

Abstract
Transforming acidic coiled‐coil‐containing (TACC) family members regulate mitotic spindles and have essential roles in embryogenesis. However, the functions of TACC3 in mitosis during mammalian development are not known. We have generated and characterized three mutant alleles of mouse Tacc3 including a conditional allele. Homozygous mutants of a hypomorphic allele exhibited malformations of the axial skeleton. The primary cause of this defect was the failure of mitosis in mesenchymal sclerotome cells. In vitro, 36% of primary mouse embryo fibroblasts (MEF) obtained from mutants homozygous for the hypomorphic allele and 67% of MEF from Tacc3 null mutants failed mitosis. In cloned immortalized MEF, Tacc3 depletion destabilized spindles and prevented chromosomes from aligning properly. Furthermore, chromosome separation and cytokinesis were also severely impaired. Chromosomes were moved randomly and cytokinesis initiated but the cleavage furrow eventually regressed, resulting in binucleate cells that then yielded aneuploid cells in the next cell division. Thus, in addition to spindle assembly, Tacc3 has critical roles in chromosome separation and cytokinesis, and is essential for the mitosis of sclerotome mesenchymal cells during axial formation in mammals. (Cancer Sci 2007; 98: 555–562)
The transforming acidic coiled‐coil‐containing (TACC) family of proteins are characterized by an evolutionally conserved coiled‐coil domain. Members of this family are concentrated at the centrosome as cells enter mitosis.( 1 ) In Xenopus, although a TACC homolog, Maskin, was originally isolated as a binding partner of cytoplasmic polyadenylation element binding protein,( 2 ) recent studies indicate that Makin is also important for mitotic spindle assembly.( 3 , 4 ) D‐TACC, the only TACC family member identified in Drosophila, also influences the stability of centrosomal microtubules in a dose‐dependent manner. Reducing D‐TACC function either by mutation or antibody injection results in shorter and weaker microtubles. Conversely, the number and length of microtubles are increased in embryos that overexpress D‐TACC.( 5 , 6 ) Similarly, dose‐dependent effects in early embryogenesis have been reported for Caenorhabditis elegans TACC, TAC‐1.( 7 , 8 , 9 ) Therefore, across a broad spectrum of species TACC family members have conserved roles in mitosis, and the dose of TACC protein may critically influence mitotic spindle formation.
In mammals, three members of the TACC family have been identified and all human TACC family members are implicated in tumorigenesis. TACC1, a founding member of the TACC family, was originally isolated as a gene located at the 8p11 locus, a region frequently amplified in breast cancer. Overexpression of TACC1 induces cellular transformation.( 10 ) Two other family members, TACC2 and TACC3, are also located in genomic regions that are rearranged in certain cancer cells.( 11 ) In addition, alterations in the expression of these two genes has been reported for various cancers,( 11 , 12 , 13 , 14 ) raising the possibility that the expression levels of TACC family members are critical for tumorigenesis.( 15 ) More recently, TACC3 was reported to be a novel independent prognostic marker in non‐small cell lung cancer( 16 ) and a potential biomarker in ovarian cancer.( 17 ) Human TACC are concentrated at the centrosome at varying times throughout the cell cycle.( 1 ) Human TACC1 and TACC3 are enriched at centrosome during mitosis, whereas TACC2 is present at the centrosome throughout the cell cycle. Despite this subcelluar localization, the functional differences among these family members has not been elucidated. Suppressing TACC3 protein expression with small interfering RNA in HeLa cells leads to the partial destabilization of mitotic microtubules,( 18 ) consistent with the essential roles of this protein in mitotic spindle assembly. However, cells obtained from Tacc3 knockout mice do not exhibit any mitotic phenotype,( 19 ) although disruption of the Tacc3 gene results in embryonic lethality at midgestation. Thus, the role of TACC3 in mitosis during mammalian embryogenesis is not well understood.
In the present study, we generated conditional (Tacc3 S ), null (Tacc3 D ) and hypomorphic (Tacc3 CN ) alleles of mouse Tacc3. Using these mutants, we explored the function of Tacc3 in mouse embryogenesis and discovered that Tacc3 is required for the formation of the axial skeleton. Studies in vitro with primary mouse embryo fibroblasts (MEF) as well as an immortalized cell line revealed that Tacc3 is required for mitotic spindle assembly and chromosomal alignment. In addition, Tacc3 has critical roles in chromosome separation and cytokinesis. These results indicate Tacc3 is essential for the proper cell division of sclerotomal mesenchymal cells during formation of the axial skeleton.
Materials and methods
Construction of Tacc3 conditional targeting vector and generation of Tacc3 mutant mice. A genomic fragment encompassing Tacc3 was isolated from a 129 mouse embryonic stem (ES) cell lambda genomic library using Tacc3 cDNA as a probe. For the Tacc3 conditional targeting vector (Tacc3 CN ), a floxed neomycin cassette was introduced into the HindIII site of intron 5 and another loxP sequence was inserted into the PvuII site of intron 4. Of 243 neomycin‐resistant clones, eight clones had undergone homologous recombination, and two clones were injected into C57BL/6 blastocysts to generate chimeric mice. Germ‐line transmission of the Tacc3 CN allele was achieved by mating with C57BL/6 females.
