

Abstract
Mouse endothelial TKD2 cells in monolayers were cocultured with various human cell lines for 24 h, and the expression of several secreted matrix metalloproteinases (MMP) and cell adhesion molecules was examined by real‐time reverse transcription–polymerase chain reaction using mouse‐specific primers. Coculture with normal fibroblasts did not elicit the expression of these molecules, but coculture with cancer cells induced the expression of MMP‐3, MMP‐9 and MMP‐10 mRNA in endothelial cells, and in normal mouse embryonic fibroblasts. The induction of MMP mRNA was dependent on direct cell adhesion, as separate culture of A549 cells in Boyden chambers did not induce MMP mRNA, and neutralizing antibody against VLA‐4 abolished the induction. An inhibitor of phosphatidylinositol‐3‐phosphate kinase strongly suppressed the induction of MMP‐3, MMP‐9 and MMP‐10 mRNA, and expression of the dominant‐negative mutant of phosphatidylinositol‐3‐phosphate kinase also decreased the induction. It was suggested that intracellular reactive oxygen species (ROS) levels were increased in TKD2 cells following adhesion to cancer cells. ROS scavengers decreased the levels of MMP induction, and roterone, an inhibitor of mitochondrial complex I, strongly suppressed the induction of MMP‐3, MMP‐9 and MMP‐10. The depletion of mitochondria in TKD2 cells decreased the induction of MMP‐9, but the induction of MMP‐3 and MMP‐10 was not affected. These results indicate that the adhesion of cancer cells to endothelial cells activates several distinct signaling pathways to induce MMP gene expression, and the pathways for MMP‐3, MMP‐9 and MMP‐10 are partly different. For the induction of MMP‐9, mitochondria participate in induction, possibly through the production of ROS. (Cancer Sci 2007; 98: 58–67)
Abbreviations:
- DMEM
Dulbecco's modified Eagle's medium
- DMTU
dimethyl thiourea
- DPI
diphenyleneiodonium
- ECM
extracellular matrix
- ICAM
intercellular adhesion molecule
- MMP
matrix metalloproteinase
- PI3K
phosphatidylinositol‐3‐phosphate kinase
- ROS
reactive oxygen species
- RT‐PCR
reverse transcription–polymerase chain reaction
- VCAM
vascular cell adhesion molecule.
Metastasis is a complex process, but the adhesion of cancer cells to surrounding normal cells or endothelial cells is one of the critical steps for metastasis. Within blood vessels, circulating tumor cells interact with humoral and cellular constituents of blood and with the endothelial cells of blood vessels. These interactions have a significant influence on the fate of metastatic cells and on the outcome of the metastatic process. Initial interactions involve mechanical contact and transient adhesion, mediated by endothelial selectins and their ligands on neoplastic cells. This contact initiates the sequence of activation pathways that involve cytokines, growth factors, bioactive lipids and ROS produced by either cancer cells or the endothelium. These molecules elicit the expression of integrin adhesion molecules in cancer cells and the endothelium, MMP and chemotactic factors that promote the attachment of tumor cells to the vessel wall and transvascular penetration. In carcinomas, the influences of the microenvironment are mediated, to a large degree, by bidirectional interactions (adhesion, survival, proteolysis, migration, immune escape mechanisms, angiogenesis, and homing in on target organs) between epithelial tumor cells and neighboring stromal cells, such as fibroblasts, as well as endothelial and immune cells. Affected proteins include growth factor signaling molecules, chemokines, cell–cell adhesion molecules (cadherins, integrins) as well as extracellular proteases (MMP).( 1 , 2 )
The adhesion of cancer cells to endothelial cells is a critical step for tumor invasion. Among the adhesion molecules on the surface of endothelial cells, VCAM‐1 is known to be induced by several cytokines such as tumor necrosis factor, interleukins‐1 and ‐4 and lipopolysaccharide, and this induction seems to be involved in adhesion to lymphocytes and leukocytes.( 3 , 4 ) VLA‐4 (integrin α4β1) is a receptor for VCAM‐1. The VCAM‐1–VLA‐4 ligand–receptor pair may play a major role in the recruitment of mononuclear leukocytes to inflammatory sites in vivo. Some types of melanoma cells expressing VCAM‐1 ligand (VLA‐4) had stronger activity to adhere to endothelial cells.( 5 ) These interactions are thought to be important as a first step in tumor invasion.
Endothelial cells adhere to each other, forming tight junctions that prohibit the penetration of cells present in blood,( 6 ) and injury to this structure allows leukocytes to penetrate into surrounding tissues.( 7 ) Degradation of the basement membrane is catalyzed by MMP secreted from macrophage or cancer cells, but recent reports indicate that MMP are also produced from surrounding normal cells.( 8 , 9 , 10 , 11 ) MMP are critical for tumor invasion by degrading the ECM and for angiogenesis.( 12 , 13 ) They also regulate cellular motility, growth and differentiation through the degradation of ECM molecules, and soluble MMP have a wide range of biological activities.( 13 , 14 ) Among soluble the MMP, MMP‐9 (gelatinase) degrades type IV collagen, and has important functions in tumor invasion.( 15 , 16 ) Endothelial cells produce MMP, but signals to induce MMP gene expression remain unknown.( 17 ) In the present study, we examined changes in endothelial cell phenotypes after adhesion to human cancer cells to reveal the functional significance of endothelial cells in tumor invasion.
Materials and Methods
Chemicals. Tiron, diphenyleneiodonium (DPI) sulfate and rotenone were from Sigma (St Louis, MO, USA) and DMTU was from Aldrich Chemicals (Milwaukee, WI, USA). PD98059, PP1(4‐amino‐5‐(4‐methylphenyl)‐7‐(t‐butyl)pyrazolo[3,4‐d]‐pyrimidine), SB203580 and API‐2 Triciribine were from Calbiochem (La Jolla, CA, USA). LY294002 was from LC Laboratories (Woburn, MA, USA). Mouse control IgG1 was from DakoCytomation (Glostrup, Denmark), and monoclonal antihuman integrin α2 and α4 were from Sigma.
