

Abstract
Tumor lymphangiogenesis is now known to play a causal role in lymph node metastasis, and thus its inhibition would have great significance for the prevention of lymph node metastasis in cancer therapy. VEGF‐C has recently been identified as a key molecule that involved in tumor lymphangiogenesis and lymphatic metastasis. However, the expressional regulation of VEGF‐C is not fully understood. We investigated the role of mTOR, which is a downstream kinase of the phosphatidylinositol 3‐kinase/Akt pathway, and the MAPK family (MEK1/2, p38, and JNK) in the regulation of VEGF‐C and VEGF‐A expression in B13LM cells, a lymphatic metastasis‐prone pancreatic tumor cell line. We also investigated the antilymphangiogenic effect of rapamycin, a specific inhibitor of mTOR in vivo using male BALB/c nu/nu mice. VEGF‐C expression was inhibited by the inhibitors for mTOR, p38, and JNK, but not by the inhibitor for MEK1/2, whereas VEGF‐A expression was inhibited by all four of these inhibitors. The serum starvation‐induced expression of VEGF‐C was inhibited by rapamycin, whereas that of VEGF‐A was incompletely inhibited. The metastatic experiment in vivo demonstrated that the number and the area of lymphatic vessels in the primary tumors were significantly decreased by rapamycin. Finally, the lymph node metastasis was significantly suppressed in rapamycin‐treated mice. Our results suggest that mTOR, p38, and JNK play important roles in VEGF‐C expression, and that rapamycin has an antilymphangiogentic effect and exerts the expected inhibition of lymphatic metastasis. (Cancer Sci 2007; 98: 726–733)
Abbreviations:
- 4E‐BP1
initiation factor 4E‐binding protein 1
- EDTA
ethylenediaminetetraacetic acid
- EGF
epidermal growth factor
- eIF4E
initiation factor 4E
- ELISA
enzyme‐linked immunosorbent assay
- FBS
fetal bovine serum
- HIF
hypoxia‐inducible factor
- FKBP
FK506‐binding protein
- IGF
insulin‐like growth factor
- IGF‐1R
insulin‐like growth factor receptor‐1
- PBS
phosphate buffered saline
- PDGF
platelet‐derived growth factor
- PI3K
phosphatidylinositol 3‐kinase
- MAPK
mitogen‐activated protein kinase
- mTOR
mammalian target of rapamycin
- RT‐PCR
reverse transcriptase polymerase chain reaction
- SDS
sodium dodecyl sulfate
- SDS‐PAGE
sodium dodecylsulfate–polyacrylamide gel electrophoresis
- TGF‐β tumor growth factor‐β VEGF
vascular endothelial growth factor.
Metastaticdissemination is the final process in the progression of malignant tumors. Therefore, prevention of metastasis must be an ultimate goal for the treatment of malignant tumors. Spread of a malignant tumor from its primary site occurs by three main routes: vascular, lymphatic and transcoelomic. Among them, metastasis to the regional lymph nodes is often the earliest appearing metastasis, which significantly affects the prognosis of patients. In conjunction with recent advances in our understanding of the lymphatic system, accumulated experimental data have shown that tumor‐induced lymphangiogenesis is an important mechanism promoting lymphatic metastasis.( 1 , 2 , 3 , 4 ) A series of studies that investigated the relationship between the lymphatic vessel density in tumors and lymph node metastasis demonstrated that high lymphatic vessel density correlated with frequent lymph node metastasis( 5 , 6 , 7 , 8 , 9 , 10 ) and with poor survival in multiple tumor types, including breast cancer, head and neck squamous cell carcinoma, and melanoma.( 5 , 7 , 8 )
VEGF‐C has recently been identified as a key regulator in lymphangiogenesis.( 11 , 12 ) VEGF‐C has a VEGF‐homologs region in the N‐terminal and binds VEGFR‐3, an fms‐like tyrosine kinase receptor.( 13 ) VEGF‐C also binds to VEGFR‐2 and can activate angiogenesis, but the higher affinity of VEGF‐C for VEGFR‐3 than for VEGFR‐2 suggests that VEGF‐C is a biologically relevant ligand of VEGFR‐3.( 14 , 15 , 16 ) Overexpression of VEGF‐C in breast cancer cells promotes tumor lymphangiogenesis and increased lymph node metastasis.( 1 ) The correlation between the expression of VEGF‐C in tumor cells and lymph node metastasis is significant for a variety of tumor types, and the VEGF‐C level in the primary tumor positively correlates with poor prognosis of patients.( 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 ) Thus, the inhibition of VEGF‐C expression in tumor cells could be a potential strategy for preventing lymph node metastasis.
The expressional regulation of VEGF‐A has been well investigated. Hypoxia induces VEGF‐A expression in an Akt‐dependent pathway with downstream activation of HIF‐1.( 25 ) The MAPK family also mediates the signal transduction of VEGF‐A expression in response to cell stresses.( 26 ) Extracellular stimulation by growth factors has been shown to induce VEGF‐C( 27 ) and VEGF‐A( 28 ) at similar levels. However, the signal transduction involved in the expressional regulation of VEGF‐C has not been fully established. PI3K and the MAPK family are involved in the signal transduction pathways of IGF‐1‐induced VEGF‐C expression in lung carcinoma cells.( 29 ) In contrast, IGF‐1R signaling negatively regulates VEGF expression in prostatic cancer cells under conditions of androgen depletion.
Rapamycin is a lipophilic macrolide antibiotic that was initially developed as a fungicide and immunosuppressant.( 30 ) Rapamycin acts as a specific inhibitor of mTOR, a serine/threonine kinase, that appears to be downstream of the PI3K/Akt signal pathway.( 31 ) A complex of rapamycin and the FKBP‐12 binds to mTOR and inhibits its activity.( 32 ) The signaling through mTOR regulates phosphorylation and activation of its two major downstream components, p70S6K and eIF4E‐binding protein 1 (4E‐BP1).( 33 , 34 ) Phosphorylation of p70S6K allows translation of ribosomal proteins.( 35 ) Phosphorylation of 4E‐BP1 regulates cap‐dependent translation by enabling the formation of an active eIF4E complex.( 36 ) mTOR plays a pivotal role in regulating the transcription initiation of many genes related to the process of oncogenic transformation and cancer progression.( 37 , 38 )
It has been noted that rapamycin and its derivatives exert a potent antitumor action on a variety of solid tumors.( 39 , 40 , 41 , 42 ) The antiangiogenic effect of rapamycin is one of the mechanisms responsible for suppressing the tumor progression.( 31 , 42 ) The mechanism responsible for the antiangiogenic effect of rapamycin is the inhibition of VEGF‐A expression that is regulated by the Akt/mTOR pathway.( 43 ) However, neither the effect of rapamycin in regulating VEGF‐C expression nor the effect of rapamycin on tumor‐associated lymphangiogenesis has been determined. We investigated the role of signal pathways, including the mTOR pathway and MAPK pathways, in regulating the VEGF‐C expression in tumor cells, and also investigated whether rapamycin reduces lymphatic metastasis using lymphatic‐metastasis‐prone murine pancreatic tumor cells. The main purpose of this study was to determine the effect of rapamycin on lymphangiogenesis and lymph node metastasis.