To obtain the Tacc3 conditional silent allele (Tacc3 S ), a Cre‐recombinase expression plasmid was electroporated into ES cells that contained the Tacc3 CN allele, and cells were selected in medium containing puromycin for 48 h.( 20 ) Colonies were picked and clones harboring the Tacc3 S allele were identified by Southern blot analysis and polymerase chain reaction. Two recombinants were injected into blastocysts and F1 mice were obtained as described above. F1 mice with the Tacc3 D allele were generated by crossing Tacc3 CN chimeric mice with CAG‐Cre mice as described previously.( 21 )
Antibodies, immunoblotting and immunofluorescence analysis. Antibodies against Tacc3 were generated by immunizing rabbits against the N peptide (CSQKENVPPQSQAKATNVTF) or C peptide (CLVTPPIEPVLEPSHQGLEP). DM1A anti α‐tubulin was purchased from Sigma and antipericentrin was obtained from Convergence. Immunoblotting and immunofluorescence analyses were carried out as described previously.( 21 )
Phenotype analysis. Skeletal staining was carried out with two dyes, Alizarin red for bone and Alcian blue for cartilage, as described previously.( 22 ) In situ hybridization of whole‐mount embryos and sections was carried out with digoxigenin‐labeled RNA probes specific for Uncx4.1,( 23 ) Pax1, Pax9,( 24 ) col2a1 and col9a2 ( 25 ) according to the method of Wilkinson.( 26 ) Apoptosis was detected by terminal deoxynucleotidyltransferase‐mediated dUTP nick‐end labelling (TUNEL) staining (Apoptag Plus Detection Kit; Oncor) according to the manufacturer's instructions.
Time‐lapse microscopy. Primary MEF were isolated from E14.5 mouse embryo and cultured in Dulbecco's modified Eagle's medium (Sigma) containing 10% fetal bovine serum. All experiments were carried out using cells between passages 1 and 3. Cells were plated onto six‐well plates 12 h before infection and AxCAH‐GFP was infected at an multiplicity of infection (MOI) of 10. Similarly, AxCANCre was applied at the indicated MOI. Multiple fields were imaged with a DMIRE2 (Leica) microscope equipped with an XY stage and 20× N Plan objective for 20 h. Alternatively, prophase cells were identified and imaged for 3 h with a 63X Fluotar objective. To analyze 13SD‐2G3 cells, four Z sections spaced by 5 µm were collected and the images were processed using a maximal intensity projection. Fluorescence and phase‐contrast images were collected with a Coolsnap HQ (Roper Scientific) controlled by AS MDW software (Leica).
Results
Generation and characterization of Tacc3 mutant alleles. Mice lacking a functional gene for Tacc3 die at the mid‐to‐late gestation stage because of deficits in the development of several cell lineages.( 19 ) To evaluate more precisely the functions of Tacc3, we generated three alleles of Tacc3. The Tacc3 CN allele contained a neomycin cassette in addition to loxP sites flanking exon 5. The neomycin cassette was removed by the transient expression of Cre recombinase in ES cells to generate the Tacc3 S allele. The Tacc3 D allele was obtained by crossing Tacc3 CN heterozygous animals to a mouse strain that expresses Cre recombinase.( 27 ) Immunoblotting analysis revealed that no Tacc3 protein was expressed from the Tacc3 D allele and the Tacc3 S allele expressed Tacc3 protein at a level comparable to the wild‐type allele (Fig. 1a). Importantly, the expression level of Tacc3 protein from the Tacc3 CN allele was ∼5% of the wild‐type allele, indicating that Tacc3 CN is a hypomorphic allele.
Figure 1.
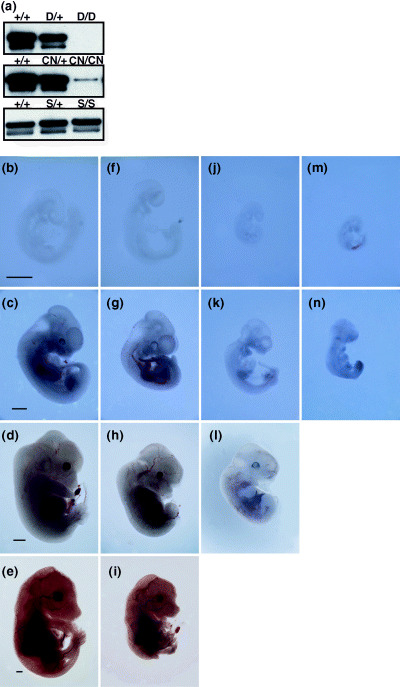
(a) Immunoblotting analysis of Tacc3 expression in an F2 litter carrying Tacc3 mutant alleles. Total cell lysates from E9.5 embryos were analyzed using an anti‐Tacc3 antibody. (b–n) Tacc3 dose‐dependent morphological defects in mouse embryos. Embryos were dissected at (b,f,j,m) E9.0, (c,g,k,n) E11.5, (d,h,l) E13.5 and (e,i) E15.5. (b–e) The left panel represents the morphology of wild‐type embryos. (f–i) Although Tacc3 CN/CN embryos were morphologically indistinguishable from Tacc3 W/W embryos until E9.0, growth retardation became apparent at E11.5. (j–l) Tacc3 CN/D embryos exhibited morphological defects as early as E9.0, and (m,n) Tacc3 D/D embryos were more severely affected. Scale bar = 1 mm.
To investigate the effects of different expression levels of Tacc3 in mouse embryogenesis, we generated various Tacc3 mutant mice. Tacc3 S/S and Tacc3 S/D mice were born at the expected Mendelian ratio, showed no overt abnormalities and were fertile as predicted for a conditional silent allele. Tacc3 CN/+ mice produced Tacc3 CN/CN offspring at a frequency lower than the expected Mendelian ratio (supplementary Table S1). All Tacc3 CN/CN animals died immediately after birth, most likely due to respiratory defects (data not shown). Analysis of Tacc3 CN/CN embryos revealed a normal gross morphology until E9.0, followed by gradual growth retardation (Fig. 1f–i). No Tacc3 D/D offspring were obtained from the Tacc3 D/+ matings as all embryos died by E12.5. Morphologically, severe growth retardation was observed in these Tacc D/D embryos as early as E9.0 (Fig. 1m). We also mated Tacc3 CN/+ mice to Tacc3 D/+ mice to produce Tacc3 CN/D compound heterozygous mice but they were not viable as neonates. The morphology of Tacc3 CN/D compound heterozygous embryos resembled Tacc3 D/D embryos, but the developmental abnormalities were milder (Fig. 1j–l). Tacc3 CN/D embryos died by E14.5 and growth retardation was less severe than that observed in Tacc3 D/D embryos at E9.5. These results indicate that Tacc3 is required for mouse embryogenesis in a dose‐dependent manner.
