krause G, Scheffner M et al . Hakai, a c‐Cbl‐like protein, ubiquitinates and induces endocytosis of the E‐cadherin complex. Nat Cell Biol 2002; 4: 222–31. [DOI] [PubMed] [Google Scholar]
- 102. Yang JY, Zong CS, Xia W et al . MDM2 promotes cell motility and invasiveness by regulating E‐cadherin degradation. Mol Cell Biol 2006; 26: 7269–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Toyoshima M, Tanaka N, Aoki J et al . Inhibition of tumor growth and metastasis by depletion of vesicular sorting protein Hrs: its regulatory role on E‐cadherin and β‐catenin. Cancer Res 2007; 67: 5162–71. [DOI] [PubMed] [Google Scholar]
- 104. Carlton JG, Martin‐Serrano J. Parallels between cytokinesis and retroviral budding. a role for the ESCRT machinery. Science 2007; 316: 1908–12. [DOI] [PubMed] [Google Scholar]
- 105. Morita E, Sandrin V, Chung HY et al . Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J 2007; 26: 4215–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kamura T, Burian D, Khalili H et al . Cloning and characterization of ELL‐associated proteins EAP45 and EAP20: a role for yeast EAP‐like proteins in regulation of gene expression by glucose. J Biol Chem 2001; 276: 16 528–33. [DOI] [PubMed] [Google Scholar]
- 107. Shilatifard A. Identification and purification of the Holo‐ELL complex. Evidence for the presence of ELL‐associated proteins that suppress the transcriptional inhibitory activity of ELL. J Biol Chem 1998; 273: 11 212–17. [DOI] [PubMed] [Google Scholar]
- 108. Irion U, St Johnston D. Bicoid RNA localization requires specific binding of an endosomal sorting complex. Nature 2007; 445: 554–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol 2002; 2: 569–79. [DOI] [PubMed] [Google Scholar]
- 110. De Gassart A, Geminard C, Hoekstra D, Vidal M. Exosome secretion: the art of reutilizing nonrecycled proteins? Traffic 2004; 5: 896–903. [DOI] [PubMed] [Google Scholar]
- 111. Taylor DD, Gercel‐Taylor C. Tumour‐derived exosomes and their role in cancer‐associated T‐cell signalling defects. Br J Cancer 2005; 92: 305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Von Schwedler UK, Stuchell M, Muller B et al . The protein network of HIV budding. Cell 2003; 114: 701–13. [DOI] [PubMed] [Google Scholar]
- 113. Li L, Cohen SN. Tsg101: a novel tumor susceptibility gene isolated by controlled homozygous functional knockout of allelic loci in mammalian cells. Cell 1996; 85: 319–29. [DOI] [PubMed] [Google Scholar]
- 114. Xu Z, Liang L, Wang H, Li T, Zhao M. HCRP1, a novel gene that is downregulated in hepatocellular carcinoma, encodes a growth‐inhibitory protein. Biochem Biophys Res Commun 2003; 311: 1057–66. [DOI] [PubMed] [Google Scholar]
- 115. Gayther SA, Barski P, Batley SJ et al . Aberrant splicing of the TSG101 and FHIT genes occurs frequently in multiple malignancies and in normal tissues and mimics alterations previously described in tumours. Oncogene 1997; 15: 2119–26. [DOI] [PubMed] [Google Scholar]
- 116. Lee MP, Feinberg AP. Aberrant splicing but not mutations of TSG101 in human breast cancer. Cancer Res 1997; 57: 3131–4. [PubMed] [Google Scholar]
- 117. Sun Z, Pan J, Bubley G, Balk SP. Frequent abnormalities of TSG101 transcripts in human prostate cancer. Oncogene 1997; 15: 3121–5. [DOI] [PubMed] [Google Scholar]
- 118. Steiner P, Barnes DM, Harris WH, Weinberg RA. Absence of rearrangements in the tumour susceptibility gene TSG101 in human breast cancer. Nat Genet 1997; 16: 332–3. [DOI] [PubMed] [Google Scholar]
- 119. Ruland J, Sirard C, Elia A et al . p53 accumulation, defective cell proliferation, and early embryonic lethality in mice lacking tsg101. Proc Natl Acad Sci USA 2001; 98: 1859–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Carstens MJ, Krempler A, Triplett AA, Van Lohuizen M, Wagner KU. Cell cycle arrest and cell death are controlled by p53‐dependent and p53‐independent mechanisms in Tsg101‐deficient cells. J Biol Chem 2004; 279: 35 984–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Li L, Liao J, Ruland J, Mak TW, Cohen SN. A TSG101/MDM2 regulatory loop modulates MDM2 degradation and MDM2/p53 feedback control. Proc Natl Acad Sci USA 2001; 98: 1619–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Oh KB, Stanton MJ, West WW, Todd GL, Wagner KU. Tsg101 is upregulated in a subset of invasive human breast cancers and its targeted overexpression in transgenic mice reveals weak oncogenic properties for mammary cancer initiation. Oncogene 2007; 26: 5950–9. [DOI] [PubMed] [Google Scholar]