Materials and Methods
Mice. Male BALB/C nu/nu mice, 4 weeks old, were obtained from Japan SLC, Hamamatsu, Japan. All mice were maintained under specific pathogen‐free conditions at the Center for Animal Experimentation, Chiba University Graduate School of Medicine. Regular laboratory food and tap water were made available ad libitum. All animal experiments were carried out under the guidelines of Chiba University.
Cell lines and culture conditions. Lymphatic metastasis‐prone cells were established by in vivo selection of cells of a rat pancreatic tumor cell line, AR42J‐B13, which were kindly provided by Professor Kojima (Gummma University, Maebashi, Japan). AR42J‐B13 cells were inoculated subcutaneously into nude mice. After 3 weeks, mice were killed and the metastatic lymph nodes were resected. The resected lymph nodes were cut into small fragments in vitro. The fragmented tissues were then incubated with collagenase (1 mg/mL)(Wako, Osaka, Japan) for 24 h and washed with PBS. Harvested cells were cultured in vitro and re‐inoculated into the nude mice for the second round of selection. The lymphatic metastasis‐prone cell line, which was designated as B13LM, was obtained by repeating the procedure 10 times. B13LM cells were cultured in Dulbecco's modified Eagle's medium (Sigma, St. Louis, MO, USA) containing penicillin (50 unit/mL), streptomycin (50 µg/mL) (Invitrogen, Carlsbad, CA, USA), L‐glutamine (2 mM), and 10% FBS (Biological Industries, Kibbutz Beit Haemek, Israel) at 37°C in 5%CO2. For serum starvation, B13LM cells were cultured in the medium without FBS for 24 h.
Inhibition of signal transduction kinases. B13LM cells were cultured with serial concentrations of inhibitors for signal transduction kinases. Rapamycin (Cell Signaling Technology, Beverly, MA, USA) was added to the medium at a concentration of 0 nM, 1 nM, 10 nM, or 100 nM. U0126 (an inhibitor of MEK1/2) (Calbiochem, La Jolla, CA, USA), SB202190 (an inhibitor of p38) (Calbiochem), and SP600125 (an inhibitor of JNK) (Calbiochem) were added to the medium at a concentration of 0 µM, 1 µM, or 25 µM. Cells were treated by each inhibitor for 48 h and then prepared for Western blot or quantitative RT‐PCR analysis.
Enzyme‐linked immunosorbent assay. Concentrations of VEGF‐C in the medium of the cultured B13LM cells were determined by ELISA (Bender MedSystems, Burlingame, CA, USA). The immunoassays were carried out and analyzed according to the manufacturer's instructions. B13LM cells were cultured in the medium with rapamycin (100 nM), then conditioned medium was used for ELISA. All samples and standards were run in duplicate.
Quantitative RT‐PCR analysis. Total RNAs from B13LM cells were isolated using an RNeasy Mini kit (Quiagen, Tokyo, Japan) according to the manufacturer's instructions. One microgram of total RNA was subjected to a reverse transcription reaction, using a Ready To Go T‐primed 1st strand cDNA synthesis kit (Amersham Pharmacia Biotech, Buckinghamshire, UK). The cDNA from 33 ng of total RNA was used as a template. VEGF‐A and VEGF‐C mRNA levels were quantified by means of a LightCycler (Roche Diagnostics, Mannheim, Germany), using the double‐strand‐specific dye SYBE Green I and a HybProbe LightCycler RNA amplification kit specifically adapted for one‐step RT‐PCR in glass capillaries with a LightCycler instrument (Roche Diagnostics). The primer sequences used in this study were as follows: for rat VEGF‐A, 5′‐TATATCTTCAAGCCGTCCTG‐3′ (forward) and 5′‐TTGGTCTGCATTCACATCTG‐3′ (reverse); rat VEGF‐C, 5′‐TGTCCAGCAAACTACGTGTG‐3′ (forward) and 5′‐ACTGGCAGGTGTCTTCATCC‐3′ (reverse); rat β‐actin, 5′‐CTCCAGGATCTCACGCTCTA‐3′ (forward) and 5′‐AGAAGAAGCTGGGAAGAGAC‐3′ (reverse). The cycling conditions were as follows: initial reverse transcription at 61°C for 30 min, denaturation at 95°C for 30 s, and 45 cycles of denaturation at 95°C for 1 s, annealing at 60°C for 15 s, and elongation at 65°C for 1 min with a ramp of 5°C/s (with fluorescence acquisition at the end of each elongation stage). The expression level of each mRNA was adjusted using the level of β‐actin mRNA, and expressed as the ratio to β‐actin mRNA.
Western blot analysis. B13LM cells were cultured in fully supplemented medium for 24 h, then cultured for 48 h with medium containing kinase inhibitors or cultured for 1 h, 2 h, 4 h, and 12 h without FBS. After this conditioning period, cells were homogenized in lysis buffer (1 mL RIPA buffer [10 mM Tris‐HCl (pH 7.4), 100 mM NaCl, 5 mM (EDTA), 1% TritonX‐100, 1% sodium deoxycholate, 0.1% SDS], 100 µL Protease Inhibitor Cocktail [Sigma], and 10 µL Phosphatase Inhibitor Cocktail [Sigma]) and put on ice for 2 h. Protein concentration of each sample was determined by using a Bio‐Rad Protein Assay kit (Bio‐Rad, Laboratories, Hercules, CA, USA) according to manufacturer's instructions. Lysates containing 50 µg of protein were separated by SDS‐PAGE and transferred to nitrocellulose membranes (Nihon Eido, Tokyo, Japan). Nonspecific reactions were blocked for 4 h with TBS‐T (10 mM Tris [pH 7.4], 100 mM NaCl, 0.1% Tween‐20) containing 5% non‐fat dry milk. Then membranes were incubated overnight at 4°C with each antibody. After being washed with TBS‐T containing non‐fat dry milk, the membranes were incubated with the horseradish peroxidase‐conjugated secondary antibodies. The protein blots were visualized by chemiluminescence using ECL (Amersham).