Skeletal defects in Tacc3CN/CN mice. Limited expression of Tacc3 can partially rescue the developmental defects caused by Tacc3 mutation. Because Tacc3 CN/CN neonates were the only viable mutants, we further examined the phenotype of Tacc3 CN/CN mice to investigate the role of Tacc3 in late gestation. Tacc3 CN/CN neonates were easily identified because they had a shortened body, a short tail and exhibited hindlimb paralysis (Fig. 2a,b). Skeletal staining revealed apparent malformation of the axial skeleton (Fig. 2c–f). These morphological abnormalities were more prominent in the caudal region, and the caudal vertebrae were lost and the sacral vertebrae were extensively malformed (Fig. 2g–j). The lumber vertebrae were fused and neural arches were disorganized. The bones derived from the rostral somites, including the exoccipital and basioccipital bones as well as the atlas and axis, were morphologically less affected (Fig. 2k–n). The ossification centers were lost in caudal vertebrae (Fig. 2j) and the ossification of the hindlimbs and ribs was also impaired (Fig. 2e,f). These results indicate that Tacc3 plays critical roles in the formation of the axial skeleton.
Figure 2.
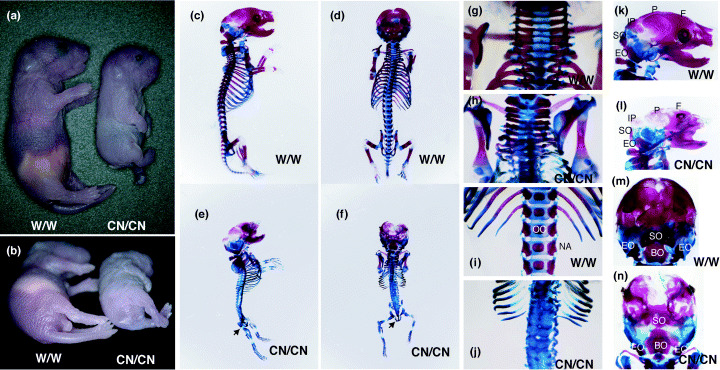
Skeletal malformation in newborn Tacc3 CN/CN mice. (a) Lateral and (b) caudal views of wild‐type (WT) and Tacc3 CN/CN (CN/CN) mice. Tacc3 CN/CN mice exhibited an overall shortening of the a–p axis. Note the abnormal outgrowth of the face in addition to the short tail. Tacc3 CN/CN mice also display paralyzed limbs. The skeletal morphology of (c,d,g,i,k,m) wild‐type and (e,f,h,j,l,n) Tacc3 CN/CN mice analyzed by staining bone with alizarin red and cartilage with alcian blue revealed a series of axial skeletal defects seen in Tacc3 CN/CN mice. Posterior regions of Tacc3 CN/CN mice were severely affected and caudal vertebra were lost (arrows in e,f). (i,j) Other vertebral defects, including loss of ossification centers (OC), malformation of neural arch (NA) and fusions of vertebra columns, were evident in the lumber regions of Tacc3 CN/CN mice. (g,h) The cervical vertebrae were also affected, but abnormalities were less severe. (k–n) Similarly, exoccipital bone (EO), basioccipital bone (BO) and superoccipital bone (SO) were less affected, but parietal (P) and interparietal (IP) bones failed to ossify.
Mitotic defects and apoptosis of sclerotomal mesenchymal cells. The axial skeleton is derived from the sclerotome. Signals from the notochord induce cells in the ventral half of the somites to detach from the somites. These cells undergo mitosis and become sclerotome mesenchymal cells. To identify and characterize the onset of phenotypic abnormalities in Tacc3 CN/CN embryos, we examined the formation of somites and sclerotomes. At E9.5–10.0, when growth retardation begins in the Tacc3 CN/CN embryos, control embryos had 25–30 pairs of somites, whereas Tacc3 CN/CN embryos contained only 20–25 pairs of somites. Despite this retardation, Uncx4.1, a marker for the posterior nascent somite and the posterior lateral sclerotome,( 23 ) was expressed in a repeated metameric pattern (Fig. 3a,b), suggesting that somite polarity was properly established. Similarly, Pax1, a marker that is expressed in the posterior ventoromedial compartment of sclerotome (the region of the future vertebral bodies), and Pax9, a marker that is expressed in the posterior ventrolateral compartment of sclerotome (the future neural arches and the proximal part of the ribs), were expressed with a clear periodicity in Tacc3 CN/CN embryos (Fig. 3c–f). Thus we conclude that somite formation and the subsequent generation of the mesenchymal sclerotome occurs normally in Tacc3 CN/CN embryos.
Figure 3.
