

Abstract
Tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL) is a promising candidate for the treatment of cancer because it elicits cell death in many tumor cells while sparing most normal cells. Liver cancer, however, is largely resistant to TRAIL and, thus, requires sensitization for TRAIL‐mediated cytotoxicity. Sensitization may be achieved by cotreatment with chemotherapeutic agents. In this study, we comparatively investigated the treatment efficacy of TRAIL in combination with histone deacetylase inhibitors (HDI) versus TRAIL in combination with conventional cytostatics in the hepatocellular carcinoma cell line HepG2 and in the childhood hepatoblastoma cell line Huh6. We found that TRAIL resistance could be overcome by cotreatment with the HDI vorinostat, sodium butyrate and MS‐275, but not by cotreatment with the cytostatics carboplatin and etoposide. However, TRAIL combination treatment bears the risk of sensitizing otherwise TRAIL‐resistant normal cells. We thus explored a potential cytotoxic effect of combined HDI/TRAIL treatment in normal hepatocytes: TRAIL in conjunction with HDI did not impose any cytotoxicity on the non‐malignant cells. In searching for the determinants of HDI‐mediated TRAIL sensitization in hepatoma cells, we observed that HDI treatment did not increase cell‐surface expression of proapoptotic TRAIL receptors. Instead, HDI treatment enhanced TRAIL‐induced cleavage of Bid. In conclusion, our data suggest that HDI are potent sensitizers to TRAIL in hepatoma cells and that the combination of HDI and TRAIL is selectively active in hepatoma cells without affecting normal hepatocytes, indicating that the combination of HDI and TRAIL may be an effective approach for the treatment of advanced liver cancer. (Cancer Sci 2008; 99: 1685–1692)
Abbreviations:
- ActD
actinomycin D
- HCC
hepatocellular carcinoma
- HDI
histone deacetylase inhibitors
- LDH
lactate dehydrogenase
- NaB
sodium butyrate
- PHH
primary human hepatocytes
- PI
propidium iodide
- PRH
primary rat hepatocytes.
Liver cancer is the sixth most common cancer and the third most common cause of death from cancer worldwide.( 1 ) The prognosis is very poor, which is reflected by the fact that the numbers of cases and deaths are almost the same; survival rates are 3% to 5%. The high lethality of liver cancer stems from the fact that presently the only curative treatments are surgical resection or liver transplantation but most patients are diagnosed at advanced stages, excluding them from these therapeutic options.( 2 ) Unfortunately, owing to resistance to existing anticancer agents, chemotherapy offers only symptomatic relief.( 3 ) More effective therapeutic strategies, thus, are urgently needed.
Tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL; Apo‐2L) has great potential for cancer treatment since it induces cell death in a wide variety of cancer cells while leaving normal cells unscathed.( 4 , 5 ) As a result, clinical trials with recombinant TRAIL and agonistic anti‐TRAIL receptor antibodies have been initiated.( 6 ) Some cancers, however, fail to respond to TRAIL's cytotoxic effects. For example, in a study on primary cells from 53 leukemia patients, TRAIL induced relevant apoptosis in only 25% of samples,( 7 ) and in a study on primary cells from 13 glioma patients, all displayed resistance to TRAIL‐mediated cell death.( 8 ) In particular, hepatoma cells have been found to be highly resistant to TRAIL.( 9 , 10 , 11 , 12 , 13 , 14 , 15 ) The cytotoxic activity of TRAIL alone therefore may be insufficient for cancer therapy, but may be augmented by coadministration of chemotherapeutic agents.( 16 ) In hepatoma cell lines, TRAIL‐induced cytotoxicity could be enhanced by cotreatment with the conventional cytostatics doxorubicin, cisplatin, camptothecin and 5‐fluorouracil,( 9 , 10 , 13 , 17 ) the histone deacetylase inhibitors (HDI) valproate and ITF2357,( 14 , 18 ) the proteasome inhibitors bortezomib, MG132 and MG115( 19 , 20 , 21 ) and the anticancer cytokines interleukin‐2 and interferon‐α.( 12 , 13 , 22 ) However, the combination of TRAIL and chemotherapy bears the risk of eliciting cell death in otherwise TRAIL‐resistant normal cells.( 16 ) The same mechanism that protects cancer cells against TRAIL may also protect normal cells and, hence, the same agents that sensitize cancer cells to TRAIL may also sensitize normal cells. For example, doxorubicin and cisplatin were found to render osteoblasts susceptible to TRAIL,( 23 ) cycloheximide and MG115 were shown to sensitize keratinocytes to TRAIL( 24 , 25 ) and, in normal urothelial cells, TRAIL resistance could be overcome by inhibiting NF‐κB.( 26 ) In hepatocytes, sensitization to TRAIL was observed for cisplatin( 27 , 28 ) and for high concentrations of bortezomib.( 21 ) Thus, potential TRAIL combination therapies have to be carefully evaluated in order to ensure both effective and safe application.
In the current study, we systematically tested the cytotoxic actvity of different TRAIL combination treatments in two liver cancer cell lines, i.e. in the hepatocellular carcinoma (HCC) cell line HepG2 and in the childhood hepatoblastoma cell line Huh6, and in normal hepatocytes, i.e. in the ‘normal’ mouse hepatocyte cell line AML12 and in primary rat (PRH) and human hepatocytes (PHH). We combined TRAIL with HDI belonging to three structural classes, the hydroxamic acid vorinostat (suberoylanilide hydroxamic acid, SAHA), the short‐chain fatty acid sodium butyrate (NaB) and the benzamide derivative MS‐275. HDI appeared to be especially interesting since they induce apoptosis, growth arrest and differentiation in cancer cells without causing significant toxicity to normal cells.( 29 , 30 , 31 ) The clinical applicability of vorinostat administration in cancer patients is highlighted by the fact that it has recently been approved by the US Food and Drug Administration for treatment of cutaneous T‐cell lymphoma. The combination of TRAIL with HDI was compared with the combination of TRAIL with cytostatics, which have shown efficacy in the management of advanced childhood hepatoblastoma, carboplatin and etoposide.( 32 ) We found that HDI were superior sensitizers to TRAIL in HepG2 and Huh6 cells. Importantly, we also found that normal hepatocytes remained resistant to TRAIL in the presence of HDI.