Antibodies against Akt, phosphor‐Akt (Ser473), eIF4E, phospho‐eIF4E (Ser209), 4E‐BP1, phospho‐4E‐BP1 (Thr70), p70S6K, phospho‐p70S6K (Thr421/Ser424), mTOR, phospho‐mTOR (Ser2448) were purchased from Cell Signaling Technology. Antibodies against VEGF‐A and VEGF‐C were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Experimental metastatic model. To establish subcutaneous tumors in mice, 5.0 × 106 B13LM cells were injected subcutaneously into the left inferior limb. Tumors were allowed to grow for 7 days and the establishment of subcutaneous tumors was confirmed before rapamycin treatment. Rapamycin (1.5 mg/kg per day)(n = 10) or vehicle alone (n = 11) was intraperitoneally administrated to the mice every day from 8 days after the injection of tumor cells. The tumor volume ([major axis] × [minor axis]2× π/6) was measured every other day. Three weeks from the start of treatment, mice were killed. The subcutaneous tumors were removed and prepared for histological analysis. Lymph node metastasis was investigated, and the occurrence of metastasis was confirmed by microscope.
Histological and immunohistochemical analysis. Formalin‐fixed, paraffin‐embedded sections were stained with hematoxylin and eosin and these sections were also used for immunohistochemical analysis. Immunostaining was carried out using the labeled streptoavidin‐biotin‐peroxidase (Dako Cytomation, Kyoto, Japan) and microwave antigen retrieval techniques. Goat polyclonal anti‐LYVE‐1 (1:100) was obtained from Santa Cruz Biotechnology. Diaminobenzidine tetrahydrochloride substrate was used to visualize the positive staining.
Evaluation of lymphatic vessels. Evaluation of lymphatic vessels in the vicinity of the subcutaneous tumors was carried out using LYVE‐1 stains and computer‐assisted quantitative analysis. Sections were scanned at low magnification, and 10 hot spot areas with the greatest numbers of positively stained vessels were identified in each section. Each hot spot was examined in turn at 400 × magnification and captured using an AxioCam MRc5 digital camera system (Carl Zeiss, Tokyo, Japan). The number of lymphatic vessels was counted, and the mean value for the 10 hot spot areas was determined for each section. The area of lymphatic vessel lumen was measured using an AxioVision 4.4 image analysis system (Carl Zeiss) by tracing the lymphatic vessel walls on a computer monitor. The mean value of the 10 hot spot areas was calculated and used as the value for each section.
Statistical analysis. Data are given as the mean ± standard deviation in quantitative experiments. The significance of the data was determined by unpaired Student's t‐test for the evaluation of lymphangiogenesis, by a repeated‐measure anova test for the evaluation of tumor size, and by χ2 test for the evaluation of lymph node metastasis. Values of P of less than 0.05 were considered statistically significant.
Results
Expression of VEGF‐A and VEGF‐C in B13LM cells. To investigate the role of the mTOR pathway in tumor lymphangiogenesis, we used cells of a lymphatic‐metastasis prone tumor cell line, B13LM. We investigated the expression of VEGF‐A and VEGF‐C in B13LM cells by quantitative RT‐PCR and Western blot analysis. B13LM cells constitutively expressed both VEGF‐A and VEGF‐C under normal culture conditions. To evaluate whether the expressional regulation of VEGF in response to cell stress remained intact in B13LM cells, we examined the expression of VEGF‐A and VEGF‐C under serum‐starved conditions. The expressions of both VEGF‐A and VEGF‐C were up‐regulated when the cells were cultured in serum‐free medium for 24 h (Fig. 1a,b). Phosphorylation of Akt was increased in B13LM cells by the withdrawal of FBS from the cultured medium (Fig. 1c). Increased phosphorylation of Akt under the serum‐starved conditions suggested that the PI3K/Akt pathway, which is upstream of mTOR, was activated by serum starvation in the B13LM cells.
Figure 1.
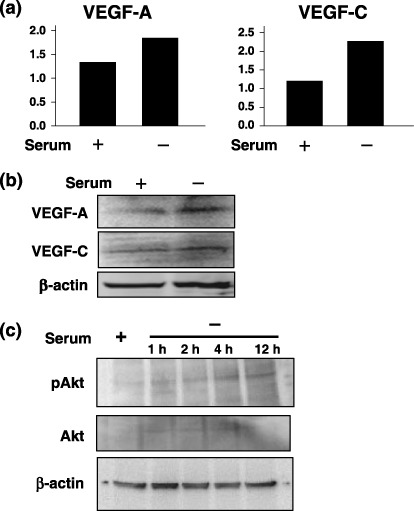
The expression of vascular endothelial growth factor‐A (VEGF‐A) and VEGF‐C in B13LM cells was evaluated by quantitative reverse transcription‐polymerase chain reaction (RT‐PCR) (a) and Western blot analysis (a representative data of two experiments) (b). Constitutive expression of VEGF‐A and VEGF‐C was observed in B13LM cells under a culture condition with 10% fetal bovine serum (FBS). The expression of VEGF‐A and VEGF‐C was up‐regulated by culture in serum‐free medium for 24 h (c) Western blot analysis of Akt and phosphor‐Akt in B13LM cells. Proteins were prepared form B13LM cells that were cultured with 10% FBS (serum +) or without FBS for 1 h, 2 h, 4 h and 12 h (serum –). Phosphorylation of Akt (pAkt) was increased in serum‐free medium.
Inhibition of the mTOR pathway in B13LM cells by rapamycin. We evaluated the inhibitory effect of rapamycin, a specific inhibitor of mTOR, in B13LM cells by analyzing the phosphorylation state of mTOR and the target molecules of mTOR. mTOR and its three downstream molecules, 4E‐BP1, p70S6K, and eIF4E, were all phosphorylated in B13LM cells under the culture conditions containing 10% FBS, indicating the activity of the mTOR pathway in B13LM cells. Slight dephosphorylation of mTOR was observed in the B13LM cells treated with 1 nM of rapamycin, and mTOR appeared to be remarkably dephosphorylated in the B13LM cells treated with 100 nM of rapamycin. 4E‐BP1, eIF4E and p70S6K were also dephosphorylated in B13LM cells treated with 1 nM of rapamycin. The reductions in the phosphorylation of these target molecules were considerable, although some phosphorylation remained (Fig. 2). These results indicated the efficacy of rapamycin for inhibiting the mTOR signal pathway in B13LM cells.
Figure 2.