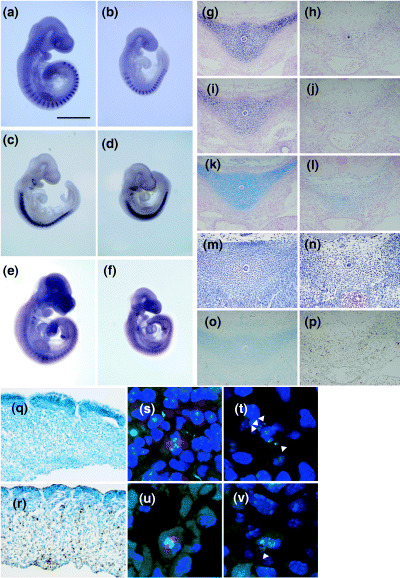
Impaired chondrocyte differentiation in Tacc3 CN/CN mice. Whole‐mount in situ hybridizations of E9.5 (a,c,e) wild‐type and (b,d,f) Tacc3 CN/CN embryos. Although Tacc3 CN/CN embryos exhibited growth retardation, the expression of (a,b) Uncx4.1, (c,d) Pax1 and (e,f) Pax9 were unaltered. (g–j) In situ hybridization and (k–p) histological analysis of E13.5 (g,i,k,m,o) wild‐type and (h,j,l,n,p) Tacc3 CN/CN embryos. The expression of (g,h) col2a1 and (i,j) col9a2 was severely impaired. (k,l) alcian blue staining was hardly detectable in Tacc3 CN/CN embryos. (m,n) hematoxylin‐eosin staining revealed that chondrocyte condensation was severely impaired. (o,p) Many apoptotic cells were observed in Tacc3 CN/CN embryos. Terminal deoxynucleotidyltransferase‐mediated dUTP nick‐end labelling analysis of E10.5 (q) wild‐type and (r) Tacc3 CN/CN embryos. Massive apoptosis was observed in sclerotome mesenchymal cells in Tacc3 CN/CN embryos. Immunofluorescence staining of (s,u) wild‐type and (t,v) Tacc3 CN/CN embryos. E10.5 sections were stained with anti‐Tacc3 (red) and anti‐AIM‐1 (green). Nuclei were stained with 4′,6‐diamidino‐2‐phenyindole (blue). (t,v) White arrowheads indicate the lagging chromosomes often observed in Tacc3 CN/CN embryos.
Mesenchymal cells from the sclerotome migrate toward the notochord, undergo cellular condensation and differentiate into chondrocytes. Therefore, we examined the expression of collagen in Tacc3 CN/CN embryos. In control embryos, col2a1 and clo9a2 were expressed in chondrocytes around the notochord at E13.5 (Fig. 3g,i). In Tacc3 CN/CN embryos, however, the expression was barely detectable (Fig. 3h,j). Similarly, the cartilage premordium of vertebral body in Tacc3 CN/CN embryos was only weakly stained by alcian blue (Fig. 3l). In addition, col10a1 was not expressed at E15.5 in Tacc3 CN/CN embryos (data not shown). Importantly, hematoxylin‐eosin (HE) staining revealed loosely scattered mesenchymal cells rather than the condensed chondrocytes typically present around the notochord (Fig. 3n). It should be noted that in spite of the reduced expression of collagen mRNA, cartilage primordia of the axial skeleton was formed in neonates (Fig. 2j). The partial formation of cartilage premordium may be a consequence of impaired cellular condensation in Tacc3 CN/CN embryos whereas the chondrocyte differentiation process itself may be normal.
Given that somite formation and the subsequent sclerotome formation occurred normally in Tacc3 CN/CN embryos, the absence of chondrocyte around notochord raised the possibility that sclerotome mesenchymal cells were lost after deepithelialization from the somites. We tested this possibility with a TUNEL assay and observed massive apoptosis (Fig. 3r). The apoptosis was most prominent around the notochord, but was also present among migrating mesenchymal cells. Because sclerotome mesenchymal cells undergo vigorous mitosis after migrating from the somites, the apoptosis observed in Tacc3 CN/CN embryos could be caused by defects in mitosis and this possibility was tested by immunofluorescence. In control embryos, mitotic cells exhibited clear metaphase plates and Tacc3 was detected at the centrosome and mitotic spindle (Fig. 3s,u, shown in red). In contrast, mitotic cells in Tacc3 CN/CN embryos, in which Tacc3 expression is severely attenuated, contained unaligned or lagging chromosomes (Fig. 3t,v, indicated by arrowhead). Although we cannot rule out the possibility that the apoptosis was induced by other processes such as migration toward the notochord, these results support the hypothesis that the vertebral defects in Tacc3 CN/CN neonates are caused by the abnormal mitosis of mesenchymal sclerotome cells. This defect in mitosis prevents these migrating cells from condensing around the notochord and then differentiating into chondrocytes.
Mitotic failure in primary cells from Tacc3 mutant mice. To characterize further the mitotic defects of mesenchymal sclerotome cells in Tacc3 CN/CN embryos, we studied MEF in vitro because MEF can complete a program of chondrocyte maturation in micromass culture that mimics the cellular condensation that occurs in vivo. ( 28 ) First we tested whether Tacc3 CN/CN MEF could differentiate into chondrocytes in this culture system. MEF from Tacc3 CN/CN mutants express approximately 5% of the Tacc3 protein expressed by wild‐type MEF (supplementary Fig. S2a) but displayed normal chondrocyte maturation. As our histological study of Tacc3 CN/CN mice suggested that aberrant mitosis by mesenchymal cells caused the vertebral defects, we next examined mitosis in these cells. To visualize chromosome behavior during mitosis, we transduced Tacc3 CN/CN MEF with an adenovirus expressing histone‐green fluorescent protein (GFP) (AxCAH‐GFP) and monitored cell division from 24 h after infection. Under these conditions, 93.1% of the wild‐type MEF divided normally (Fig. 4a,e). In these cells, chromosomes condensed, aligned to the metaphase plate, separated and migrated toward the opposite poles. After cytokinesis, chromosomes decondensed and the nuclear membrane formed. The process was completed within approximately 40–50 min. However, in Tacc3 CN/CN MEF, the percentage of cells that exhibited normal mitosis was reduced to 63.9% as mitosis was aberrant in 36.1% of these cells (Fig. 4e). In most of these abnormal mitotic events (30.6%), the metaphase plate formed but chromosomes did not align properly. In addition, after the onset of anaphase, separated chromosomes exhibited the unusual migration that is characteristic of Tacc3‐deficient cells (Fig. 4b). In these cells, chromosomes fail to migrate toward the poles but instead move back and forth rapidly. These movements appeared random and were not synchronized between chromosomes. After approximately 30–50 min of this activity, cytokinesis initiated but was unable to proceed to completion, yielding binucleate cells. In addition to these abnormal mitotic cells, 6.9% of Tacc3 CN/CN MEF displayed a distinct mitotic deficiency. These cells did not exhibit the characteristic random chromosome movement but multiple cytokinesis (Fig. 4d). Most of these cells were binuceate and divided into three or four cells. These cells were likey tetraploid cells and arose from previously aberrant rounds of mitosis that failed to complete cytokinesis.