Cell culture. Mouse TKD2 endothelial cells were established from transgenic mice of temperature‐sensitive SV‐40T,( 18 ) and were cultured in DMEM supplemented with 10% fetal bovine serum at 33°C in 5% CO2 in humidified air. Primary mouse embryonic fibroblasts were prepared as described previously.( 19 ) Human hepatoma cells (HepG2), lung cancer cells (A549) and normal human fibroblasts (TIG‐3) were cultured in DMEM. HeLa S3 cells and human gastric cancer cell lines (KATO‐III and TMK‐1) were cultured in RPMI‐1640 medium supplemented with 10% fetal bovine serum. Human sarcoma cells (HT1080) were cultured in Eagle's MEM supplemented with 10% fetal bovine serum. Cells were cultured at 37°C in 5% CO2 in humidified air. These cell lines were supplied by the Riken Bioresource Center Cell Bank.
Establishment of TKD2 cells expressing a dominant negative form of p85a. Cells were transfected either with pSG5‐p85αWT or pSG5‐p85αΔiSH2‐N,( 20 ) together with the hygromycin‐resistance gene (pTK‐Hyg), and hygromycin‐resistant clones were isolated. Stable lines that expressed p85α, as estimated by western blotting, were used.
Establishment of pseudo ρ0 cells. TKD2 cells were cultured in the presence of ethidium bromide (250 ng/mL) and uridine (50 µg/mL) for 7 days. Levels of mRNA for complex I or cytochrome b were determined by real‐time RT‐PCR. For the isolation of revertants, ρ0 cells were cultured in the absence of ethidium bromide and uridine for 14 days.( 21 )
RNA analysis. Total RNA was extracted with TRIzol reagent (Life Technologies, Rockville, MD, USA). For quantitative RT‐PCR, 1 µg of total RNA was reverse‐transcribed with random hexamers (Takara Shuzoh, Kyoto, Japan) and SuperScript II (Invitrogen, San Diego, CA, USA). Subsequently, a fragment of MMP cDNA was amplified by PCR with 40 cycles of denaturing (90°C, 20 s), annealing (55°C, 20 s) and extension (72°C, 30 s) using SYBR Green PCR master mix (PE Biosystems, Foster City, CA, USA). Monitoring and quantitative analysis of PCR products were carried out using a GeneAmp 5700 (PE Biosystems). All PCR primers were designed using PrimerExpress 1.0 (PE Biosystems).
Polymerase chain reaction primers. Primers of mouse‐specific MMP that did not detect human MMP were designed as follows: MMP‐2 forward 5′‐TGCACCACCTTAACTGTTGCTT and reverse 5′‐GGAAATGCAGTGGAGTGGAAA; MMP‐3 forward 5′‐GGAGATGCTCACTTTGACGATG and reverse 5′‐CAGCAACCAGGAATAGGTTGG; MMP‐9 forward 5′‐AGCGTCATTCGCGTGGATA and reverse 5′‐GGAAACTCACACGCCAGAAGA; MMP‐10 forward 5′‐CCTGTGTTGTCTGTCTCTCCAAGA and reverse 5′‐CGTGCTGACTGAATCAAAGGAC; VCAM‐1 forward CAAGGACTATTTTTCGCCCG and reverse ATCCCGATGGCAGGTATTACC; ICAM‐1 forward GTGGTGAAGTCTGTCAAACAGGAG and reverse ATTCCTGGTGACATTCCCATG; integrin α4 forward GCAAGGAAGTTCCAGGTTACATTG and reverse CACACCTGTATGCTTCCTGTAATCAC. Mitochondrial markers were: complex I (forward 5′‐TGCACCTACCCTATCACTCACACT, reverse 5′‐GGCTCATCCTGATCATAGAATGG) and cytochrome b (forward 5′‐CCGATTCTTCGCTTTCCACT and reverse 5′‐CTGTTTCGTGGAGGAAGAGGAG). Glyceraldehyde‐3‐phosphate dehydrogenase was used as a reference (forward 5′‐ATGTGTCCGTCGTGGATCTGA and reverse 5‐ATGCCTGCTTCACCACCTTCT).
Measurement of redox state. We used Redox Sensor Red CC‐1 (Molecular Probes, Eugene, OR, USA), a dye oxidized in the cytoplasm before it becomes localized to the mitochondria,( 22 ) and the mitochondria‐selective dye MitoTracker Green FM (Molecular Probes) to measure the intracellular redox state and mitochondrial membrane potential, respectively. TKD2 cells were cultured in 60‐mm plastic dishes for 24 h, and an approximately equal number of A549 cells were inoculated. At 30 min after coculture, Red CC‐1 was added at a concentration of 5 µM. Cells were rinsed with phosphate‐buffered saline after 10 min, and fluorescence (568 nm) was observed with a microscope (Eclipse TE2000‐E; Nikon, Tokyo, Japan).
Mitochondrial membrane potential. Membrane potential was measured using JC‐1 (BioVision Research Products, Palo Alto, CA, USA). TKD2 cells were seeded on 35‐mm plastic dishes overnight, and JC‐1 was added at a concentration of 10 µg/mL. After 15 min, cells were rinsed with Hanks’ balanced salt solution (HBSS), and A549 cells were added. Green (527 nm) and red (590 nm) fluorescence was measured with a fluorescent microscope and analyzed using Aquacosmos version 2.5 (Hamamatsu Photonics, Hamamatsu, Shizuoka, Japan). Mitochondrial membrane potential was estimated by the intensity ratio of red and green fluorescence.
Results
Induction of adhesion‐related genes in mouse TKD2 cells cocultured with human cancer cells. Human cancer cells were added to a monolayer of TKD2 cells (6 × 105 cells/60‐mm dish) at a ratio of 1:1 and cultured for 24 h at 33°C. Total RNA was extracted, and levels of MMP mRNA in TKD2 cells were examined by real‐time RT‐PCR. The primers did not detect human MMP (data not shown). The results shown in Fig. 1a indicate that coculture with normal human fibroblasts did not affect the levels of MMP mRNA significantly, but cultivation with cancer cells induced the expression of MMP‐3, MMP‐9 and MMP‐10. Levels of MMP‐2 also increased but to a lesser extent. Expression of several cell adhesion molecules, such as VCAM‐1 and ICAM‐1, was also increased in TKD2 cells cocultured with tumor cells. The effect of A549 seemed most prominent and we concentrated on A549 in later experiments.