Materials and Methods
Reagents. TRAIL was purchased from Peprotech (Hamburg, Germany). Vorinostat and MS‐275 were purchased from Alexis (Grünberg, Germany). NaB, carboplatin, etoposide and actinomycin D (ActD) were purchased from Sigma (Deisenhofen, Germany).
Cell lines. HepG2 HCC cells and AML12 non‐transformed mouse hepatocytes( 33 ) were obtained from ATCC (Manassas, USA). Huh6 hepatoblastoma cells were a gift from Dr S. Warmann (Tübingen, Germany). HepG2 and Huh6 cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM), AML12 cells were maintained in a 1:1 mixture of DMEM/Ham's F12 medium. Media were supplemented with 10% fetal calf serum, 2 mM L‐glutamine, 100 units/mL penicillin G sodium, and 100 µg/mL streptomycin sulfate. The medium for HepG2 cells was additionally supplemented with 0.1 mM nonessential amino acids (all media and supplements were purchased from Biochrom, Berlin, Germany). The medium for AML12 cells was additionally supplemented with 40 ng/mL dexamethasone (Sigma) and a mixture of 5 µg/mL insulin, 5 µg/mL transferrin and 5 ng/mL selenium (Roche, Germany). Cells were cultivated at 37°C in a humidified 5% CO2 incubator and routinely passaged when 90% confluent. Cell viability was determined by the trypan blue exclusion test. Cells were regularly inspected to be free of mycoplasma with mycoplasma detection reagents from Roche (Mannheim).
Isolation of primary rat hepatocytes. PRH were isolated from freshly killed female Wistar rats by a modified collagenase perfusion method.( 34 ) Briefly, the livers were perfused first with an ethyleneglycoltetraacetic acid (EGTA)‐containing N‐(2‐hydroxyethyl)‐piperazine‐N′‐2‐ethanesulfonic acid (HEPES) buffer and then with calcium‐ and type‐IV collagenase‐(0.5 mg/mL; Sigma) containing HEPES buffer. The liver lobes were then cut into slices and filtered through 80 µm sterile cotton gauze to release the hepatocytes. The resultant cell suspension was washed three times by centrifugation at 35 × g for 5 min. Hepatocytes were then cultured in Williams E medium (Biochrom) containing 219 I.E./L human insulin (Sigma), 16 µg/L glucagon (Sigma), 910 µg/L prednisolone (Sigma), 4.6 mM L‐glutamine, 228 mg/L penicillin/streptomycin and 5% fetal calf serum in tissue culture flasks coated with rat tail collagen (5 µg/cm2; Roche). After 5 h, medium was changed to contain 2% fetal calf serum. All experiments were performed in accordance with The Guide for the Care and Use of Laboratory Animals (National Academy Press, Washington, DC, 1996). The experimental protocols used in this study were approved by the local authorities of the state of Mecklenburg‐Vorpommern.
Treatment of cells. The cells were plated at 2 × 105 cells in six‐well plates and treated with chemotherapeutics for 24 h or left untreated before application of TRAIL. TRAIL was added directly to the culture medium containing chemotherapeutics without a medium change. Cells were then exposed to increasing concentrations of TRAIL for an additional 24 h.
Primary human hepatocytes. PHH from two donors were obtained commercially from Lonza (Verviers, Belgium), plated as confluent monolayers in collagen‐coated 96‐well plates with only the inner 60 wells seeded with cells. PHH were cultivated in hepatocyte culture medium consisting of human blood medium (HBM) supplemented with hepatocyte basal medium Singlequots (Lonza).
Determination of cytotoxicity.
Flow cytometric analysis of DNA content To determine DNA fragmentation, ethanol‐fixed cells were analyzed for propidium iodide (PI) incorporation into DNA. Cells were harvested after the indicated treatments, washed twice with phosphate‐buffered saline (PBS) and fixed in 70% ethanol at –20°C for at least 30 min. After centrifugation, cells were resuspended in PBS containing 1% glucose, 50 µg/mL RNase A (Roche) and 50 µg/mL PI (Sigma) and incubated in the dark at room temperature for 30 min. Flow cytometry analysis was performed on a Becton Dickinson FACSCalibur (Heidelberg, Germany). 20 000 cells were analyzed in each sample; data were gated to exclude debris. sub‐G1 phase cells were calculated from the DNA content histograms.
Flow cytometric analysis of PI uptake Cell death was assessed by determining the integrity of the cell membrane by flow cytometric analysis of PI uptake. Cells were harvested after the indicated treatments, followed by a 5‐min incubation in 2 µg/mL PI in PBS at 4°C in the dark. PI uptake was measured by flow cytometry analysis on a FACSCalibur. 10 000 cells were analyzed in each sample; data were gated to exclude debris.
Lactate dehydrogenase (LDH) activity Cell death was assessed by determining LDH release into the extracellular medium. After the indicated treatments, LDH activities in the medium and in the vital cells were measured spectrophotometrically at the Institute for Clinical Chemistry and Laboratory Medicine of the University of Greifswald. Cell death was calculated as the ratio of extracellular LDH activity to total LDH activity.
Alamar Blue assay The assays were done in triplicate in 96‐well plates. At the end of the incubation period, 1/10 volume of Alamar Blue (Biosource, Solingen, Germany) solution was added and cells incubated at 37°C for an additional 4 h. The absorbance of the wells was measured at 560/595 nm using a Wallac Victor (Perkin Elmer, Rodgau‐Jügesheim, Germany) fluorometer. Results are expressed as a percentage of relative cell numbers of untreated control cells.
Caspase‐3 activity. Caspase‐3 activity was measured using the synthetic fluorogenic substrate Ac‐DEVD‐AFC (Bachem, Weil am Rhein, Germany). After the indicated treatments, cells were lyzed in 10 mM TRIS‐HCl, 10 mM NaH2PO4/NaHPO4 (pH 7.5), 130 mM NaCl, 1% Triton‐X‐100, and 10 mM Na4P2O7 and then incubated with 20 mM HEPES (pH 7.5), 10% glycerol, 2 mM dithiothreitol (DTT) and 25 µg/mL Ac‐DEVD‐AFC at 37°C for 2 h. The release of trifluoromethylcoumarin (AFC) was analyzed on a Wallac Victor fluorometer (Perkin Elmer, Rodgau‐Jügesheim, Germany) using an excitation/emission wavelength of 390/510 nm. Relative caspase‐3 activities were calculated as a ratio of emission of treated cells to untreated cells.