Figure 4.
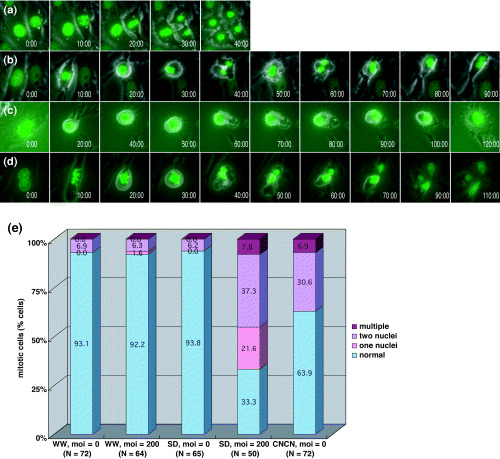
Abnormal mitosis in mouse embryo fibroblasts (MEF) from Tacc3 mutant mice. (a–d) Primary MEF from (a) wild‐type or (b–d) Tacc3 S/D mice were infected with AxCAH‐GFP and AxCANCre. Mitosis was followed by phase‐contrast and fluorescence microscopy. Selected frames from a time‐lapse recording are shown. The elapsed time in min is shown at the lower right. (e) Quantitation of the defects observed in cells from Tacc3 mutant mice. The percentage of cells exhibiting a particular phenotype is shown. Blue, pink, red and dark red represent normal mitosis (as shown in a), abnormal mitosis resulting in binucleate cells (as shown in b), mononucleate cells (as shown in c), and aberrant cytokinesis (as shown in d), respectively. At least 50 mitotic cells were monitored for each of the cells indicated.
We next examined the mitosis of Tacc3 null cells compared with Tacc3 CN/CN cells. As Tacc3 D/D embryos die by E12.5, we prepared MEF from Tacc3 S/D embryos and transduced these cells with an adenovirus expressing Cre recombinase (AxCANCre) to eliminate expression of the Tacc3 gene. Tacc3 protein levels decreased in a time‐ and dose‐dependent manner; at a MOI of 200, no Tacc3 protein was detectable 2 days after infection (supplementary Fig. S2b,c). Mitosis in MEF procured from wild‐type mice was unaffected by infection with AxCANCre. In contrast, mitosis in Tacc3 S/D MEF was severely perturbed by infection with AxCANCre. In cells that were not infected most of the Tacc3 S/D MEF (93.8%) carried out cell division normally whereas this fraction was reduced to 33% in infected cells. Among these infected cells, 37% exhibited the characteristic random chromosomal movement observed in Tacc3 CN/CN MEF, and produced binucleate cells. Furthermore, 7.8% of cells carried out cytokinesis that resulted in three or four cells. In addition to this aberrant cell division, 22% of cells failed to separate sister chromosomes (Fig. 4c). In these cells, chromosomes also exhibited the characteristic random movement, cytokinesis failed, and resulting cells were binucleate. Thus, a low level of Tacc3 expression may be sufficient to initiate separation of sister chromatids, as this cellular phenotype was not observed in MEF from Tacc3 CN/CN mice. These abnormalities were not seen in Tacc3 S/D MEF that were not infected with AxCANCre, confirming that this phenotype was specific to the disruption of the Tacc3 gene. We conclude from these results that the complete depletion of Tacc3 protein raises the frequency of the mitotic defects as well as perturbs chromosome behavior during mitosis. It should be noted that although Tacc3 S/D MEF infected with AxCANCre completely depleted Tacc3 protein, 37% of these cells completed mitosis. This may indicate that in some cell types, normal mitosis occurs even in the absence of Tacc3 protein.
Complete depletion of Tacc3 protein caused defects in spindle stability, alignment and separation of chromosomes, and cytokinesis. The primary MEF showed mitotic defects in the absence of Tacc3. However, these cells presented a spectrum of phenotypes that were a consequence of the heterogeneity of primary culture. This phenotypic variation complicates our quantitative analysis of the DNA content and mitosis by primary cells. To overcome this problem, we established a cloned immortalized cell line, 13SD‐2G3, from primary Tacc3 S/D MEF. Tacc3 was completely depleted in 13SD‐2G3 cells by AxCANCre infection at 2 days after infection (supplementary Fig. S2d). We first examined the performance of this cell line in micromass culture to test if Tacc3 is required for chondrocyte differentiation and found that bone morphogenetic protein (BMP)‐2, ‐4 or ‐6 induced alkaline phosphatase and these cells stained with alcian blue, indicating that the 13SD‐2G3 cells differentiate into chondrocytes in response to BMP (supplementary Fig. S3). Consistent with the results we obtained with primary MEF, chondrocyte differentiation was not impaired by the loss of Tacc3. This finding further supports the notion that once mesenchymal cells undergo cellular condensation, Tacc3 is not required for the subsequent chondrocyte differentiation process.
Next, we examined the role of Tacc3 in mitosis. After disrupting the Tacc3 gene with AxCANCre, the ploidy was determined with laser scanning microscopy (LSC) (Fig. 5a–d). Three days after infection, the fraction of cells with 2C DNA content decreased and the fraction of 4C and 8C cells increased. Furthermore, the number of cells with aberrant DNA content was dramatically increased, indicating that depletion of Tacc3 induced aneuploidy. Consistent with this, immunofluorescence staining revealed that most of them were multinucleate cells (mostly binucleate cells) and micronuclei were also observed (Fig. 5h). These defects were not seen in uninfected 13SD‐2G3 cells or in wild‐type cells infected with AxCANCre (Fig. 5e–g).