Figure 1.
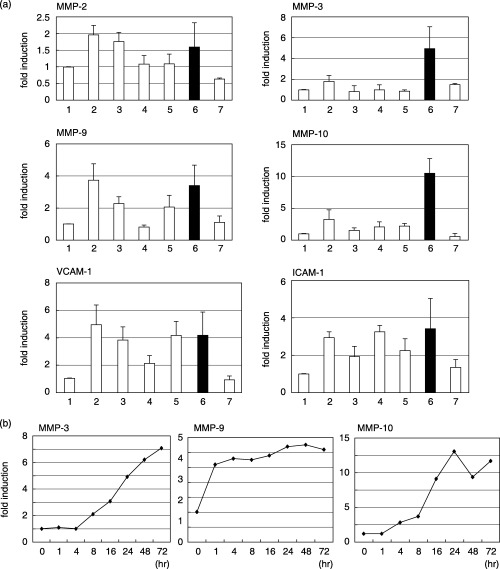
(a) Change in matrix metalloprotease (MMP) mRNA in TKD2 cells cultured with human cells. 1, TKD2 alone; 2, TKD2 + HT1080; 3, TKD2 + TMK‐1; 4, TKD2 + HepG2; 5, TKD2 + HelaS3; 6, TKD2 + A549; 7, TKD2 + TIG3. Cells were seeded onto TKD2 confluent monolayer cultures at 6 × 105 cells/60‐mm dish and incubated for 24 h. Total RNA was extracted and the levels of MMP mRNA were determined by real‐time reverse transcription–polymerase chain reaction (RT‐PCR) with mouse‐specific primers. Values were normalized using glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) mRNA levels as a reference. (b) Time course of induction of MMP mRNA. TKD2 cells were cocultured with A549 cells for the indicated times, and levels of MMP mRNAs in TKD2 cells were determined by RT‐PCR. ICAM, intercellular adhesion molecule; VCAM, vascular cell adhesion molecule.
A549 cells were seeded on a monolayer of TKD2 cells, and RNA was extracted at various times after starting coculture. The MMP‐9 mRNA level increased rapidly and reached a plateau at 1 h. Induction of MMP‐3 and MMP‐10 was slower (Fig. 1b), starting after 4 h and reaching a plateau at around 24–48 h.
Requirement of cell adhesion for induction. Cancer cells secrete various cytokines that could induce MMP in surrounding cells. To examine whether direct cell contact was necessary for induction, cells were cocultured in a Boyden chamber. TKD2 cells were seeded in the lower chamber at 6 × 104 cells/well in a 24‐well plate, and A549 cells were seeded in the upper chamber at the same concentration. RNA levels were examined at 24 h, and the results shown in Fig. 2a clearly indicate that separated A549 cells did not affect MMP mRNA levels of TKD2 cells. Conditioned medium of A549 also did not influence MMP mRNA levels (data not shown).
Figure 2.

(a) Requirement of direct contact between TKD2 and A549 cells. 1, TKD2 cells alone; 2, TKD2 cells were cultured on the bottom of Boyden chambers, and A549 cells were cultured on the chamber membrane for 24 h; 3, TKD2 cells were cocultured with A549 cells. Matrix metalloprotease (MMP) mRNA levels of TKD2 cells were determined by reverse transcription–polymerase chain reaction (RT‐PCR). (b) Effect of neutralizing antibody against human integrins. Total RNA was extracted from TKD2 cells alone (lane 1), or cocultured with A549 cells (lanes 2–5); 2, without antibody; 3, mouse control IgG (10 µg/mL); 4, antihuman integrin α2 (10 µg/mL); 5, antihuman integrin α4 (10 µg/mL). Antibody was added together with A549 seeded onto the monolayer of TKD2, and incubated for 24 h. MMP mRNA levels of TKD2 cells were determined by real time RT‐PCR. (c) Expression levels of integrin α4 mRNA. Total RNA was prepared from: 1, TIG3; 2, HT1080; 3, A549; 4, HeLaS3; and 5, TMK‐1 cells. Integrin α4 mRNA levels were determined by real time RT‐PCR. (d) Phase‐contrast microscopic observation of A549 cells cocultured on TKD2 cells (1) in the presence of antihuman integrin α4 antibody (1 µg/mL) for 24 h (2).
Many types of tumor cells express VLA‐4 (integrin α4β1), which binds to VCAM‐1 associated with the endothelial surface.( 3 ) To furhter examine the role of cell–cell contact, the effect of neutralizing antibody against human integrins was tested. A549 cells were seeded on a monolayer of TKD2 cells, and anti‐integrin α2 antibody or anti‐integrin α4 antibody (10 µg/mL each) was added at the same time. MMP mRNA levels were quantitated 24 h after starting coculture. The main ligand of integrin α2 is collagen, and that of integrin α4 is VCAM‐1. Anti‐integrin α4 inhibited the induction of MMP‐3, MMP‐9 and MMP‐10, whereas anti‐integrin α2 inhibited the induction of only MMP‐9 (Fig. 2b). Expression of integrin α4 mRNA was examined (Fig. 2c), but the expression levels of integrin α4 and induction of MMP (Fig. 1) did not correlate. Cell–cell interaction seemed to be altered in the presence of anti‐integrin α4, as observed under a phase‐contrast microscope (Fig. 2d). Coculture of human tumor cells with normal mouse embryonic fibroblasts also induced MMP‐9 (Fig. 3). Thus, the phenotypes of normal cells surrounding tumor cells are thought to be affected by the interaction of tumor cells.
Figure 3.
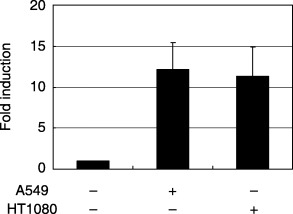
Induction of matrix metalloprotease (MMP)‐9 mRNA in normal mouse embryonic fibroblasts cocultured with human tumor cells. TKD2 cells were cocultured with A549 or HT1080 cells for 24 h, and MMP‐9 mRNA levels were determined as in Fig. 1.