Western blot analysis. Cell were lyzed on ice for 15 min in 40 mM TRIS‐HCl (pH 7.4), 150 mM NaCl, 1% Triton X‐100, 0.5% sodium deoxycholate and 0.1% sodium dodecylsulfate (SDS) supplemented with a protease inhibitor cocktail (Roche) followed by brief sonification. Protein concentration was assayed using bicinchoninic acid (Pierce, Rockford, IL, USA) according to the manufacturer's instructions. For immunoblotting, 30 µg of total cellular protein per lane were separated by standard SDS‐polyacrylamide gel electrophoresis (PAGE) and electrophoretically transferred to PVDF membranes (Millipore, Eschborn, Germany). After blocking in PBS containing 5% dry milk and 0.05% Tween‐20, PARP‐1 and Bid cleavage was immunodetected using mouse anti‐PARP‐1 monoclonal (dilution 1:200; Biomol, Hamburg, Germany) and rabbit anti‐Bid polyclonal (dilution 1:1000; Cell Signaling Technology, Danvers, MA, USA) antibodies, respectively. Equal loading of protein was verified by detection of glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) using mouse anti‐GAPDH monoclonal antibody (dilution 1:10 000; Biodesign International, Saco, ME, USA). Peroxidase‐conjugated goat antimouse or antirabbit immunoglobulin G (IgG; dilution 1:25 000; Dianova, Hamburg, Germany) were used as secondary antibodies followed by enhanced chemiluminescence detection (Amersham Biosciences, Freiburg, Germany) of specific signals.
Flow cytometric detection of cell‐surface TRAIL receptors. After 24 h‐treatment with HDI, cells were harvested with 0.2% ethylenediaminetetraacetic acid (EDTA) and washed twice with isotonic PBS. Cells (5 × 105) were incubated with mouse anti‐TRAIL‐R1 and anti‐TRAIL‐R2 monoclonal antibodies (2 µg/mL; BioLegend, San Diego, CA, USA) for 45 min at 4°C. Cells were washed twice and stained with a fluorescein isothiocyanate (FITC)‐conjugated goat antimouse IgG antibody (2 µg/mL; BioLegend) for 45 min at 4°C. After washing, 50 000 cells were analyzed using a FACSCalibur. A purified mouse IgG1 (2 µg/mL; eBioscience, San Diego, CA, USA) was used as isotype‐matched control.
Results
Toxicity of TRAIL combined with HDI or cytostatics in HepG2 and Huh6 cells. Previously, we found that HDI interacted synergistically with TRAIL to induce cell death in lung and prostate cancer cell lines.( 35 ) This finding prompted us to examine the combinatorial application of HDI and TRAIL in liver cancer cell lines and to compare it with (i) the combination of TRAIL with standard cytostatics and (ii) the combination of HDI and TRAIL in normal hepatocytes. First, cytotoxic effects were monitored by assessing DNA fragmentation. To begin with, we established the concentrations of HDI that produced slight cytotoxic effects (i.e. 7–21% cell death after 2 µM vorinostat, 2 mM NaB or 2 µM MS‐275 in HepG2 cells, 5–10% cell death after 1 µM vorinostat, 1 mM NaB or 1 µM MS‐275 in Huh6 cells). These doses were then used as maximum concentrations in the combination experiments. As shown in Fig. 1(a), HepG2 cells were susceptible to TRAIL alone, but their susceptibility could be strongly increased by pretreatment with HDI. Remarkably, even non‐toxic concentrations of HDI (0.5 µM vorinostat, 0.5 mM NaB, 0.5 µM MS‐275) potently enhanced TRAIL‐induced cell death. For example, 10 ng/mL of TRAIL caused death in 16% of non‐pretreated cells, but in 60% of cells pretreated with 0.5 µM MS‐275. Huh6 cells were found to be marginally responsive to TRAIL even at a concentration of 300 ng/mL. However, when combined with HDI, TRAIL elicited death in up to 49% of cells. To evaluate the favorable cooperation of HDI and TRAIL by an independent read‐out, we examined cell death by determining LDH release; LDH release serves as surrogate marker for severe cellular damage. This assay also revealed that HDI significantly augmented TRAIL killing in HepG2 and Huh6 cells (Fig. 1b; for the differential effects of TRAIL alone in HepG2 cells in the two assays, see Discussion). In contrast, the combination of TRAIL with either carboplatin or etoposide produced only additive or – in case of etoposide in Huh6 – slightly supra‐additive effects (Fig. 1a).