Figure 5.
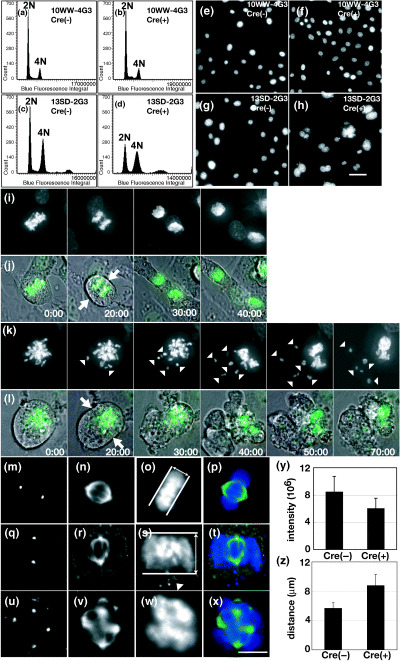
Mitotic abnormalities in cloned immortalized fibroblasts. (a–d) Two cell lines, 10WW‐4G3 and 13SD‐2G3, were established from littermate embryos with the Tacc3 W/W and Tacc3 S/D genotypes, respectively. Cells on chamber slides were left untreated (Cre[–]) or infected with AxCANCre at a multiplicity of infection (MOI) of 200. DNA contents were analyzed by laser scanning microscopy (LSC) 3 days after infection. (e–h) Cells were plated and infected as described above. Three days after infection, cells were fixed and stained with 4′,6‐diamidino‐2‐phenyindole (DAPI). (i–l) Time‐lapse images of 13SD‐3G3. 13SD‐2G3 cells infected with AxCAH‐GFP. Mitosis was followed by phase‐contrast and fluorescence microscopy. (k,l) AxCANCre was coinfected. Fluorescence images (i,k) are shown as merged images (j,l). Elapsed time in min is shown at the lower right. (m–x) Immunofluorescence of 13SD‐2G3 cells. Cells left untreated (m–p) or infected with AxCANCre at an MOI of 200 (q–x) were fixed and stained with anti‐pericentrin (m,q,u) and anti‐α‐tubulin (n,r,v). (o,s,w) DNA was stained with DAPI. (p,t,x) Merged images are shown. (y) Quantitation of integral fluorescence intensity of mitotic spindles. At least 50 mitotic cells were examined by LSC. (z) Quantitation of metaphase plates. At least 50 metaphase plates were imaged and the metaphase plates were measured.
We then examined chromosome behavior during mitosis in greater detail in 13SD‐2G3 cells expressing histone‐GFP (Fig. 5i–l). In contrast to uninfected cells, 13SD‐2G3 cells lacking Tacc3 exhibited aberrant mitosis. Chromosomes condensed and cytokinesis initiated with a time course similar to uninfected cells (Fig. 5j,l and 20:00, indicated by arrows). The clear metaphase plate, however, did not form, and chromosomes failed to align at the metaphase plate (Fig. 5k and 20:00, indicated by arrowheads). Chromosomes did not separate and cells eventually formed an irregularly shaped nucleus (Fig. 5k and 70:00). Unaligned chromosomes showed rapid and apparently random movement, and by the end of mitosis formed micronuclei (Fig. 5k and 30:00–70:00, indicated by arrowheads). Cells attempted cytokinesis, which was accompanied by intense blebbing of the plasma membrane. The cleavage furrow ultimately regressed to yield a single cell (Fig. 5l and 30:00–60:00). Thus, Tacc3 has essential roles in chromosome separation and cytokinesis.
Last, we examined the mitotic spindle by immunofluorescence and quantified integral fluorescence intensity with LSC. In Tacc3 mutant 13SD‐2G3 cells, the localization of pericentrin and γ‐tubulin was not disrupted (Fig. 5q), but the number and intensity of mitotic spindles was significantly reduced (Fig. 5r). The integral intensity of the bipolar mitotic spindle of Tacc3 null cells was significantly reduced (6.0 × 106 ± 1.5 × 106 compared with 8.4 × 106 ± 2.3 × 106 in controls; Fig. 5y). Furthermore, the metaphase plates of Tacc3 null cells were thicker than those of control cells (8.8 ± 1.5 µm compared with 5.7 ± 0.8 µm in controls; Fig. 5z) and lagging chromosomes were often observed (Fig. 5s, indicated by arrowhead). The number of mitotic cells containing multipolar spindles increased with time after infection (Fig. 5u–x). At 2 days after infection, 62.5% of mitotic cells infected with AxCAN‐Cre contained multipolar spindles. By comparison, over the same timecourse only 13.3% of control cells contained multiple spindles. Based on these observations, we conclude that the defects observed in the absence of Tacc3 result, at least in part, from the lack of proper tension of the mitotic spindle.
Discussion
In the present study, we demonstrate that depletion of Tacc3 in mammalian cells causes mitotic defects both in vivo and in vitro. Importantly, we determined for the first time that complete depletion of Tacc3 leads to the failure of chromosome separation at anaphase and cytokinesis. In MEF mutant for Tacc3, cytokinesis was initiated but intense blebbing of the plasma membrane occurred and the cleavage furrow eventually regressed. Depletion of TACC3 protein in HeLa cells by RNA interference results in spindle formation defects and the failure of chromosome alignment at the metaphase plate in TACC3‐depleted cells.( 18 ) This study did not report defects in chromosome separation and cytokinesis, and this phenotypic difference may be cell‐type specific. Alternatively, as in a preceding study, (18) the levels of TACC3 protein level were only reduced to ∼20% of that found in untreated cells, the differences in phenotype we observe may be a consequence of dosage. Specifically, chromosomal separation and cytokinesis may require less Tacc3 than spindle formation and chromosome alignment. Thus, in the presence of limited Tacc3, these processes maybe remain unaffected. As Tacc3 localizes to the cleavage furrow during cytokinesis,( 1 ) it may have a direct role in cytokinesis.