Signals from adhesion to MMP induction. Integrins activate intracellular signals that regulate cellular phenotypes, including nuclear functions. The expression of MMP is reported to be dependent on PI3K or AKT, and p38.( 23 , 24 , 25 ) To estimate the signaling cascade from cell contact to MMP expression, we examined the effects of several signal inhibitors.
Inhibitors of PI3K (LY294002), Src (PP1), MEK (PD98059) or p38 (SB203580) were added when A549 cells were seeded onto the TKD2 monolayer. As summarized in Fig. 4, LY294002 significantly inhibited the induction of MMP‐3, MMP‐9 and MMP‐10. PD98059 inhibited the induction of MMP‐9 almost completely, MMP‐3 only partially, and had no effect on MMP‐10.
Figure 4.
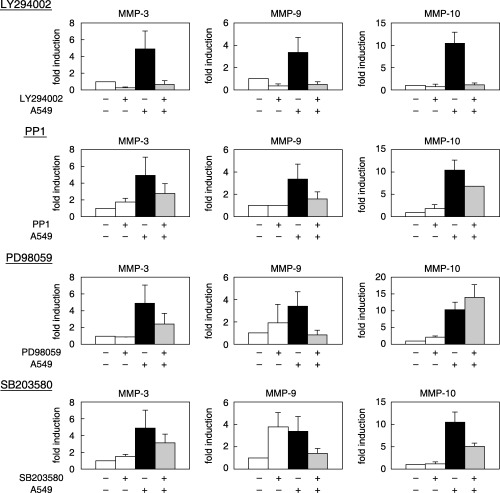
Effect of inhibitors of signals on matrix metalloprotease (MMP) mRNA induction. A549 cells were seeded on the monolayer of TKD2 cells with and without inhibitors. Total RNA was extracted, and MMP mRNA levels of TKD2 cells were determined by reverse transcription–polymerase chain reaction. LY294002 (phosphatidylinositol‐3‐phosphate kinase inhibitor: 20 µM), PP1 (Src family inhibitor: 5 µM), PD98059 (MEK inhibitor: 30 µM), SB203580 (p38 inhibitor: 10 µM).
As the PI3K pathway was suggested to be important for induction, the involvement of PI3K was further examined. p85α is a regulatory subunit of PI3K and ΔiSH2 is a dominant‐negative form.( 20 ) Stable cell lines were established by transfection with an expression vector of wild‐type p85α or dominant‐negative form of p85α (ΔiSH2) together with the hygromycin‐resistance gene. Three ΔiSH2‐expressing cells isolated independently did not respond to added A549 cells, whereas MMP mRNA were induced in cells expressing wild‐type p85α to a similar extent as non‐transfected control cells (Fig. 5b).
Figure 5.
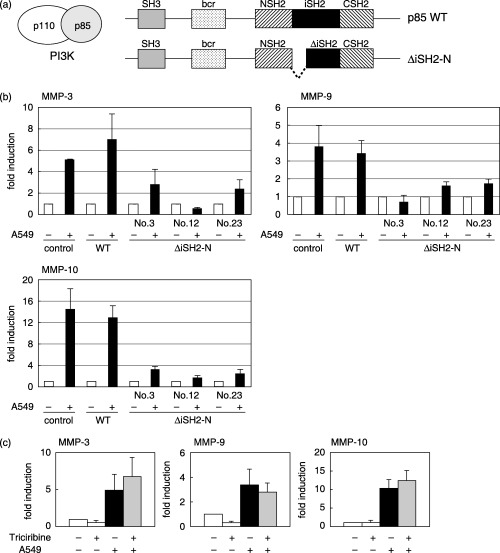
Involvement of phosphatidylinositol‐3‐phosphate kinase (PI3K) in matrix metalloprotease (MMP) mRNA induction. (a) Schematic representation of wild‐type (p85WT) and dominant‐negative (ΔiSH2‐N) regulatory subunit (p85α) of PI3K. (b) TKD2 cells were transfected with expression vectors for wild‐type p85α (WT) or dominant‐negative type (ΔiSH2‐N) together with the hygromycin‐resistance gene. Stable transformants expressing either WT or dominant‐negative type (nos 3, 12 and 23) were isolated. A549 cells were cocultured for 24 h, and MMP mRNA levels were determined as in Fig. 5. (c) Effect of Akt inhibitor (Triciribine) on MMP mRNA induction. TKD2 cells were cocultured with A549 cells in the absence or presence of triciribine (1 µM). Total RNA was extracted, and MMP mRNA levels of TKD2 cells were determined by reverse transcription–polymerase chain reaction.
Akt is a downstream effector of PI3K, and triciribine is reported to be a specific inhibitor of Akt.( 26 ) The results shown in Fig. 5c indicate that triciribine did not affect the induction of MMP.
Involvement of ROS in MMP induction. Recent reports suggest that ROS were produced when VCAM‐1 was activated by adhesion between endothelial cells and leukocytes, and this production was catalyzed by NADPH oxidase, a target of VCAM‐1 signaling.( 27 , 28 , 29 ) Adhesion of cancer cells to endothelial cells is also known to activate ROS production.( 30 , 31 ) These observations prompted us to examine the involvement of ROS in MMP induction following the adhesion of TKD2 cells to A549 cells.
A fluorescent redox sensor, Red CC‐1, was incorporated into TKD2 cells, and A549 cells were seeded. Figure 6 shows that the fluorescence intensity of TKD2 cells increased 30 min after the addition of A549 cells, indicating that the intracellular ROS level was elevated after adhesion of TKD2 cells to A549 cells.
Figure 6.

Increase in reactive oxygen species (ROS) levels in TKD2 cells cocultured with A549 cells. Redox sensor (Red CC‐1) was incorporated for 15 min into TKD2 cells. Cells were either (a,d) untreated, (b,e) treated with hydrogen peroxide (0.5 mM) or (c,f) cocultured with A549 cells. (a–c) Phase‐contrast microscopy. (d–f) Fluorescence microscopy. Photographs were taken 30 min later.
The importance of ROS in MMP induction was estimated by examining the effects of ROS scavengers and inhibitors of ROS production. An antioxidant that scavenges hydroxyradicals (DMTU) inhibited the induction of MMP‐3, MMP‐9 and MMP‐10. A non‐specific inhibitor of flavin‐containing enzymes (DPI) and an inhibitor of mitochondrial electron transporter complex I (rotenone) strongly inhibited MMP induction (Fig. 7).