Figure 1.
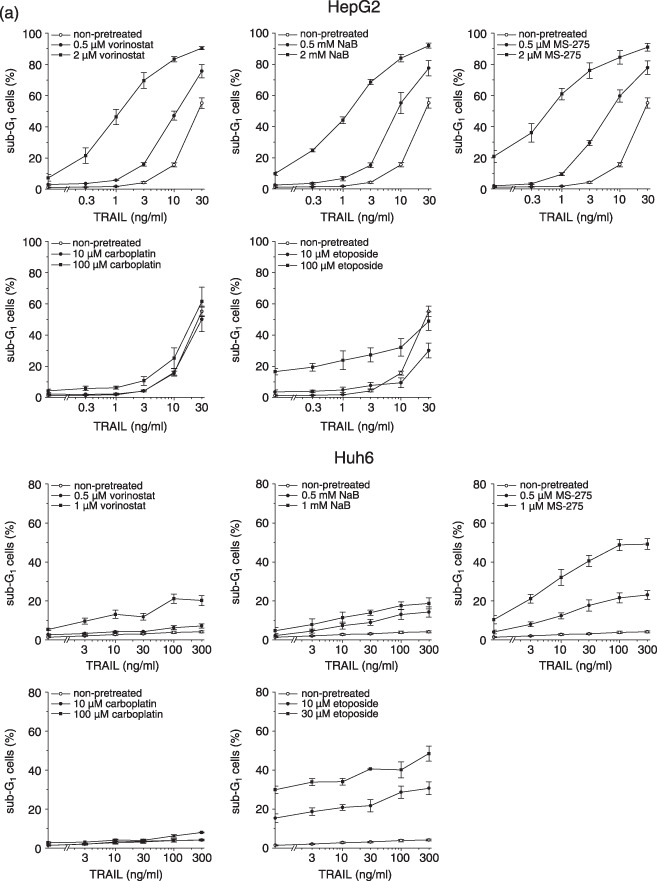
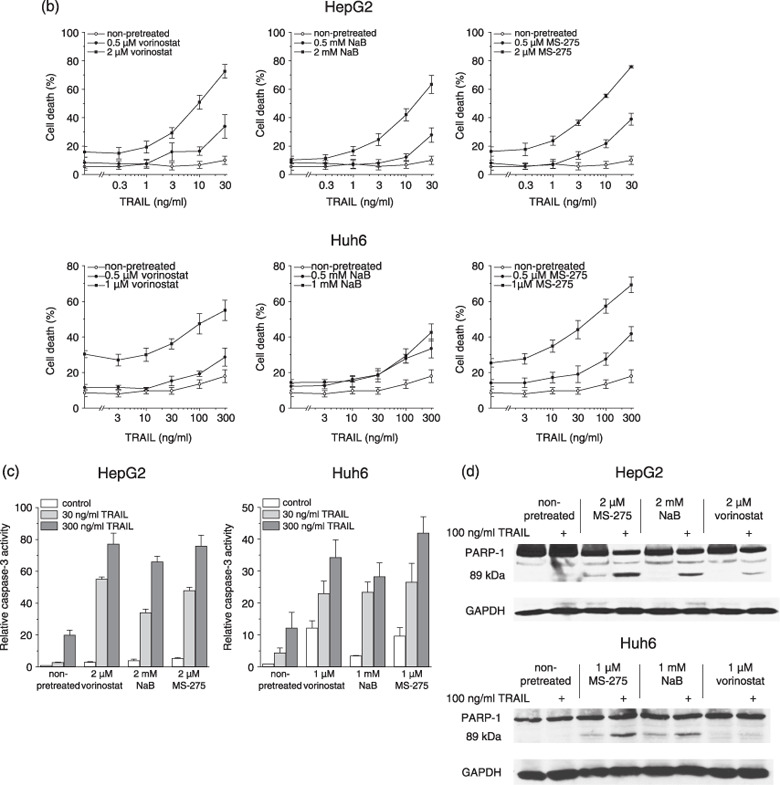
Induction of cell death in hepatoma cells by tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL) in combination with histone deacetylase inhibitors (HDI) or cytostatics. Twenty‐four hours after administration of cytotoxic compounds, cells were exposed to TRAIL for another 24 h. (a) DNA fragmentation was determined by flow cytometric cell cycle analysis; apoptotic cells were detected as sub‐G1 peak. (b) Cell death was determined by lactate dehydrogenase (LDH) release assay. (c) Caspase‐3 activity was determined using the fluorogenic substrate Ac‐DEVD‐AFC; relative caspase‐3 activities are the ratio of treated cells to untreated cells. (d) PARP‐1 cleavage was determined by Western blotting using an anti‐PARP‐1 monoclonal antibody; equal protein loading was confirmed with an anti‐glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) monoclonal antibody. (a–c) Means ± SEM of each three separate experiments are shown. (d) Results are representative of two separate experiments.
To analyze whether the cooperative action of HDI and TRAIL in hepatoma cells stemmed from the cooperative induction of apoptosis, we assessed the activation of caspase‐3 and the cleavage of poly (ADP‐ribose) polymerase (PARP‐1). The activation of caspase‐3 is a typical feature of apoptotic cell death and thus considered a suitable measure of apoptotic responsiveness.( 36 ) As shown in Fig. 1(c), TRAIL alone led to a maximum 20‐fold raise in caspase‐3 activity, whereas in cells pre‐exposed to HDI, TRAIL activated caspase‐3 by up to 77‐fold. Cleavage of PARP‐1 into fragments of 89 and 24 kDa is another useful hallmark of apoptotic cell death.( 37 ) Treatment with TRAIL alone provoked the appearance of a very faint band at 89 kDa in HepG2 cells (Fig. 1d). In Huh6 cells, TRAIL did not cause detectable PARP‐1 cleavage, consistent with its marginal cytotoxic effect in these cells. However, TRAIL‐induced cleavage of PARP‐1 was observed in both cell lines after pretreatment with HDI.
Toxicity of TRAIL combined with HDIs in normal hepatocytes. Since the combination of TRAIL with antineoplastic agents bears the risk of eliciting cell death in otherwise resistant normal cells, we investigated whether HDI also sensitized normal hepatocytes to TRAIL. We employed the differentiated, non‐transformed mouse hepatocyte cell line AML12, hepatocytes freshly isolated from rat liver, and normal human hepatocytes. The basic functionality of human TRAIL in mouse hepatocytes is evident from the ActD experiment (see below), in rat hepatocytes it has been demonstrated by Malhi et al.( 38 ) As depicted in Fig. 2, non‐malignant hepatocytes were completely resistant to TRAIL and also remained resistant in the presence of HDI. Even after pretreatment with a toxic dose of NaB (i.e. 38% cell death at 5 mM NaB), AML12 cells remained refractory to TRAIL. In sharp contrast, pretreatment with a toxic dose of ActD (i.e. 17% cell death at 200 µM ActD), a potent TRAIL sensitizer of hepatoma cells,( 9 ) effectively sensitized AML12 cells to TRAIL‐induced death (i.e. 70% cell death at 200 µM ActD/300 ng/mL TRAIL).
Figure 2.
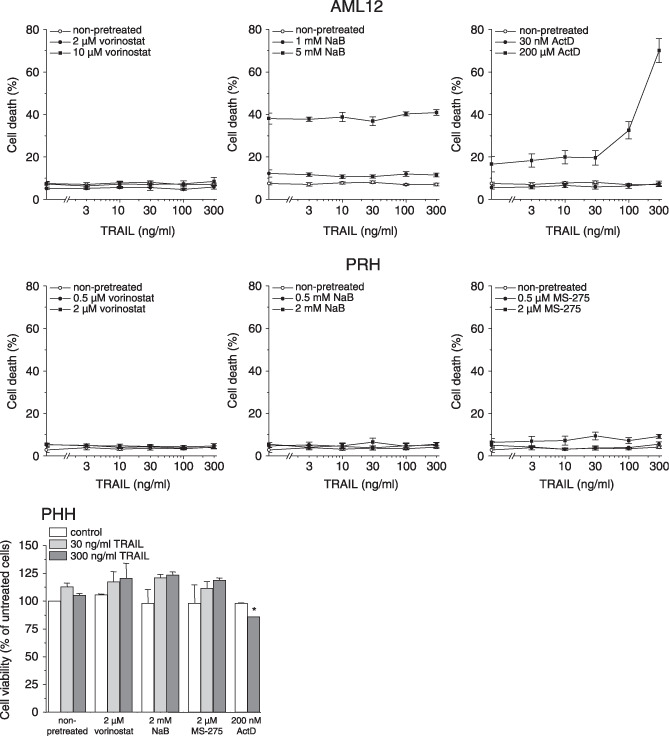
Induction of cell death in rodent and human hepatocytes by tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL) in combination with histone deacetylase inhibitors (HDI) or actinomycin D (ActD). Twenty‐four hours after administration of HDI or 1 h after administration of ActD, cells were exposed to TRAIL for another 24 h. AML12, cell death was determined by flow cytometric analysis of propidium iodide (PI) uptake; primary rat hepatocytes (PRH), cell death was determined by lactate dehydrogenase (LDH) release assay; primary human hepatocytes (PHH), cell viability was determined by Alamar Blue assay. Means ± SEM of two (PHH) or three (AML12, PRH) separate experiments are shown (*P < 0.05).