In a complementary study, Piekorz et al. (19) disrupted exon3 of Tacc3 and observed growth retardation and embryonic lethality in their homozygous mutants. Although these results are consistent with our findings, there are some phenotypic differences. For example, a significant fraction of their mutants survived until E18–19, whereas all of our Tacc3 D/D embryos died by E12.5. Importantly, they observed no phenotypic alterations in MEF, whereas Tacc3 D/D MEF exhibited clear mitotic defects. These differences are most likely due to allelic differences. If this is the case, it may suggest that Tacc3 has multiple functions. Consistent with this hypothesis, Tacc3 has been reported to contribute to transcriptional( 29 , 30 ) and translational( 2 ) regulation. Understanding the relationship between structure and function for this important protein, especially deducing the domains of Tacc3 required for these disparate activities, mitosis, transcription and translation, could yield important insights into the mechanisms governing cellular homeostasis and division.
Tacc3 is expressed in various tissues, including neuroepithelium and epithelial cells of intestines, kidney and lung in late gestation( 31 ) as well as erythroid and Sertoli cells.( 32 ) Although we did not do a detailed examination of these tissues, certain cell types do not require Tacc3 for normal development as Tacc3 CN/CN embryos survived until birth. These results may suggest that other family members could compensate for Tacc3. However, the expression patterns of Tacc1 and Tacc2 are distinct from that of Tacc3. Furthermore, Tacc2 is dispensable for normal development.( 33 ) Thus we believe that Tacc3 has a distinct role that is required in a cell‐type specific manner. Because screlotome mesenchymal cells undergo several biological processes during axial skeleton formation, such as epithelial–mesenchymal transition, migration and cellular condensation, a unique function of Tacc3 may be necessary to permit these functions to occur contemporaneously.
Three human TACC family members have been implicated in carcinogenesis, but their precise mechanisms have not been elucidated. In the present study, we have demonstrated that Tacc3 is required for proper mitosis. Cells lacking Tacc3 fail to complete cytokinesis and produce tetraploid cells, which become aneuploid after further division. This two‐step process of producing aneuploid cells is consistent with that reported by Shi and King who described that chromosome non‐disjunction yields tetraploid cells.( 34 ) As aneuploidy and chromosomal instability are characteristic of many cancer cells and are linked to the progressive development of high‐grade invasive tumors,( 35 ) Tacc3 may have a direct role in tumorigenesis and progression. The Tacc3 conditional mutant mice generated in this study will provide an excellent opportunity to test this hypothesis.
Acknowledgments
We thank H. Yamanaka and S. Kawashima for technical assistance with gene targeting. We gratefully acknowledge the gifts of clones from S. Takada (Okazaki Institute for Integrative Bioscience) and H. Akazawa (Chiba University Graduate School of Medicine). We also thank Y. Saga (National Institute of Health Science) for critical reading of the manuscript. This work was supported in part by grant‐in‐aid for Scientific Research on Priority Arias from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1. Gergely F, Karlsson C, Still I, Cowell J, Kilmartin J, Raff JW. The TACC domain identifies a family of centrosomal proteins that can interact with microtubules. Proc Natl Acad Sci USA 2000; 97: 14 352–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Groisman I, Huang YS, Mendez R, Cao Q, Theurkauf W, Richter JD. CPEB, maskin, and cyclin B1 mRNA at the mitotic apparatus: implications for local translational control of cell division. Cell 2000; 103: 435–47. [DOI] [PubMed] [Google Scholar]
- 3. O’Brien LL, Albee AJ, Liu L et al. The xenopus TACC homologue, maskin, functions in mitotic spindle assembly. Mol Biol Cell 2005; 16: 2836–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kinoshita K, Noetzel TL, Pelletier L et al. Aurora A phosphorylation of TACC3/maskin is required for centrosome‐dependent microtubule assembly in mitosis. J Cell Biol 2005; 170: 1047–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee MJ, Gergely F, Jeffers K, Peak‐Chew SY, Raff JW. Msps/XMAP215 interacts with the centrosomal protein D‐TACC to regulate microtubule behaviour. Nat Cell Biol 2001; 3: 643–9. [DOI] [PubMed] [Google Scholar]
- 6. Gergely F, Kidd D, Jeffers K, Wakefield JG, Raff JW. D‐TACC: a novel centrosomal protein required for normal spindle function in the early Drosophila embryo. EMBO J 2000; 19: 241–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Srayko M, Quintin S, Schwager A, Hyman AA. Caenorhabditis elegans TAC‐1 and ZYG‐9 form a complex that is essential for long astral and spindle microtubules. Curr Biol 2003; 13: 1506–11. [DOI] [PubMed] [Google Scholar]
- 8. Le Bot N, Tsai MC, Andrews RK, Ahringer J. TAC‐1, a regulator of microtubule length in the C. elegans embryo. Curr Biol 2003; 13: 1499–505. [DOI] [PubMed] [Google Scholar]
- 9. Bellanger JM, Gonczy P. TAC‐1 and ZYG‐9 form a complex that promotes microtubule assembly in C. elegans embryos. Curr Biol 2003; 13: 1488–98. [DOI] [PubMed] [Google Scholar]