Figure 7.
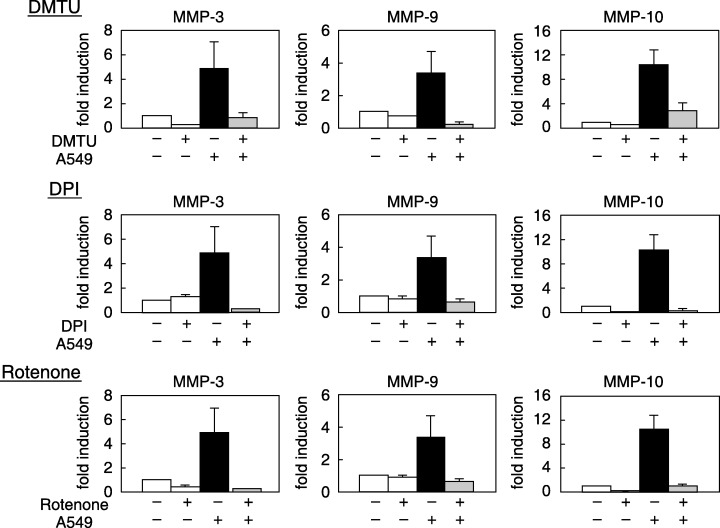
Effect of antioxidants and inhibitors for reactive oxygen species (ROS) production on matrix metalloprotease (MMP) mRNA induction. TKD2 cells were either cultured alone or cocultured with A549 cells in the absence or presence of each inhibitor for 24 h. Tiron (5 mM), dimethyl thiourea (DMTU; 20 mM), diphenyleneiodonium (DPI; 0.5 µM), rotenone (5 µM). Total RNA was extracted, and MMP mRNA levels of TKD2 cells were determined by reverse transcription–polymerase chain reaction.
Importance of mitochondria in MMP induction. To address the question of whether mitochondria were involved in MMP induction, we first examined the activity of mitochondria after adhesion. The fluorescence intensity of JC‐1 is known to reflect the membrane potential of mitochondria. TKD2 cells that incorporated JC‐1 were cocultured with A549, and red fluorescence was measured. Figure 8a shows that membrane potential was increased following adhesion to A549 cells. This increase was abrogated by the addition of neutralizing antibody against integrins. The increase in mitochondrial membrane potential was also inhibited by LY294002 (Fig. 8b). The involvement of PI3K in the activation of mitochondria was further confirmed from the effect of the dominant‐negative form of PI3K (Fig. 8c). The importance of mitochondria in MMP induction was examined using mitochondria‐depleted cells (pseudo ρ0 cells). Pseudo ρ0 cells were prepared by the cultivation of TKD2 cells in the presence of ethidium bromide or chloramphenicol. The extent of mitochondria depletion was confirmed by the decrease in complex I and cytochrome b mRNA (Fig. 9a). When cocultured with A549 cells, MMP‐9 induction was significantly decreased in pseudo ρ0 cells, whereas that of MMP‐3 and MMP‐10 was observed as in control cells (Fig. 9b).
Figure 8.
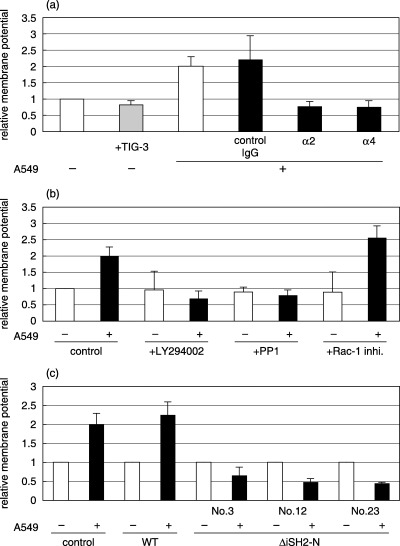
Change in mitochondrial membrane potential in TKD2 cells. Fluorescent dye, JC‐1 (10 µg/mL), was incorporated into TKD2 cells for 15 min. (a) TKD2 cells were either cultured alone, cocultured with TIG‐3 cells or with A549 cells in the absence or presence of control IgG, antihuman integrin α2, or antihuman integrin α4. Fluorescence intensity was measured 5 min after cell addition. (b) TKD2 cells were treated for 3 h with inhibitors (LY294002, 20 µM; PP1, 5 µM; Rac‐1 inhibitor, 100 nM), and JC‐1 was incorporated for 15 min. A549 cells were seeded, and fluorescence intensity was measured 5 min later. (C) JC‐1 was incorporated into control TKD2 cells, or stable transformants of wild‐type p85α (WT) or dominant‐negative p85α (nos 3, 12 and 23) used in Fig. 6. Fluorescence intensity was measured 5 min after coculture with A549 cells.
Figure 9.
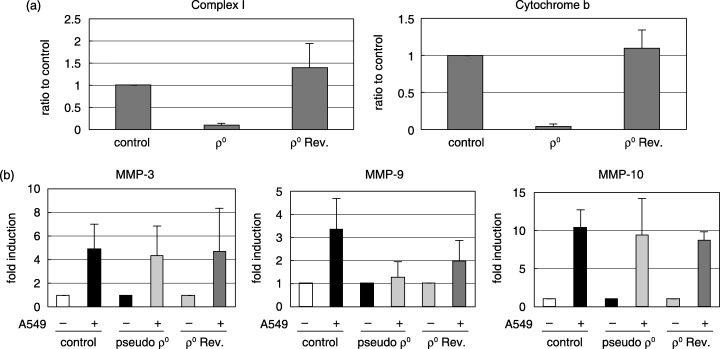
(a) Construction of mitochondria‐depleted cells (pseudo ρ0). (a) TKD2 cells were cultured in the presence of ethidium bromide (250 ng/mL) for 1 week (pseudo ρ0). Pseudo ρ0 cells were cultured in the absence of ethidium bromide for 1 week to make revertants (ρ0 rev). Levels of mRNA for mitochondrial markers (complex I, and cytochrome b) were measured by real‐time reverse transcription–polymerase chain reaction (RT‐PCR). (b) Matrix metalloprotease (MMP) mRNA induction in mitochondria‐depleted cells. Control TKD2, pseudo ρ0 or ρ0Rev cells were cocultured with A549 cells, and total RNA was extracted after 24 h. Levels of MMP mRNA were determined by real‐time RT‐PCR.