The role of TRAIL receptors and Bid cleavage in the HDI‐mediated sensitization to TRAIL‐induced cell death. HDI‐mediated sensitization to TRAIL may be the result of HDI‐induced up‐regulation of TRAIL receptors TRAIL‐R1 and/or TRAIL‐R2.( 29 ) To analyze whether vorinostat, NaB or MS‐275 would elevate the expression of TRAIL receptors in hepatoma cells, the expression of TRAIL‐R1 and ‐R2 on the cell surface was measured by flow cytometric analysis. Both HepG2 and Huh6 cells showed cell‐surface expression of TRAIL‐R1, whereas TRAIL‐R2 could only be detected on the surface of HepG2 cells (Fig. 3a). However, HDI did not increase cell‐surface expression of TRAIL‐R1 or ‐R2 in either of the cell lines.
Figure 3.
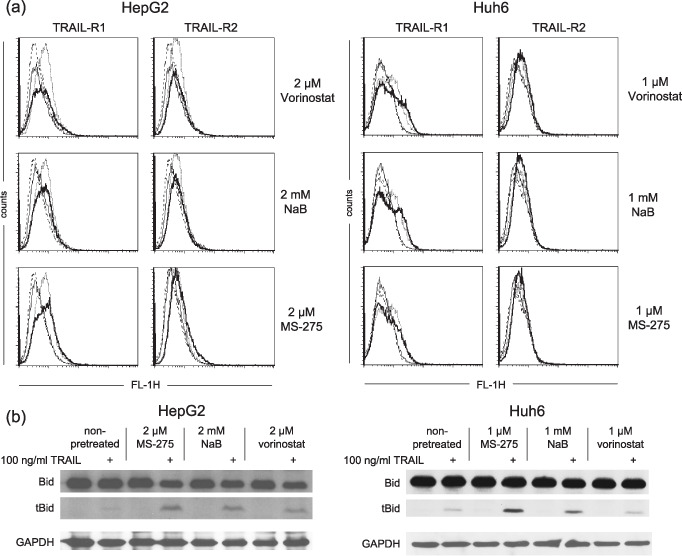
Effects of histone deacetylase inhibitors (HDI) on tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL) receptor expression and Bid cleavage in hepatoma cells. (a) Cells were exposed to HDI for 24 h. Cell‐surface expression of TRAIL‐R1 and ‐R2 was measured by flow cytometric analysis using monoclonal antibodies against TRAIL‐R1 and ‐R2, respectively, followed by a fluorescein isothiocyanate (FITC)‐conjugated antimouse immunoglobulin G antibody. Dotted line, untreated cells stained with anti‐TRAIL‐R1/R2 antibodies; solid line, HDI‐treated cells stained with anti‐TRAIL‐R1/R2 antibodies; dashed line, untreated cells stained with isotype‐matched control antibody; thin line, HDI‐treated cells stained with isotype‐matched control antibody. (b) Twenty‐four hours after administration of HDI, cells were exposed to TRAIL for another 2 h. Bid cleavage was determined by Western blotting using an anti‐Bid polyclonal antibody; equal protein loading was confirmed with an anti‐ glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) monoclonal antibody. (a, b) Results are representative of each two separate experiments.
TRAIL activates the death receptor (extrinsic) pathway of apoptosis, but may also activate the mitochondrial (intrinsic) one. The latter is initiated via caspase‐8/caspase‐10‐mediated activating cleavage of Bid to tBid.( 4 ) In order to disclose whether HDI facilitated the intrinsic pathway, we examined TRAIL‐induced Bid cleavage in HepG2 and Huh6 cells. As shown in Fig. 3(b), treatment with TRAIL alone led to faint Bid cleavage, whereas the treatment with HDI alone had no effect. The TRAIL‐induced Bid cleavage could be strongly enhanced by pretreatment with HDI.