Inhibition of the mammalian target of the rapamycin signal pathway by rapamycin in B13LM cells. Proteins were prepared from cells that were treated with a concentration gradient of rapamycin (0 nM, 1 nM, 10 nM, 50 nM, and 100 nM) for 48 h 50 mg of each protein was evaluated by sodium dodecylsulfate–polyacrylamide gel electrophoresis (SDS‐PAGE) for the expression of the mammalian target of rapamycin, phosphor‐mammalian target of rapamycin, initiation factor 4E‐binding protein 1, phosphor‐initiation factor 4E‐binding protein 1, initiation factor 4E, phosphor‐initiation factor 4E, p70S6K, and phospho‐p70S6K. β‐actin was also evaluated as a control. Slight dephosphorylation of mammalian target of rapamycin (mTOR) was observed in the B13LM cells treated with 1 nM of rapamycin. initiation factor 4E‐binding protein 1, initiation factor 4E and p70S6K were also dephosphorylated in B13LM cells treated with 1 nM of rapamycin.
Inhibition of the expression of VEGF‐C by rapamycin. We investigated whether the expressions of VEGF‐A and VEGF‐C were inhibited by rapamycin in B13LM cells in vitro. Dose‐dependent reductions of VEGF‐A and VEGF‐C expression were observed when the cells were cultured with rapamycin for 48 h. The reduction of VEGF‐C expression was more significant than that of VEGF‐A expression (Fig. 3a). The secretion of VEGF‐C was also reduced when B13LM cells were treated with rapamycin (Table 1). Next we investigated whether rapamycin can inhibit the serum starvation‐induced induction of VEGFs. The mRNA expression of both VEGF‐A and VEGF‐C was increased under the normal culture conditions (Fig. 1a). Rapamycin repressed mRNA expression of both VEGF‐A and VEGF‐C of B13LM cells in normal culture conditions. Moreover, rapamycin repressed the serum starvation‐induced expression of both of VEGF‐A and VEGF‐C, although the inhibition of VEGF‐A mRNA was incomplete (Fig. 3b).
Figure 3.

Inhibition of vascular endothelial growth factor‐A (VEGF‐A) and vascular endothelial growth factor‐C in B13LM cells by rapamycin. (a) Proteins were prepared from cells that were treated with a concentration gradient of rapamycin (0 nM, 1 nM, 10 nM, 50 nM, and 100 nM) for 48 h 50 µg of each protein was evaluated by sodium dodecylsulfate–polyacrylamide gel electrophoresis (SDS‐PAGE) for the expression of VEGF‐A and VEGF‐C. β‐actin was also evaluated as a control. The expressions of VEGF‐A and VEGF‐C were reduced by rapamycin in a dose‐dependent manner. (a) Representative data of two experiments). (b) Cells were cultured in the medium with 10% (serum +), or with 0% (serum –) fetal bovine serum (FBS) for 24 h. The mRNA expressions of VEGF‐A and VEGF‐C were evaluated by quantitative reverse transcription‐polymerase chain reaction (RT‐PCR) analysis. Serum starvation‐induced expression of VEGF‐A was observed, and was partially repressed by rapamycin treatment. In contrast, the serum starvation‐induced expression of VEGF‐C was almost completely inhibited by rapamycin treatment. Note that rapamycin inhibited the constitutive expression of VEGF‐A and VEGF‐C in the culture condition with 10% serum. (*P < 0.05).
Table 1.
Inhibition of vascular endothelial growth factor‐C (VEGF‐C) secretion by rapamycin
| Rapamycin | ||
|---|---|---|
| (+) | (–) | |
| VEFG‐C (pg/mL) | 510.6 | 131.8 |
VEGF‐C expression was inhibited by the inhibitors of p38 and JNK but not by the inhibitor of MEK1/2. We investigated the role of signal transduction pathways via three MAPK family members – MAPK kinase (MEK)1/2, p38, and c‐Jun N‐terminal kinase (JNK) – in the regulation of VEGF‐C expression. We also investigated the regulation of VEGF‐A expression by MAPK signaling, because MAPK signaling is known to be involved in the regulation of VEGF‐A expression. The inhibitors of MEK1/2, p38, and JNK inhibited the expression of VEGF‐A mRNA in a dose‐dependent manner in B13LM cells. The inhibitors of p38 and JNK also inhibited the expression of VEGF‐C mRNA, although a higher concentration of inhibitor was needed for effective inhibition of VEGF‐C expression than for effective inhibition of VEGF‐A. The inhibitory effect of the MEK1/2 inhibitor on the expression of VEGF‐C mRNA was not clear (Fig. 4).
Figure 4.

mRNA expression of vascular endothelial growth factor‐A (VEGF‐A) (a, c, e) and VEGF‐C (b, d, f) in B13LM cells treated by the MEK1/2 inhibitor (U0126) (a, b), p38 inhibitor (SB202190) (c, d), and JNK inhibitor (SP600125) (e, f). mRNA were prepared from cells treated with a concentration gradient of each inhibitor (0 nM, 1 nM, 25 nM) for 48 h. The expression level of each mRNA was adjusted using the level of β‐actin mRNA, and expressed as ratio to β‐actin mRNA. The inhibitors of MEK1/2, p38, and JNK inhibited VEGF‐A expression in a dose‐dependent manner. The inhibitors of p38 and JNK inhibited VEGF‐C expression at their highest concentration (25 nM). However, the inhibitory effect of the MEK1/2 inhibitor was ambiguous. (*P < 0.05)
Rapamycin inhibited the intratumor lymphangiogenesis and the lymphatic metastasis in nude mice. We evaluated whether rapamycin plays a role in lymphangiogenesis and lymphatic metastasis in vivo using a nude mouse model. The size of subcutaneous tumors in the rapamycin‐treated mice became smaller than that of control mice at 20 days after the initiation of rapamycin treatment (P = 0.009), although it increased at the same rate as in the control mice until 16 days (Fig. 5a). To identify tumor‐associated lymphatic vessels, we used immunohistochemistry using an antibody specific for mouse LYVE‐1, which is a highly specific marker for mouse lymphatic vessels( 44 ) (Fig. 5b). The computer‐assisted quantitative analysis of lymphatic vessels revealed that rapamycin treatment reduced lymphangiogenesis in the xenografted tumors in nude mice. The number of lymphatic vessels was significantly decreased in the tumors of the rapamycin‐treated mice compared with the control mice (P < 0.001). In addition, when we calculated the area of the lymphatic vessel lumen, it was significantly smaller in the tumors of the rapamycin‐treated mice than in those of control mice (P = 0.033) (Fig. 5c). Finally, the occurrence of lymphatic metastasis was significantly decreased in the rapamycin‐treated nude mice (P = 0.049). All control mice had one or two metastatic lymph nodes in the para‐aortic area, whereas, three of 10 mice that were treated with rapamycin were free from lymph node metastasis and another one of the 10 had only one metastatic lymph node. Metastasis was not observed in lymph nodes in other areas, with the exception of the para‐aortic area. No metastasis was observed in organs other than the lymph nodes. The mean size of the metastatic lymph nodes in mice with and without rapamycin treatment was 26.7 ± 21.3 mm3 and 47.4 ± 77.8 mm3, and the difference was not statistically significant (Table 2).