Discussion
Many types of tumor cells express high levels of MMP, which have critical functions in tumor metastasis by facilitating penetration into blood vessels.( 32 ) Circulating tumor cells adhere to endothelial cells and invade the surrounding tissues to grow. In the present study, we presented evidence that adhesion to tumor cells activates the induction of MMP in endothelial cells in a cocultivation system. Cocultivation with normal fibroblasts did not affect MMP levels (Fig. 1). Experiments with Boyden chambers indicated that this induction depended on direct contact between tumor cells and endothelial cells, and induction was abrogated when cells were treated with anti‐integrin α4 (Fig. 2). VLA‐4 integrin α4β1 binds to ECM molecules, such as VCAM‐1, and transmits signals into the cytoplasm.( 33 ) These results indicate that direct cell–cell interaction through VCAM‐1 and VLA‐4 is necessary for the induction of MMP‐3, MMP‐9 and MMP‐10 mRNA in TKD2 cells, but integrin α2 was not critical for the induction of MMP‐3 and MMP‐10 (Fig. 2b). Tumor cells are reported to express both integrin α4 and α2,( 34 , 35 ) and the induction of MMP in endothelial cells through interactions with tumor cells can be important for invasion and tumor progression. The effect of A549 on MMP induction was the most prominent, but the pattern of induction differed among tumor cell lines (Fig. 1). Expression levels of integrin α4 in various tumor cells (Fig. 2c) and induction of MMP (Fig. 1) did not correlate. Even integrin α4 was important for the induction (Fig. 1b), but other factors, such as intracellular association with other factors,( 36 ) may affect MMP induction.
Next, we examined signaling pathways elicited by adhesion between TKD2 and A549 cells using specific inhibitors. PI3K inhibitor (LY294002) decreased the induction of MMP‐3, MMP‐9 and MMP‐10, whereas inhibitors of Src (PP1), MEK (PD98059) or p38 (SB203580) decreased induction only partially (Fig. 4). It is known that the ERK1/2 pathway is necessary for MMP‐9 induction by homocysteine,( 37 )α3β1 integrin( 38 ) and ras oncogene,( 39 ) and that MMP‐9 induction by platelet‐activating factor iss dependent on PI3K.( 40 ) Integrin signals are thought to stimulate focal adhesion kinase, a tyrosine kinase that leads to activation of PI3K,( 41 , 42 ) and the PI3K/Akt pathway modulates mitochondrial functions.( 43 ) The results shown in Fig. 4 indicate that signal pathways after PI3K activation are different, at least in part, between the induction of MMP‐9, and MMP‐3 and MMP‐10 after the adhesion of endothelial cells to cancer cells. Experiments using inhibitors did not reveal which cells were affected but as seen in Fig. 5, the dominant‐negative form expressed in TKD2 cells abolished the induction of MMP. PI3K is composed of an 85‐kDa regulatory subunit and a 110‐kDa catalytic subunit, and the inter‐SH2 domain of p85 associates with p110 to be activated.( 19 ) The involvement of PI3K was confirmed using dominant‐negative form p85 expressed in TKD2 cells. The induction of MMP‐3, MMP‐9 and MMP‐10 by A549 was suppressed almost completely in TKD2 cells expressing the dominant‐negative form of p85, indicating that PI3K activity of TKD2 cells was necessary for induction. Akt/PKB is known to be a downstream effector of PI3K,( 44 ) but triciribine, an inhibitor of Akt inhibitor, did not affect induction (Fig. 5c). The signaling pathways downstream of PI3K should be investigated in further studies.
Reactive oxygen species are known to work in the signal transduction of growth factors and cell surface receptors,( 45 , 46 ) and their production is regulated by Rac‐1.( 45 ) Adhesion of A549 cells to TKD2 cells also increased intracellular ROS levels (Fig. 6). ROS scavengers (DMTU) and inhibitors of ROS production, such as DPI and rotenone, suppressed the induction of MMP (Fig. 7). VCAM‐1 stimulation was reported to activate NADPH oxidase.( 27 , 28 , 29 ) It is therefore suggested that ROS generated by NADPH oxidase is critical for induction. As for the source of ROS, mitochondria seemed to be important as induction was significantly decreased in mitochondria‐depleted TKD2 cells (Fig. 9). It was reported that ROS produced from mitochondria participate in the induction of MMP,( 47 , 48 ) and in the present study, direct interaction with tumor cells was shown to be necessary for the activation of ROS production from mitochondria (Fig. 8a). Mitochondrial activity seemed to be stimulated in TKD2 cells cocultured with A549 cells, and this stimulation was inhibited by LY294002 (Fig. 8b) or the dominant‐negative form of PI3K (Fig. 8c), indicating that PI3K activation following cell–cell interaction is required for the activation of mitochondria. MMP‐9 induction was significantly decreased in pseudo ρ0 cells, and recovered at least in part in revertants (Fig. 9b). The findings in the present study indicate that mitochondria are an important target for the development of antimetastatic drugs, and further studies are necessary to clearly understand the precise mechanisms.
Acknowledgments
This work was supported in part by Grants‐in‐Aid for Scientific Research, a Showa University Special Grant‐in‐Aid for Innovative Collaborative Research Projects, a Special Research Grant‐in‐Aid for development of Characteristic education, and the High‐Technology Research Center Project from the Japanese Ministry of Education, Culture, Sports, Science and Technology from the ministry for Education, Science, Sports, and Culture of Japan.