Discussion
TRAIL is considered a promising anticancer agent because of its tumor cell selectivity and its potent cytotoxic action on the majority of cancer cell types. Unfortunately, hepatoma has been found to be largely resistant to TRAIL‐induced death.( 9 , 10 , 11 , 12 , 13 , 14 , 15 , 39 ) In the present work, we have thus studied whether the antineoplastic activity of TRAIL against hepatoma cells could be enhanced by combination with various chemotherapeutic drugs. In particular, we have investigated the sensitizing potential of HDI versus conventional chemotherapeutics. To the best of our knowledge, this is the first study that directly compares HDI and standard cytostatics for their TRAIL‐sensitizing effects in hepatoma cells. For our analyses, we employed HepG2 cells, a well studied HCC cell line, and Huh6 cells, a childhood hepatoblastoma cell line. The latter were of particular interest because hepatoblastoma cells have not yet been studied for their susceptibility to TRAIL‐induced death. Though the overall survival rate of children with hepatoblastoma has been remarkably improved during recent years, patients suffering from advanced staged hepatoblastoma still have a dismal prognosis, and more effective therapeutic strategies for these patients are required.( 40 )
The two cell lines differed in their responsiveness to treatment with TRAIL alone: Huh6 hepatoblastoma cells were resistant and HepG2 HCC cells were sensitive. However, with respect to the latter, we noted that the TRAIL effect depended on the assay applied: we observed a much stronger effect in the DNA fragmentation assay than in the LDH release assay. This difference may be attributable to not fully equivalent end points. During the apoptotic process, DNA fragmentation precedes cell death. Hence, after a certain period of time, not all cells that have undergone DNA fragmentation must also have undergone cell death. This may also explain the discrepancy of the TRAIL effects in HepG2 between different studies. HepG2 cells have repeatedly been reported to be TRAIL resistant( 9 , 10 , 11 , 14 , 15 ) or to be TRAIL sensitive.( 17 , 19 , 20 , 27 , 41 ) In any case, although HepG2 and Huh6 cells displayed differential susceptibility to TRAIL alone, we observed a highly favorable interaction of HDI with TRAIL in both the cell lines. Consistent with previous reports,( 14 , 18 ) HDI significantly enhanced TRAIL‐induced cell death in HepG2 cells. In Huh6 cells, we even found HDI to overcome resistance to TRAIL; particularly MS‐275 had a strong sensitizing effect. In contrast, conventional chemotherapeutic agents produced a largely additive effect in both cell lines; a slight sensitization in Huh6 cells occurred only with etoposide at concentrations that were themselves toxic. Taken together, these results suggest that HDI are significantly better TRAIL sensitizers than cytostatics in hepatoma‐derived cells. However, HDI may promote TRAIL‐induced death not only in hepatoma cells, but also in normal hepatocytes. We thus explored a potential cytotoxic effect of combined HDI/TRAIL treatment in rodent and human hepatocytes. Our findings demonstrate that normal hepatocytes were resistant to TRAIL both when applied alone or in conjunction with HDI, in concordance with a PHH study on the combinatorial application of HDI and TRAIL.( 14 ) But since a recent article reported that TRAIL induced cytotoxicity in tissue explants of human liver when combined with the HDI depsipeptide,( 42 ) the clinical applicability of a combinatorial HDI/TRAIL administration still needs to be tested in more depth.
Our finding that the killing efficiency of TRAIL could be increased by HDI, but not by conventional cytostatics, implies that this effect was not due to a mere cytotoxic activity, but rather depended on the mode of action of HDI. So, how may HDI second TRAIL to induce death in hepatoma cells? HDI function by inhibiting histone deacetylases, resulting in the accumulation of acetylated histones, in turn leading to an increase in transcriptionally active chromatin( 29 ) (though many non‐histone proteins have also been shown to be modified by acetylation and thus are potential targets for HDI).( 31 ) In so doing, HDI have been found to regulate the expression of up to more than 40% of genes.( 43 ) Unfortunately, the key determinants of TRAIL responsiveness are not consistently defined, and the concomitant alteration of several factors may account for a cell's resistance to TRAIL. Fortunately, however, the effects of HDI go beyond the modulation of single genes and HDI, thus, have the potential to overcome the resistant phenotype, even if the latter is the result of multiple gene expression changes.( 44 ) Among the factors that have been proposed for TRAIL resistance are the down‐regulation of the agonistic receptors TRAIL‐R1 or ‐R2, the expression of the ‘decoy’ receptors TRAIL‐R3 or ‐R4, the silencing of caspase‐8, the up‐regulation of c‐FLIP, the up‐regulation of antiapoptotic or the down‐regulation of proapoptotic Bcl‐2 family members, the up‐regulation of inhibitor of apoptosis proteins (IAPs) or the TRAIL‐induced activation of NF‐κB.( 4 , 5 , 39 ) In this study, we examined the relevance of agonistic TRAIL receptors and Bid cleavage for HDI‐induced TRAIL sensitization.
Several studies on HDI have suggested that increased sensitivity to TRAIL stemmed from elevated expression of TRAIL receptors.( 45 , 46 , 47 , 48 ) Others, however, found no association of enhanced TRAIL responsiveness with altered TRAIL receptor expression,( 49 , 50 , 51 , 52 ) including one on HCC cells.( 14 ) Our study did also not reveal an HDI‐triggered overexpression of TRAIL receptors in HepG2 and Huh6 cells, indicating that HDI mediate TRAIL sensitization by mechanisms different from TRAIL receptor up‐regulation, at least in hepatoma‐derived cells. Instead, we found that HDI enhanced TRAIL‐induced cleavage of the proapoptotic Bcl‐2 relative Bid. The binding of TRAIL to its receptors leads to the activation of caspase‐8, which in turn may lead to the direct activation of the caspase cascade or the cleavage of Bid to tBid; tBid then triggers the mitochondrial pathway of apoptosis.( 4 , 5 ) The finding that HDI promoted Bid cleavage implicates that HDI mediated sensitization to TRAIL through the activation of the mitochondrial pathway, in concordance with previous reports.( 35 , 45 , 46 , 48 , 49 , 50 ) Hence, the amplification of TRAIL's cytotoxic effect appears to be the result of the enhancement of the intrinsic pathway by HDI. Of course, this finding does not rule out the possibility that HDI also enhance TRAIL‐induced apoptosis by regulating the expression of proteins pertinent to the extrinsic pathway.
In advanced liver cancer, currently applied cytotoxic chemotherapies are basically limited to a palliative benefit without a clear improvement in overall survival.( 3 ) Clearly, new approaches are required. Both TRAIL and HDI hold promise to become potent anticancer agents. Here, we have demonstrated that their antineoplastic efficacy can be considerably increased by cotreatment. We have shown that the HDI vorinostat, NaB and MS‐275, but not the cytostatics carboplatin and etoposide, sensitized HCC and hepatoblastoma cells to TRAIL‐induced death. We have also shown that normal hepatocytes remained resistant to TRAIL in the presence of HDI. To sum up, our study suggests that the combination of HDI and TRAIL may be an attractive new treatment option for advanced HCC and childhood hepatoblastoma.
Acknowledgments
We thank Thomas Krieg (Department of Cardiology, University of Greifswald) for providing us with rat livers and Markus Roser (Institute for Clinical Chemistry and Laboratory Medicine, University of Greifswald) for his help with LDH activity assays. This work was supported by a grant from the ‘Wilhelm Sander‐Stiftung, Neustadt/Donau’.