Figure 5.

Evaluation of the effect of rapamycin on intratumoral lymphangiogenesis and lymphatic metastasis using an experimental metastatic model in nude mice. 5.0 × 10.06 B13LM cells were injected subcutaneously into the left inferior limb. Tumors were allowed to grow for 7 days and then rapamycin (1.5 mg/kg per day) (n = 10) or the vehicle alone (n = 11) was intraperitoneally given to the mice every day. The subcutaneous tumor volume ([major axis] × [minor axis]2 × π/6) was measured every other day (a). Inhibition of the tumor growth was observed in rapamycin‐treated mice at day 20 (P = 0.009). After 3 weeks from the start of treatment, mice were killed. Lymphatic vessels in the subcutaneous tumors were evaluated using LYVE‐1 immunostains and computer‐assisted quantitative analysis. (b) Representative examples of LYVE‐1 immunostains of the subcutaneous tumors (left, control mice; right, rapamycin‐treated mice). The lymphatic vessels were less conspicuous in the tumor of rapamycin‐treated mice than that of control mice. (c) Ten hot spots having the greatest number of LYVE‐1‐positive vessels were selected in each slide and evaluated. The number (left) and the area (right) of lymphatic vessels were significantly decreased in rapamycin‐treated mice (P < 0.001 and P = 0.033, respectively).
Table 2.
Results of the experimental metastasis model
| Lymph node | Lymph node metastasis | Size of metastatic lymph node (mm3) |
|---|---|---|
| Rapamycin | 7/10†, * | 26.7 ± 21.3 |
| Control | 11/11 | 47.4 ± 77.8 |
The numbers of metastatic lymph nodes in each mouse with and without rapamycin treatment were 0, 0, 0, 1, 1, 1, 1, 1, 1, 1 and 1, 1, 1, 1, 1, 1, 1, 1, 1, 2, 2, respectively. The size of the metastatic lymph node is given as the mean ± SD;
P = 0.049.
Discussion
Our experimental metastatic study using nude mice demonstrated that rapamycin has the potential to inhibit lymphatic metastasis of tumor cells, because the development of lymph node metastasis of B13LM cells was significantly reduced by intraperitoneal administration of rapamycin. Both the number and area of lymphatic vessels in the subcutaneous tumor were significantly smaller in the rapamycin‐treated mice than in the control mice, indicating that in vivo rapamycin treatment inhibited the tumor‐associated lymphangiogenesis.
VEGF‐C and its receptor, VEFGR‐3, are considered to be potent targets for the prevention of lymphatic metastasis, because the proliferating signals via VEGFR‐3 promote lymphangiogenesis, and clinicopathological studies have indicated that VEGF‐C/VEGFR‐3 signal transduction has an important role in lymphatic metastasis.( 1 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 45 , 46 ) Studies using experimental animal models have demonstrated that a decrease in lymphatic metastasis could be achieved by inhibiting VEGF‐C/VEGFR‐3 signal transduction.( 47 , 48 , 49 ) Inhibition of the VEGF‐C/VEGFR‐3 interaction was experimentally realized by inhibitors including a soluble VEGFR‐3 decoy receptor( 47 ) and antagonistic antibody.( 48 ) Tumor‐derived VEGF‐C is another therapeutic target, and VEGF‐C‐specific inhibition by small interfering RNA‐mediated gene silencing has been shown to decrease lymphatic metastasis.( 49 ) These results provide direct evidence of the significant role of the VEGF‐C/VEGFR‐3 signal transduction pathway in tumor‐associated lymphangiogenesis and lymphatic metastasis. However, the methods of VEGF‐C/VEGFR‐3 inhibition in these studies were not immediately applicable to clinical therapy. The observed antimetastatic effect of rapamycin in our study thus has great meaning from a clinical point of view, because the analogs of rapamycin, such as CCI‐779, have already progressed to the Phase III evaluation of antitumor activity.( 50 ) In addition, it has been demonstrated that rapamycin and its derivatives have a potent antitumor action on a variety of solid tumors.( 39 , 40 , 41 ) Furthermore, rapamycin and its analogs have been reported to increase the efficacy of a variety of chemotherapeutic agents, including cisplatin, doxorubicin, camptothecin, 5‐fluorouracil, gemcitabin, and tamoxifen, in several types of cancers.( 51 , 52 , 53 )
The inhibitory effect of rapamycin on VEGF‐C expression in B13LM cells is thought to be one of the mechanisms involved in the antilymphangiogenetic effect of rapamycin in vivo. There is a substantial body of evidence that tumor‐derived VEGF‐C plays a causal role in lymphangiogenesis and lymphatic metastasis.( 1 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 45 , 46 ) However, because we used systemic administration of rapamycin in the present study, we cannot rule out the possibility that rapamycin affected the lymphatic endothelial cells. The decrease in the number of lymphatic vessels in the rapamycin‐treated tumor could be mediated by a direct effect of rapamycin on lymphatic endothelial cells, because the growth signal via VEGFR‐3 activates the Akt‐dependent pathway and promotes the proliferation of lymphatic endothelial cells.( 54 , 55 ) However, it remains to be determined whether rapamycin directly inhibits the proliferation of lymphatic endothelial cells.
The mTOR pathway in B13LM cells was activated under a normal culture condition, which was not surprising because up‐regulation of the mTOR pathway is observed in many human cancers.( 56 , 57 , 58 ) Inhibition of the mTOR pathway by rapamycin reduced the expression of VEGF‐C and, furthermore, inhibited the VEGF‐C expression that was induced by serum starvation. Increased phosphorylation by withdrawal of FBS suggested that the Akt pathway in B13LM cells was activated by serum starvation, although PI3K/Akt pathway would be usually inactivated under the serum starved‐conditions. These results indicate the involvement of the mTOR pathway in the expressional regulation of VEGF‐C in B13LM cells. This is not in conflict with the results of a previous study that showed that the PI3K pathway plays an important role in IGF‐1‐induced VEGF‐C expression,( 29 ) because activation of the PI3K/Akt pathway leads to mTOR signaling.