References
- 1. Bogenrieder T, Herlyn M. Axis of evil: molecular mechanisms of cancer metastasis. Oncogene 2003; 22: 6524–36. [DOI] [PubMed] [Google Scholar]
- 2. Orr FW, Wang HH, Lafrenie RM, Scherbarth S, Nance DM. Interactions between cancer cells and the endothelium in metastasis. J Pathol 2000; 190: 310–29. [DOI] [PubMed] [Google Scholar]
- 3. Elices MJ, Osborn L, Takada Y et al. VCAM‐1 on activated endothelium interacts with the leukocyte integrin VLA‐4 at a site distinct from the VLA‐4/fibronectin binding site. Cell 1990; 60: 577–84. [DOI] [PubMed] [Google Scholar]
- 4. Haraldsen G, Kvale D, Lien B et al. Cytokine‐regulated expression of E‐selectin, intercellular adhesion molecule‐1 (ICAM‐1), and vascular cell adhesion molecule‐1 (VCAM‐1) in human microvascular endothelial cells. J Immunol 1996; 156: 2558–65. [PubMed] [Google Scholar]
- 5. Garofalo A, Chirivi RG, Foglieni C et al. Involvement of the very late antigen 4 integrin on melanoma in interleukin 1‐augmented experimental metastases. Cancer Res 1995; 55: 414–19. [PubMed] [Google Scholar]
- 6. Oshima T, Blaschuk O, Gour B et al. Tight junction peptide antagonists enhance neutrophil trans‐endothelial chemotaxis. Life Sci 2003; 73: 1729–40. [DOI] [PubMed] [Google Scholar]
- 7. Muller WA, Randolph GJ. Migration of leukocytes across endothelium and beyond: molecules involved in the transmigration and fate of monocytes. J Leukoc Biol 1999; 66: 698–704. [DOI] [PubMed] [Google Scholar]
- 8. Westerlund A, Hujanen E, Puistola U, Turpeenniemi‐Hujanen T. Fibroblasts stimulate human ovarian cancer cell invasion and expression of 72‐kDa gelatinase A (MMP‐2). Gynecol Oncol 1997; 67: 76–82. [DOI] [PubMed] [Google Scholar]
- 9. Himelstein BP, Lee EJ, Sato H, Seiki M, Muschel RJ. Tumor cell contact mediated transcriptional activation of the fibroblast matrix metalloproteinase‐9 gene: involvement of multiple transcription factors including Ets and an alternating purine–pyrimidine repeat. Clin Exp Metastas 1998; 16: 169–77. [DOI] [PubMed] [Google Scholar]
- 10. Bair EL, Massey CP, Tran NL et al. Integrin‐ and cadherin‐mediated induction of the matrix metalloprotease matrilysin in cocultures of malignant oral squamous cell carcinoma cells and dermal fibroblasts. Exp Cell Res 2001; 270: 259–67. [DOI] [PubMed] [Google Scholar]
- 11. Jodele S, Chantrain CF, Blavier L et al. The contribution of bone marrow‐derived cells to the tumor vasculature in neuroblastoma is matrix metalloproteinase‐9 dependent. Cancer Res 2005; 65: 3200–8. [DOI] [PubMed] [Google Scholar]
- 12. Lynch CC, Matrisian LM. Matrix metalloproteinases in tumor–host cell communication. Differentiation 2002; 70: 561–73. [DOI] [PubMed] [Google Scholar]
- 13. Egeblad M, Werb A. New functions for the matrix metalloproteinases in cancer progression. Nature Rev Cancer 2002; 2: 161–74. [DOI] [PubMed] [Google Scholar]
- 14. Seiki M. The cell surface: the stage for matrix metalloproteinase regulation of migration. Curr Opin Cell Biol 2002; 14: 624–32. [DOI] [PubMed] [Google Scholar]
- 15. Bjorklund M, Koivunen E. Gelatinase‐mediated migration and invasion of cancer cells. Biochim Biophys Acta 2005; 1755: 37–69. [DOI] [PubMed] [Google Scholar]
- 16. Liabakk NB, Talbot I, Smith RA et al. Matrix metalloprotease 2 (MMP‐2) and matrix metalloprotease 9 (MMP‐9) type IV collagenases in colorectal cancer. Cancer Res 1996; 56: 190–6. [PubMed] [Google Scholar]
- 17. Taraboletti G, D’Ascenzo S, Borsotti P et al. Shedding of the matrix metalloproteinases MMP‐2, MMP‐9, and MT1‐MMP as membrane vesicle‐associated components by endothelial cells. Am J Pathol 2002; 160: 673–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yanai N, Satoh T, Kyo S, Abe K, Suzuki M, Obinata M. A tubule cell line established from transgenic mice harboring temperature‐sensitive simian virus 40 large T‐antigen gene. Jpn J Cancer Res 1991; 82: 1344–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ishino K, Kaneyama J‐K, Shibanuma M, Nose K. Specific decrease in the level of Hic‐5, a focal adhesion protein, during immortalization of mouse embryonic fibroblasts, and its association with focal adhesion kinase. J Cell Biochem 2000; 76: 411–19. [DOI] [PubMed] [Google Scholar]
- 20. Jimenz C, Jones DR, Rodriguerz‐Viciana P et al. Identification and characterization of a new oncogene derived from the regulatory subunit of phosphoinositide 3‐kinase. EMBO J 1998; 17: 743–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kennedy ED, Maechler P, Wollheim CB. Effects of depletion of mitochondrial DNA in metabolism secretion coupling in INS‐1 cells. Diabetes 1998; 47: 374–80. [DOI] [PubMed] [Google Scholar]
- 22. Tanaka H, Matsumura I, Ezoe S et al. E2F1 and c‐Myc potentiate apoptosis through inhibition of NF‐B activity that facilitates MnSOD‐mediated ROS elimination. Mol Cell 2002; 9: 1017–29. [DOI] [PubMed] [Google Scholar]
- 23. Ruhul Amin AR, Senga T, Oo ML et al. Secretion of matrix metalloproteinase‐9 by the proinflammatory cytokine, IL‐1β: a role for the dual signalling pathways, Akt and Erk. Genes Cells 2003; 8: 515–23. [DOI] [PubMed] [Google Scholar]