References
- 1. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55: 74–108. [DOI] [PubMed] [Google Scholar]
- 2. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet 2003; 362: 1907–17. [DOI] [PubMed] [Google Scholar]
- 3. Burroughs A, Hochhauser D, Meyer T. Systemic treatment and liver transplantation for hepatocellular carcinoma: two ends of the therapeutic spectrum. Lancet Oncol 2004; 5: 409–18. [DOI] [PubMed] [Google Scholar]
- 4. Yagita H, Takeda K, Hayakawa Y, Smyth MJ, Okumura K. TRAIL and its receptors as targets for cancer therapy. Cancer Sci 2004; 95: 777–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reed JC. Drug insight: cancer therapy strategies based on restoration of endogenous cell death mechanisms. Nat Clin Pract Oncol 2006; 3: 388–98. [DOI] [PubMed] [Google Scholar]
- 6. Carlo‐Stella C, Lavazza C, Locatelli A, Vigano L, Gianni AM, Gianni L. Targeting TRAIL agonistic receptors for cancer therapy. Clin Cancer Res 2007; 13: 2313–17. [DOI] [PubMed] [Google Scholar]
- 7. Baader E, Toloczko A, Fuchs U et al . Tumor necrosis factor‐related apoptosis‐inducing ligand‐mediated proliferation of tumor cells with receptor‐proximal apoptosis defects. Cancer Res 2005; 65: 7888–95. [DOI] [PubMed] [Google Scholar]
- 8. Koschny R, Holland H, Sykora J et al . Bortezomib sensitizes primary human astrocytoma cells of WHO grades I to IV for tumor necrosis factor‐related apoptosis‐inducing ligand‐induced apoptosis. Clin Cancer Res 2007; 13: 3403–12. [DOI] [PubMed] [Google Scholar]
- 9. Yamanaka T, Shiraki K, Sugimoto K et al . Chemotherapeutic agents augment TRAIL‐induced apoptosis in human hepatocellular carcinoma cell lines. Hepatology 2000; 32: 482–90. [DOI] [PubMed] [Google Scholar]
- 10. Shin EC, Seong YR, Kim CH et al . Human hepatocellular carcinoma cells resist to TRAIL‐induced apoptosis, and the resistance is abolished by cisplatin. Exp Mol Med 2002; 34: 114–22. [DOI] [PubMed] [Google Scholar]
- 11. Kim YS, Schwabe RF, Qian T, Lemasters JJ, Brenner DA. TRAIL‐mediated apoptosis requires NF‐kappaB inhibition and the mitochondrial permeability transition in human hepatoma cells. Hepatology 2002; 36: 1498–508. [DOI] [PubMed] [Google Scholar]
- 12. Shigeno M, Nakao K, Ichikawa T et al . Interferon‐alpha sensitizes human hepatoma cells to TRAIL‐induced apoptosis through DR5 upregulation and NF‐kappa B inactivation. Oncogene 2003; 22: 1653–62. [DOI] [PubMed] [Google Scholar]
- 13. Chen XP, He SQ, Wang HP, Zhao YZ, Zhang WG. Expression of TNF‐related apoptosis‐inducing ligand receptors and antitumor tumor effects of TNF‐related apoptosis‐inducing ligand in human hepatocellular carcinoma. World J Gastroenterol 2003; 9: 2433–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pathil A, Armeanu S, Venturelli S et al . HDAC inhibitor treatment of hepatoma cells induces both TRAIL‐independent apoptosis and restoration of sensitivity to TRAIL. Hepatology 2006; 43: 425–34. [DOI] [PubMed] [Google Scholar]
- 15. Kusaba M, Nakao K, Goto T et al . Abrogation of constitutive STAT3 activity sensitizes human hepatoma cells to TRAIL‐mediated apoptosis. J Hepatol 2007; 47: 546–55. [DOI] [PubMed] [Google Scholar]
- 16. Koschny R, Walczak H, Ganten TM. The promise of TRAIL‐potential and risks of a novel anticancer therapy. J Mol Med 2007; 85: 923–35. [DOI] [PubMed] [Google Scholar]
- 17. Ganten TM, Haas TL, Sykora J et al . Enhanced caspase‐8 recruitment to and activation at the DISC is critical for sensitisation of human hepatocellular carcinoma cells to TRAIL‐induced apoptosis by chemotherapeutic drugs. Cell Death Differ 2004; 11 (Suppl 1): S86–96. [DOI] [PubMed] [Google Scholar]
- 18. Schuchmann M, Schulze‐Bergkamen H, Fleischer B et al . Histone deacetylase inhibition by valproic acid down‐regulates c‐FLIP/CASH and sensitizes hepatoma cells towards CD95‐ and TRAIL receptor‐mediated apoptosis and chemotherapy. Oncol Rep 2006; 15: 227–30. [DOI] [PubMed] [Google Scholar]
- 19. Ganten TM, Koschny R, Haas TL et al . Proteasome inhibition sensitizes hepatocellular carcinoma cells, but not human hepatocytes, to TRAIL. Hepatology 2005; 42: 588–97. [DOI] [PubMed] [Google Scholar]
- 20. Inoue T, Shiraki K, Fuke H et al . Proteasome inhibition sensitizes hepatocellular carcinoma cells to TRAIL by suppressing caspase inhibitors and AKT pathway. Anticancer Drugs 2006; 17: 261–8. [DOI] [PubMed] [Google Scholar]
- 21. Koschny R, Ganten TM, Sykora J et al . TRAIL/bortezomib cotreatment is potentially hepatotoxic but induces cancer‐specific apoptosis within a therapeutic window. Hepatology 2007; 45: 649–58. [DOI] [PubMed] [Google Scholar]
- 22. Liedtke C, Groger N, Manns MP, Trautwein C. Interferon‐alpha enhances TRAIL‐mediated apoptosis by up‐regulating caspase‐8 transcription in human hepatoma cells. J Hepatol 2006; 44: 342–9. [DOI] [PubMed] [Google Scholar]
- 23. Van Valen F, Fulda S, Schafer KL et al . Selective and nonselective toxicity of TRAIL/Apo2L combined with chemotherapy in human bone tumour cells vs. normal human cells. Int J Cancer 2003; 107: 929–40. [DOI] [PubMed] [Google Scholar]
- 24. Leverkus M, Neumann M, Mengling T et al . Regulation of tumor necrosis factor‐related apoptosis‐inducing ligand sensitivity in primary and transformed human keratinocytes. Cancer Res 2000; 60: 553–9. [PubMed] [Google Scholar]
- 25. Leverkus M, Sprick MR, Wachter T et al . Proteasome inhibition results in TRAIL sensitization of primary keratinocytes by removing the resistance‐mediating block of effector caspase maturation. Mol Cell Biol 2003; 23: 777–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Steele LP, Georgopoulos NT, Southgate J, Selby PJ, Trejdosiewicz LK. Differential susceptibility to TRAIL of normal versus malignant human urothelial cells. Cell Death Differ 2006; 13: 1564–76. [DOI] [PubMed] [Google Scholar]