The exact mechanism of how rapamycin inhibits mTOR function is not fully understood. Evidence has been presented that kinases, including Akt, can phosphorylate mTOR at Ser2448 and that such phosphorylation is likely to have a regulatory role.( 59 , 60 , 61 ) On the other hand, the complex of rapamycin and FKBP‐12 binds directly to mTOR and inhibits the mTOR‐mediated phosphorylation of S6K1 and 4E‐BP1.( 32 ) Rapamycin also weakens the interaction of mTOR and raptor (a componet of the mTOR complex), which results in the inhibition of mTOR functions.( 62 ) In the B13LM cells treated with 1 nM of rapamycin, the downstream molecules of mTOR, such as 4E‐BP1, elF4E and p70S6K were dephosphorylated, suggested that 1 nM of rapamycin sufficiently inhibited the mTOR pathway, although phosphorylation of mTOR (at Ser2448) remained. It was suspected from these results that the dephosphorylation of mTOR was not necessarily in the inhibitory mechanism of rapamycin in B13LM cells. A similar observation was previously observed in the hepatoma cell, Hep‐G2, that was activated by insulin.( 63 )
The expression of both VEGF‐A and VEGF‐C was increased under the serum starved‐conditions. However, the contribution of the mTOR pathway in the regulation of VEGF‐A and VEGF‐C to serum‐starvation stimuli seemed not to be same because the efficacy of suppression by rapamycin was different. The serum‐starvation‐inducing expression of VEGF‐C was completely suppressed by rapamycin, whereas the inhibition of serum‐starvation‐inducing VEGF‐A by rapamycin appeared partially. Interestingly, a previous study using colon carcinoma cells suggested that extracellular signal‐regulated kinase (ERK)1/2 activation, but not Akt activation, is required for the induction of VEGF‐A by serum starvation.( 26 )
The size of the subcutaneous tumors in nude mice was reduced at day 20 by the rapamycin treatment. One possible explanation for the suppression of the growth of the tumor may be that tumor angiogenesis was inhibited, because rapamycin has an antiangiogenic effect. A previous study using an animal tumor model showed a similar inhibition of tumor growth, in which rapamycin was considered to mainly affect the endothelial cells; VEGF‐induced endothelial cell proliferation rather than tumor‐derived VEGF‐A expression was inhibited by rapamycin.( 41 ) In our previous study, rapamycin inhibited the B13LM cell proliferation in vitro. Thus, a direct effect of rapamycin on cell proliferation should be also considered.
The stimulation by growth factors, including IGF‐1, PDGF, EGF, and TGF‐β, has been shown to induce mRNA expression of VEGF‐C.( 27 ) Stimulatory signals from these growth factors activate MAPK signal pathways, which in turn control the cell regulation, including proliferation, differentiation, and apoptosis. The results of our experiment using kinase inhibitors suggest that the signal transduction pathways of VEGF‐A and VEGF‐C are similar but not identical in B13LM cells. The inhibitors for JNK and p38 inhibited the expression of both VEGF‐A and VEGF‐C. However, it is notable that the inhibition of VEGF‐C by the MEK1/2 inhibitor was not remarkable. Binding of growth factors to their receptor‐type tyrosine kinase leads to Grb2/SOS/Ras interaction, which activates MEK1/2. Activated MEK1/2 then activates ERK1/2. Interestingly, a previous study has shown that IGF‐1‐induced VEGF‐C expression in lung carcinoma cells is both PI3K‐ and ERK‐dependent, but PI3K has the predominant role.( 29 ) Another study has shown that the induction of Ras oncoprotein in fibroblasts or fibrosarcoma cells induced VEGF‐A mRNA, but did not induce VEGF‐C mRNA.( 27 , 54 )
In summary, rapamycin inhibited the expression of VEGF‐C, a potent growth factor for lymphatic endothelial cells, in vitro. Lymphatic vessels in the primary tumors were significantly reduced, and finally the lymph node metastasis was decreased in our experimental animal model using lymphatic metastatic‐prone B13LM cells. Our results provide the first preclinical data that rapamycin has the potential to suppress tumor‐related lymphangiogenesis and lymph node metastasis. Further understanding of the signal transduction of VEGF‐C/VEGFR‐3 axis could prove invaluable for improving treatments that target lymphangiogenesis.
References
- 1. Skobe M, Hawighorst T, Jackson DG et al. Induction of tumor lymphangiogenesis by VEGF‐C promotes breast cancer metastasis. Nat Med 2001; 7: 192–8. [DOI] [PubMed] [Google Scholar]
- 2. Stacker SA, Caesar C, Baldwin ME et al. VEGF‐D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med 2001; 7: 151–2. [DOI] [PubMed] [Google Scholar]
- 3. Mandriota SJ, Jussila L, Jeltsch M et al. Vascular endothelial growth factor‐C‐mediated lymphangiogenesis promotes tumour metastasis. EMBO J 2001; 20: 672–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Karpanen T, Egeblad M, Karkkainen MJ et al. Vascular endothelial growth factor C promotes tumor lymphangiogenesis and intralymphatic tumor growth. Cancer Res 2001; 61: 1786–90. [PubMed] [Google Scholar]
- 5. Nakamura Y, Yasuoka H, Tsujimoto M et al. Lymph vessel density correlates with nodal status, VEGF‐C expression, and prognosis in breast cancer. Breast Cancer Res Treat 2005; 91: 125–32. [DOI] [PubMed] [Google Scholar]
- 6. Kitadai Y, Kodama M, Cho S et al. Quantitative analysis of lymphangiogenic markers for predicting metastasis of human gastric carcinoma to lymph nodes. Int J Cancer 2005; 115: 388–92. [DOI] [PubMed] [Google Scholar]
- 7. Kyzas PA, Geleff S, Batistatou A, Agnantis NJ, Stefanou D. Evidence for lymphangiogenesis and its prognostic implications in head and neck squamous cell carcinoma. J Pathol 2005; 206: 170–7. [DOI] [PubMed] [Google Scholar]
- 8. Straume O, Jackson DG, Akslen LA. Independent prognostic impact of lymphatic vessel density and presence of low‐grade lymphangiogenesis in cutaneous melanoma. Clin Cancer Res 2003; 9: 250–6. [PubMed] [Google Scholar]
- 9. Birner P, Schindl M, Obermair A et al. Lymphatic microvessel density in epithelial ovarian cancer: its impact on prognosis. Anticancer Res 2000; 20: 2981–5. [PubMed] [Google Scholar]