- 10. Still IH, Hamilton M, Vince P, Wolfman A, Cowell JK. Cloning of TACC1, an embryonically expressed, potentially transforming coiled coil containing gene, from the 8p11 breast cancer amplicon. Oncogene 1999; 18: 4032–8. [DOI] [PubMed] [Google Scholar]
- 11. Still IH, Vince P, Cowell JK. The third member of the transforming acidic coiled coil‐containing gene family, TACC3, maps in 4p16, close to translocation breakpoints in multiple myeloma, and is upregulated in various cancer cell lines. Genomics 1999; 58: 165–70. [DOI] [PubMed] [Google Scholar]
- 12. Conte N, Charafe‐Jauffret E, Delaval B et al. Carcinogenesis and translational controls: TACC1 is down‐regulated in human cancers and associates with mRNA regulators. Oncogene 2002; 21: 5619–30. [DOI] [PubMed] [Google Scholar]
- 13. Conte N, Delaval B, Ginestier C et al. TACC1‐chTOG‐Aurora A protein complex in breast cancer. Oncogene 2003; 22: 8102–16. [DOI] [PubMed] [Google Scholar]
- 14. Chen HM, Schmeichel KL, Mian IS, Lelievre S, Petersen OW, Bissell MJ. AZU‐1: a candidate breast tumor suppressor and biomarker for tumor progression. Mol Biol Cell 2000; 11: 1357–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Raff JW. Centrosomes and cancer: lessons from a TACC. Trends Cell Biol 2002; 12: 222–5. [DOI] [PubMed] [Google Scholar]
- 16. Jung CK, Jung JH, Park GS, Lee A, Kang CS, Lee KY. Expression of transforming acidic coiled‐coil containing protein 3 is a novel independent prognostic marker in non‐small cell lung cancer. Pathol Int 2006; 56: 503–9. [DOI] [PubMed] [Google Scholar]
- 17. Peters DG, Kudla DM, Deloia JA et al. Comparative gene expression analysis of ovarian carcinoma and normal ovarian epithelium by serial analysis of gene expression. Cancer Epidemiol Biomarkers Prev 2005; 14: 1717–23. [DOI] [PubMed] [Google Scholar]
- 18. Gergely F, Draviam VM, Raff JW. The ch‐TOG/XMAP215 protein is essential for spindle pole organization in human somatic cells. Genes Dev 2003; 17: 336–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Piekorz RP, Hoffmeyer A, Duntsch CD et al. The centrosomal protein TACC3 is essential for hematopoietic stem cell function and genetically interfaces with p53‐regulated apoptosis. EMBO J 2002; 21: 653–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Taniguchi M, Sanbo M, Watanabe S, Naruse I, Mishina M, Yagi T. Efficient production of Cre‐mediated site‐directed recombinants through the utilization of the puromycin resistance gene, pac: a transient gene‐integration marker for ES cells. Nucl Acids Res 1998; 26: 679–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yao R, Ito C, Natsume Y et al. Lack of acrosome formation in mice lacking a Golgi protein, GOPC. Proc Natl Acad Sci USA 2002; 99: 11 211–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kaufman MH, ed. The Atlas of Mouse Development. London: Academic Press, 1992. [Google Scholar]
- 23. Mansouri A, Yokota Y, Wehr R, Copeland NG, Jenkins NA, Gruss P. Paired‐related murine homeobox gene expressed in the developing sclerotome, kidney, and nervous system. Dev Dyn 1997; 210: 53–65. [DOI] [PubMed] [Google Scholar]
- 24. Neubuser A, Koseki H, Balling R. Characterization and developmental expression of Pax9, a paired‐box‐containing gene related to Pax1. Dev Biol 1995; 170: 701–16. [DOI] [PubMed] [Google Scholar]
- 25. Metsaranta M, Toman D, De Crombrugghe B, Vuorio E. Specific hybridization probes for mouse type I, II, III and IX collagen mRNAs. Biochim Biophys Acta 1991; 1089: 241–3. [DOI] [PubMed] [Google Scholar]
- 26. Wilkinson DG. In Situ Hybridization, A Practical Approach. New York: Oxford University Press, 1998. [Google Scholar]
- 27. Sakai K, Miyazaki J. A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem Biophys Res Commun 1997; 237: 318–24. [DOI] [PubMed] [Google Scholar]
- 28. Lengner CJ, Lepper C, Van Wijnen AJ, Stein JL, Stein GS, Lian JB. Primary mouse embryonic fibroblasts: a model of mesenchymal cartilage formation. J Cell Physiol 2004; 200: 327–33. [DOI] [PubMed] [Google Scholar]
- 29. Garriga‐Canut M, Orkin SH. Transforming acidic coiled‐coil protein 3 (TACC3) controls friend of GATA‐1 (FOG‐1) subcellular localization and regulates the association between GATA‐1 and FOG‐1 during hematopoiesis. J Biol Chem 2004; 279: 23 597–605. [DOI] [PubMed] [Google Scholar]
- 30. Simpson RJ, Yi Lee SH, Bartle N et al. A classic zinc finger from friend of GATA mediates an interaction with the coiled‐coil of transforming acidic coiled‐coil 3. J Biol Chem 2004; 279: 39 789–97. [DOI] [PubMed] [Google Scholar]
- 31. Aitola M, Sadek CM, Gustafsson JA, Pelto‐Huikko M. Aint/Tacc3 is highly expressed in proliferating mouse tissues during development, spermatogenesis, and oogenesis. J Histochem Cytochem 2003; 51: 455–69. [DOI] [PubMed] [Google Scholar]
- 32. McKeveney PJ, Hodges VM, Mullan RN et al. Characterization and localization of expression of an erythropoietin‐induced gene, ERIC‐1/TACC3, identified in erythroid precursor cells. Br J Haematol 2001; 112: 1016–24. [DOI] [PubMed] [Google Scholar]
- 33. Schuendeln MM, Piekorz RP, Wichmann C et al. The centrosomal, putative tumor suppressor protein TACC2 is dispensable for normal development, and deficiency does not lead to cancer. Mol Cell Biol 2004; 24: 6403–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shi Q, King RW. Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature 2005; 437: 1038–42. [DOI] [PubMed] [Google Scholar]
- 35. Cahill DP, Kinzler KW, Vogelstein B, Lengauer C. Genetic instability and Darwinian selection in tumours. Trends Cell Biol 1999; 9: M57–60. [PubMed] [Google Scholar]