- 27. Ganten TM, Koschny R, Sykora J et al . Preclinical differentiation between apparently safe and potentially hepatotoxic applications of TRAIL either alone or in combination with chemotherapeutic drugs. Clin Cancer Res 2006; 12: 2640–6. [DOI] [PubMed] [Google Scholar]
- 28. Meurette O, Fontaine A, Rebillard A et al . Cytotoxicity of TRAIL/anticancer drug combinations in human normal cells. Ann NY Acad Sci 2006; 1090: 209–16. [DOI] [PubMed] [Google Scholar]
- 29. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 2006; 5: 769–84. [DOI] [PubMed] [Google Scholar]
- 30. Riester D, Hildmann C, Schwienhorst A. Histone deacetylase inhibitors – turning epigenic mechanisms of gene regulation into tools of therapeutic intervention in malignant and other diseases. Appl Microbiol Biotechnol 2007; 75: 499–514. [DOI] [PubMed] [Google Scholar]
- 31. Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors. molecular mechanisms of action. Oncogene 2007; 26: 5541–52. [DOI] [PubMed] [Google Scholar]
- 32. Fuchs J, Rydzynski J, Von Schweinitz D et al . Pretreatment prognostic factors and treatment results in children with hepatoblastoma: a report from the German Cooperative Pediatric Liver Tumor Study HB 94. Cancer 2002; 95: 172–82. [DOI] [PubMed] [Google Scholar]
- 33. Wu JC, Merlino G, Fausto N. Establishment and characterization of differentiated, nontransformed hepatocyte cell lines derived from mice transgenic for transforming growth factor alpha. Proc Natl Acad Sci USA 1994; 91: 674–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seglen PO. Preparation of isolated rat liver cells. Meth Cell Biol 1976; 13: 29–83. [DOI] [PubMed] [Google Scholar]
- 35. Sonnemann J, Gange J, Kumar KS, Muller C, Bader P, Beck JF. Histone deacetylase inhibitors interact synergistically with tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL) to induce apoptosis in carcinoma cell lines. Invest New Drugs 2005; 23: 99–109. [DOI] [PubMed] [Google Scholar]
- 36. Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 2008; 9: 231–41. [DOI] [PubMed] [Google Scholar]
- 37. Soldani C, Scovassi AI. Poly (ADP‐ribose) polymerase‐1 cleavage during apoptosis: an update. Apoptosis 2002; 7: 321–8. [DOI] [PubMed] [Google Scholar]
- 38. Malhi H, Barreyro FJ, Isomoto H, Bronk SF, Gores GJ. Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut 2007; 56: 1124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Herr I, Schemmer P, Buchler MW. On the TRAIL to therapeutic intervention in liver disease. Hepatology 2007; 46: 266–74. [DOI] [PubMed] [Google Scholar]
- 40. Warmann SW, Fuchs J. Drug resistance in hepatoblastoma. Curr Pharm Biotechnol 2007; 8: 93–7. [DOI] [PubMed] [Google Scholar]
- 41. Ohuchida T, Okamoto K, Akahane K et al . Midkine protects hepatocellular carcinoma cells against TRAIL‐mediated apoptosis through down‐regulation of caspase‐3 activity. Cancer 2004; 100: 2430–6. [DOI] [PubMed] [Google Scholar]
- 42. Volkmann X, Fischer U, Bahr MJ et al . Increased hepatotoxicity of tumor necrosis factor‐related apoptosis‐inducing ligand in diseased human liver. Hepatology 2007; 46: 1498–508. [DOI] [PubMed] [Google Scholar]
- 43. Peart MJ, Smyth GK, Van Laar RK et al . Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci USA 2005; 102: 3697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Perez‐Plasencia C, Duenas‐Gonzalez A. Can the state of cancer chemotherapy resistance be reverted by epigenetic therapy? Mol Cancer 2006; 5: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guo F, Sigua C, Tao J et al . Cotreatment with histone deacetylase inhibitor LAQ824 enhances Apo‐2L/tumor necrosis factor‐related apoptosis inducing ligand‐induced death inducing signaling complex activity and apoptosis of human acute leukemia cells. Cancer Res 2004; 64: 2580–9. [DOI] [PubMed] [Google Scholar]
- 46. Nakata S, Yoshida T, Horinaka M, Shiraishi T, Wakada M, Sakai T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL‐R2 and sensitize apoptosis induced by TRAIL/APO2‐L in human malignant tumor cells. Oncogene 2004; 23: 6261–71. [DOI] [PubMed] [Google Scholar]
- 47. Chopin V, Slomianny C, Hondermarck H, Le Bourhis X. Synergistic induction of apoptosis in breast cancer cells by cotreatment with butyrate and TNF‐alpha, TRAIL, or anti‐Fas agonist antibody involves enhancement of death receptors’ signaling and requires P21 (waf1). Exp Cell Res 2004; 298: 560–73. [DOI] [PubMed] [Google Scholar]
- 48. Singh TR, Shankar S, Srivastava RK. HDAC inhibitors enhance the apoptosis‐inducing potential of TRAIL in breast carcinoma. Oncogene 2005; 24: 4609–23. [DOI] [PubMed] [Google Scholar]
- 49. Rosato RR, Almenara JA, Dai Y, Grant S. Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cells. Mol Cancer Ther 2003; 2: 1273–84. [PubMed] [Google Scholar]
- 50. Muhlethaler‐Mottet A, Flahaut M, Bourloud KB et al . Histone deacetylase inhibitors strongly sensitise neuroblastoma cells to TRAIL‐induced apoptosis by a caspases‐dependent increase of the pro‐ to anti‐apoptotic proteins ratio. BMC Cancer 2006; 6: 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Inoue S, Twiddy D, Dyer MJ, Cohen GM. Upregulation of TRAIL‐R2 is not involved in HDACi mediated sensitization to TRAIL‐induced apoptosis. Cell Death Differ 2006; 13: 2160–2. [DOI] [PubMed] [Google Scholar]
- 52. Sonnemann J, Dreyer L, Hartwig M et al . Histone deacetylase inhibitors induce cell death and enhance the apoptosis‐inducing activity of TRAIL in Ewing's sarcoma cells. J Cancer Res Clin Oncol 2007; 133: 847–58. [DOI] [PubMed] [Google Scholar]