- 10. Zeng Y, Opeskin K, Horvath LG, Sutherland RL, Williams ED. Lymphatic vessel density and lymph node metastasis in prostate cancer. Prostate 2005; 65: 222–30. [DOI] [PubMed] [Google Scholar]
- 11. Joukov V, Pajusola K, Kaipainen A et al. A novel vascular endothelial growth factor, VEGF‐C, is a ligand for the Flt4 (VEGFR‐3) and KDR (VEGFR‐2) receptor tyrosine kinases. EMBO J 1996; 15: 290–8. [PMC free article] [PubMed] [Google Scholar]
- 12. Jeltsch M, Kaipainen A, Joukov V et al. Hyperplasia of lymphatic vessels in VEGF‐C transgenic mice. Science 1997; 277: 463. [DOI] [PubMed] [Google Scholar]
- 13. Galland F, Karamysheva A, Mattei MG, Rosnet O, Marchetto S, Birnbaum D. Chromosomal localization of FLT4, a novel receptor‐type tyrosine kinase gene. Genomics 1992; 3: 475–8. [DOI] [PubMed] [Google Scholar]
- 14. Joukov V, Sorsa T, Kumar V et al. Proteolytic processing regulates receptor specificity and activity of VEGF‐C. EMBO J 1997; 16: 3898–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Achen MG, Jeltsch M, Kukk E et al. Vascular endothelial growth factor D (VEGF‐D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc Natl Acad Sci USA 1998; 95: 548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee J, Gray A, Yuan J, Luoh SM, Avraham H, Wood WI. Vascular endothelial growth factor‐related protein: a ligand and specific activator of the tyrosine kinase receptor Flt4. Proc Nat Acad Sci USA 1996; 93: 1988–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Suzuki K, Morita T, Tokue A. Vascular endothelial growth factor‐C (VEGF‐C) expression predicts lymph node metastasis of transitional cell carcinoma of the bladder. Int J Urol 2005; 12: 152–8. [DOI] [PubMed] [Google Scholar]
- 18. Nakamura Y, Yasuoka H, Tsujimoto M, Et al Q. Clinicopathological significance of vascular endothelial growth factor‐C in breast carcinoma with long‐term follow‐up. Mod Pathol 2003; 16: 309–14. [DOI] [PubMed] [Google Scholar]
- 19. Fujimoto J, Toyoki H, Sato E, Sakaguchi H, Tamaya T. Clinical implication of expression of vascular endothelial growth factor‐C in metastatic lymph nodes of uterine cervical cancers. Br J Cancer 2004; 91: 466–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Onogawa S, Kitadai Y, Tanaka S, Kuwai T, Kimura S, Chayama K. Expression of VEGF‐C and VEGF‐D at the invasive edge correlates with lymph node metastasis and prognosis of patients with colorectal carcinoma. Cancer Sci 2004; 95: 32–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsutsumi S, Kuwano H, Shimura T, Morinaga N, Mochiki E, Asao T. Vascular endothelial growth factor C (VEGF‐C) expression in pT2 gastric cancer. Hepatogastroenterology 2005; 52: 629–32. [PubMed] [Google Scholar]
- 22. Arinaga M, Noguchi T, Takeno S, Chujo M, Miura T, Uchida Y. Clinical significance of vascular endothelial growth factor C and vascular endothelial growth factor receptor 3 in patients with non‐small cell lung carcinoma. Cancer 2003; 97: 457–64. [DOI] [PubMed] [Google Scholar]
- 23. Yokoyama Y, Charnock‐Jones DS, Licence D et al. Vascular endothelial growth factor‐D is an independent prognostic factor in epithelial ovarian carcinoma. Br J Cancer 2003; 88: 237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kurahara H, Takao S, Maemura K, Shinchi H, Natsugoe S, Aikou T. Impact of vascular endothelial growth factor‐C and ‐D expression in human pancreatic cancer: its relationship to lymph node metastasis. Clin Cancer Res 2004; 10: 8413–20. [DOI] [PubMed] [Google Scholar]
- 25. Skinner HD, Zheng JZ, Fang J, Agani F, Jiang BH. Vascular endothelial growth factor transcriptional activation is mediated by hypoxia‐inducible factor 1alpha, HDM2, and p70S6K1 in response to phosphatidylinositol 3‐kinase/AKT signaling. J Biol Chem 2004; 279: 45 643–51. [DOI] [PubMed] [Google Scholar]
- 26. Jung YD, Nakano K, Liu W, Gallick GE, Ellis LM. Extracellular signal‐regulated kinase activation is required for up‐regulation of vascular endothelial growth factor by serum starvation in human colon carcinoma cells. Cancer Res 1999; 59: 4804–7. [PubMed] [Google Scholar]
- 27. Enholm B, Paavonen K, Ristimaki A et al. Comparison of VEGF, VEGF‐B, VEGF‐C and Ang‐1 mRNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene 1997; 14: 2475–83. [DOI] [PubMed] [Google Scholar]
- 28. Poulaki V, Mitsiades CS, McMullan C et al. Regulation of vascular endothelial growth factor expression by insulin‐like growth factor I in thyroid carcinomas. J Clin Endocrinol Metab 2003; 88: 5392–8. [DOI] [PubMed] [Google Scholar]
- 29. Tang Y, Zhang D, Fallavollita L, Brodt P. Vascular endothelial growth factor C expression and lymph node metastasis are regulated by the type I insulin‐like growth factor receptor. Cancer Res 2003; 63: 1166–71. [PubMed] [Google Scholar]
- 30. Vezina C, Kudelski A, Sehgal SN. Rapamycin (AY‐22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo) 1975; 28: 721–6. [DOI] [PubMed] [Google Scholar]
- 31. Mendez R, Myers MG Jr, White MF, Rhoads RE. Stimulation of protein synthesis, eukaryotic translation initiation factor 4E phosphorylation, and PHAS‐I phosphorylation by insulin requires insulin receptor substrate 1 and phosphatidylinositol 3‐kinase. Mol Cell Biol 1996; 16: 2857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sabers CJ, Martin MM, Brunn GJ et al. Isolation of a protein target of the FKBP12‐rapamycin complex in mammalian cells. J Biol Chem 1995; 270: 815–22. [DOI] [PubMed] [Google Scholar]
- 33. Brown EJ, Beal PA, Keith CT, Chen J, Shin TB, Schreiber SL. Control of p70, s6 kinase by kinase activity of FRAP in vivo. Nature, 1995: 377: 441–6. [DOI] [PubMed] [Google Scholar]
- 34. Brunn GJ, Hudson CC, Sekulic A et al. Phosphorylation of the translational repressor PHAS‐I by the mammalian target of rapamycin. Science 1997; 277: 99–101. [DOI] [PubMed] [Google Scholar]