- 24. Johansson N, Ala‐aho R, Uitto V et al. Expression of collagenase‐3 (MMP‐13) and collagenase‐1 (MMP‐1) by transformed keratinocytes is dependent on the activity of p38 mitogen‐activated protein kinase. J Cell Sci 2000; 113 (Part 2): 227–35. [DOI] [PubMed] [Google Scholar]
- 25. Yao J, Xiong S, Klos K et al. Multiple signaling pathways involved in activation of matrix metalloproteinase‐9 (MMP‐9) by heregulin‐β1 in human breast cancer cells. Oncogene 2001; 20: 8066–74. [DOI] [PubMed] [Google Scholar]
- 26. Yang L, Dan HC, Sun M et al. Akt/protein kinase B signaling inhibitor‐2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res 2004; 64: 4394–9. [DOI] [PubMed] [Google Scholar]
- 27. Matheny HE, Deem T, Cook‐Mills JM. Lymphocyte migration through monolayers of endothelial cell lines involves VCAM‐1 signaling via endothelial cell NADPH oxidase. J Immunol 2000; 164: 6550–9. [DOI] [PubMed] [Google Scholar]
- 28. Tudor KS, Hess KL, Cook‐Mills JM. Cytokines modulate endothelial cell intracellular signal transduction required for VCAM‐1‐dependent lymphocyte transendothelial migration. Cytokine 2001; 15: 196–211. [DOI] [PubMed] [Google Scholar]
- 29. Van Wetering S, Van Buul JD, Quik S et al. Reactive oxygen species mediate Rac‐induced loss of cell–cell adhesion in primary human endothelial cells. J Cell Sci 2002; 115: 1837–46. [DOI] [PubMed] [Google Scholar]
- 30. Zhang G, Miura Y, Yagasaki K. Suppression of adhesion and invasion of hepatoma cells in culture by tea compounds through antioxidative activity. Cancer Lett 2000; 159: 169–73. [DOI] [PubMed] [Google Scholar]
- 31. Lim HW, Hong S, Jin W et al. Up‐regulation of defense enzymes is responsible for low reactive oxygen species in malignant prostate cancer cells. Exp Mol Med, 2005; 37: 497–506. [DOI] [PubMed] [Google Scholar]
- 32. Overall CM, Lüpez‐Otin C. Strategies for MMP inhibition in cancer: innovations for the post‐trial era. Nat Rev Cancer 2002; 2: 657–72. [DOI] [PubMed] [Google Scholar]
- 33. Danen EHJ, Yamada K. Fibronectin, integrins, and growth control. J Cell Physiol 2001; 189: 1–13. [DOI] [PubMed] [Google Scholar]
- 34. Holzmann B, Gosslar U, Bittner M. Alpha 4 integrins and tumor metastasis. Curr Top Microbiol Immuno 1998; 231: 125–41. [DOI] [PubMed] [Google Scholar]
- 35. Vihinen P, Riikonen T, Laine A, Heino J. Integrin α2β1 in tumorigenic human osteosarcoma cell lines regulates cell adhesion, migration, and invasion by interaction with type I collagen. Cell Growth Differ 1996; 7: 439–47. [PubMed] [Google Scholar]
- 36. Alon R, Feigelson SW, Manevich E et al. Alpha4beta1‐dependent adhesion strengthening under mechanical strain is regulated by paxillin association with the α4‐cytoplasmic domain. J Cell Biol 2005; 171: 1073–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moshal KS, Sen U, Tyagi N et al. Regulation of homocysteine‐induced MMP‐9 by ERK1/2 pathway. Am J Physiol Cell Physiol 2006; 290: C883–91. [DOI] [PubMed] [Google Scholar]
- 38. Iyer V, Pumiglia K, DiPersio CM. Alpha3beta1 integrin regulates MMP‐9 mRNA stability in immortalized keratinocytes: a novel mechanism of integrin‐mediated MMP gene expression. J Cell Sci 2005; 118: 1185–95. [DOI] [PubMed] [Google Scholar]
- 39. Shin I, Kim S, Song H, Kim HR, Moon A. H‐Ras‐specific activation of Rac–MKK3/6–p38 pathway: its critical role in invasion and migration of breast epithelial cells. J Biol Chem 2005; 280: 14 675–83. [DOI] [PubMed] [Google Scholar]
- 40. Ko HM, Kang JH, Choi JH et al. Platelet‐activating factor induces matrix metalloproteinase‐9 expression through Ca2+‐ or PI3K‐dependent signaling pathway in a human vascular endothelial cell line. FEBS Lett 2005; 579: 6451–8. [DOI] [PubMed] [Google Scholar]
- 41. Zeng ZZ, Jia Y, Hahn NJ, Markwart SM, Rockwood KF, Livant DL. Role of focal adhesion kinase and phosphatidylinositol 3′‐kinase in integrin fibronectin receptor‐mediated, matrix metalloproteinase‐1‐dependent invasion by metastatic prostate cancer cells. Cancer Res 2006; 66: 8091–9. [DOI] [PubMed] [Google Scholar]
- 42. Heit B, Colarusso P, Kubes P. Fundamentally different roles for LFA‐1, Mac‐1 and α4‐integrin in neutrophil chemotaxis. J Cell Sci 2005; 118: 5205–20. [DOI] [PubMed] [Google Scholar]
- 43. Wallace DC. Mitochondria and cancer: Warburg addressed. Cold Spring Harb Symp Quant Biol 2005; 70: 363–74. [DOI] [PubMed] [Google Scholar]
- 44. Vivanco I, Sawyers CL. The phosphatidylinositol 3‐kinase–Akt pathway in human cancer. Nature Rev Cancer 2002; 2: 489–501. [DOI] [PubMed] [Google Scholar]
- 45. Finkel T. Oxygen radicals and signaling. Curr Opin Cell Biol 1998; 10: 248–53. [DOI] [PubMed] [Google Scholar]
- 46. Nose K. Redox control of protein trafficking. Antioxidants Redox Signaling 2005; 7: 303–7. [DOI] [PubMed] [Google Scholar]
- 47. Nelson KK, Melendez JA. Mitochondrial redox control of matrix metalloproteinases. Free Radic Biol Med 2004; 37: 768–84. [DOI] [PubMed] [Google Scholar]
- 48. Woo CH, Lim JH, Kim JH. Lipopolysaccharide induces matrix metalloproteinase‐9 expression via a mitochondrial reactive oxygen species‐p38 kinase‐activator protein‐1 pathway in Raw 264.7 cells. J Immunol 2004; 173: 6973–80. [DOI] [PubMed] [Google Scholar]