- 35. Grove JR, Banerjee P, Balasubramanyam A et al. Cloning and expression of two human p70, S6 kinase polypeptides differing only at their amino termini. Mol Cell Biol 1991; 11: 5541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pause A, Belsham GJ, Gingras AC et al. Insulin‐dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′‐cap function. Nature 1994; 371: 747–8. [DOI] [PubMed] [Google Scholar]
- 37. Agbunag C, Bar‐Sagi D. Oncogenic K‐ras drives cell cycle progression and phenotypic conversion of primary pancreatic duct epithelial cells. Cancer Res 2004; 64: 5659–63. [DOI] [PubMed] [Google Scholar]
- 38. Wislez M, Spencer ML, Izzo JG et al. Inhibition of mammalian target of rapamycin reverses alveolar epithelial neoplasia induced by oncogenic K‐ras. Cancer Res 2005; 65: 3226–35. [DOI] [PubMed] [Google Scholar]
- 39. Amornphimoltham P, Patel V, Sodhi A et al. Mammalian target of rapamycin, a molecular target in squamous cell carcinomas of the head and neck. Cancer Res 2005; 65: 9953–61. [DOI] [PubMed] [Google Scholar]
- 40. Geoerger B, Kerr K, Tang CB et al. Antitumor activity of the rapamycin analog CCI‐779 in human primitive neuroectodermal tumor/medulloblastoma models as single agent and in combination chemotherapy. Cancer Res 2001; 61: 1527–32. [PubMed] [Google Scholar]
- 41. Guba M, Von Breitenbuch P, Steinbauer M et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med 2002; 8: 128–35. [DOI] [PubMed] [Google Scholar]
- 42. Bruns CJ, Koehl GE, Guba M et al. Rapamycin‐induced endothelial cell death and tumor vessel thrombosis potentiate cytotoxic therapy against pancreatic cancer. Clin Cancer Res 2004; 10: 2109–19. [DOI] [PubMed] [Google Scholar]
- 43. Phung TL, Ziv K, Dabydeen D et al. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell 2006; 10: 159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prevo R, Banerji S, Ferguson DJ, Clasper S, Jackson DG. Mouse LYVE‐1 is an endocytic receptor for hyaluronan in lymphatic endothelium. J Biol Chem 2001; 276: 19 420–30. [DOI] [PubMed] [Google Scholar]
- 45. Su JL, Yang PC, Shih JY et al. The VEGF‐C/Flt‐4 axis promotes invasion and metastasis of cancer cells. Cancer Cell 2006; 9: 209–23. [DOI] [PubMed] [Google Scholar]
- 46. Takizawa H, Kondo K, Fujino H et al. The balance of VEGF‐C and VEGFR‐3 mRNA is a predictor of lymph node metastasis in non‐small cell lung cancer. Br J Cancer 2006; 95: 75–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lin J, Lalani AS, Harding TC et al. Inhibition of lymphogenous metastasis using adeno‐associated virus‐mediated gene transfer of a soluble VEGFR‐3 decoy receptor. Cancer Res 2005; 65: 6901–9. [DOI] [PubMed] [Google Scholar]
- 48. Roberts N, Kloos B, Cassella M et al. Inhibition of VEGFR‐3 activation with the antagonistic antibody more potently suppresses lymph node and distant metastases than inactivation of VEGFR‐2. Cancer Res 2006; 66: 2650–7. [DOI] [PubMed] [Google Scholar]
- 49. Chen Z, Varney ML, Backora MW et al. Down‐regulation of vascular endothelial cell growth factor‐C expression using small interfering RNA vectors in mammary tumors inhibits tumor lymphangiogenesis and spontaneous metastasis and enhances survival. Cancer Res 2005; 65: 9004–11. [DOI] [PubMed] [Google Scholar]
- 50. Elit L. CCI‐779 Wyeth. Curr Opin Invest Drugs 2002; 3: 1249–53. [PubMed] [Google Scholar]
- 51. Wendel HG, De Stanchina E, Fridman JS et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 2004; 428: 332–7. [DOI] [PubMed] [Google Scholar]
- 52. Seeliger H, Guba M, Koehl GE et al. Blockage of 2‐deoxy‐D‐ribose‐induced angiogenesis with rapamycin counteracts a thymidine phosphorylase‐based escape mechanism available for colon cancer under 5‐fluorouracil therapy. Clin Cancer Res 2004; 10: 1843–52. [DOI] [PubMed] [Google Scholar]
- 53. Mondesire WH, Jian W, Zhang H et al. Targeting mammalian target of rapamycin synergistically enhances chemotherapy‐induced cytotoxicity in breast cancer cells. Clin Cancer Res 2004; 10: 7031–42. [DOI] [PubMed] [Google Scholar]
- 54. Salameh A, Galvagni F, Bardelli M, Bussolino F, Oliviero S. Direct recruitment of CRK and GRB2 to VEGFR‐3 induces proliferation, migration, and survival of endothelial cells through the activation of ERK, AKT, and JNK pathways. Blood 2005; 106: 3423–31. [DOI] [PubMed] [Google Scholar]
- 55. Makinen T, Veikkola T, Mustjoki S et al. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF‐C/D receptor VEGFR‐3. EMBO J 2001; 20: 4762–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Conde E, Angulo B, Tang M et al. Molecular context of the EGFR mutations: evidence for the activation of mTOR/S6K signaling Clin. Cancer Res 2006; 12: 710–7. [DOI] [PubMed] [Google Scholar]
- 57. Sun SY, Rosenberg LM, Wang X et al. Activation of Akt and eIF4E survival pathways by rapamycin‐mediated mammalian target of rapamycin inhibition. Cancer Res 2005; 65: 7052–8. [DOI] [PubMed] [Google Scholar]
- 58. Sahin F, Kannangai R, Adegbola O, Wang J, Su G, Torbenson M. Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt Activity. Clin Cancer Res 2004; 10: 8059–67. [DOI] [PubMed] [Google Scholar]
- 59. Burgering BM, Coffer PJ. Protein kinase B (c‐Akt) in phosphatidylinositol‐3‐OH kinase signal transduction. Nature 1995; 376: 599–602. [DOI] [PubMed] [Google Scholar]
- 60. Scott PH, Brunn GJ, Kohn AD, Roth RA, Lawrence JC Jr. Evidence of insulin‐stimulated phosphorylation and activation of the mammalian target of rapamycin mediated by a protein kinase B signaling pathway. Proc Natl Acad Sci USA 1998; 95: 7772–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gingras A, Kennedy SG, O’Leary MA, Sonenburg N, Hay N. 4E‐BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt (PKB) signaling pathway. Genes Dev 1998; 12: 502–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kim DH, Sarbassov DD, Ali SM et al. mTOR Interacts with Raptor to Form a Nutrient‐Sensitive Complex that Signals to the Cell Growth Machinery. Cell 2002; 110: 163–75. [DOI] [PubMed] [Google Scholar]
- 63. Varma S, Khandelwal RL. Effects of rapamycin on cell proliferation and phosphorylation of mTOR and p70S6K in HepG2 and HepG2 cells overexpressing constitutively active Akt/PKB. Biochim Biophys Acta 2007; 1770: 71–8. [DOI] [PubMed] [Google Scholar]