

Abstract
Aurora A kinase plays an essential role in the proper assembly and function of the mitotic spindle. We have shown previously that Aurora A expression is increased aberrantly in human T‐cell leukemia virus type 1 (HTLV‐1)‐infected T‐cell lines and primary adult T‐cell leukemia cells, and a pan‐Aurora kinase inhibitor, which inhibits both Aurora A and Aurora B kinases, reduces viability and induces apoptosis in these cells. However, the specific effects of Aurora A inhibition on HTLV‐1‐infected T‐cells are poorly understood. In this study, we addressed this question by comparing the effects of MLN8237, a selective inhibitor of Aurora A, on cell viability, cell cycle progression, and induction of apoptosis in HTLV‐1‐infected and ‐uninfected T‐cell lines. MLN8237 reduced the viability of HTLV‐1‐infected T‐cell lines within 24 h, but its effects on that of HTLV‐1‐uninfected T‐cell lines were moderate. MLN8237 induced early apoptosis of HTLV‐1‐infected T‐cell lines without induction of polyploidy. It induced p53 and p21 expression in HTLV‐1‐infected but not in ‐uninfected T‐cell lines, suggesting that MLN8237‐treated HTLV‐1‐infected T‐cell lines exit from mitosis and activate a p53‐dependent postmitotic G1 checkpoint, leading to G1 arrest followed by the induction of apoptosis. Our results suggest that specific inhibition of Aurora A kinase is a potentially useful therapeutic strategy in the treatment of adult T‐cell leukemia and that further in vivo exploration is warranted.
(Cancer Sci 2010; 101: 1204–1211)
Aurora A belongs to a highly conserved family of serine/threonine protein kinases that also includes Aurora B and Aurora C. Aurora A and Aurora B are structurally related. In humans, these kinases share 75% sequence homology in their kinase domains.( 1 ) Despite similarities in structure, Aurora A and Aurora B carry out distinct activities in mitosis. Aurora A localizes to centrosomes and the proximal mitotic spindle during mitosis and has a crucial role in bipolar spindle formation.( 2 ) Aurora B localizes to kinetochores in mitosis and to the midbody during cytokinesis, where it phosphorylates several proteins, including histone H3.( 2 ) It plays a role in chromosome alignment, kinetochore‐microtubule biorientation, activation of the spindle assembly checkpoint and cytokinesis. Aurora C is specifically expressed in the testis and plays a role in spermatogenesis.( 3 ) Its role in carcinogenesis remains unclear.
The Aurora A gene, which is located at chromosome 20q13.2, is frequently amplified and overexpressed in various human cancers.( 4 ) Increased Aurora A expression may lead to genomic instability, which is thought to contribute to tumor initiation and progression.( 5 ) Indeed increased Aurora A expression in experimental rodent models induces centrosome amplification, chromosome instability, and aneuploidy, resulting in malignant transformation.( 5 , 6 ) Moreover, targeted forced expression of Aurora A in the mouse mammary gland induces mitotic abnormalities that precede tumor formation.( 7 , 8 ) The oncogenic potential and essential roles in mitosis of Aurora A make it an intriguing target for anticancer therapeutic intervention.( 9 )
Small molecule inhibitors of Aurora kinases, including ZM447439,( 10 , 11 ) Hesperadin,( 12 ) VX‐680,( 13 ) PHA‐680632,( 14 ) PHA‐739358, and AZD1152,( 15 ) have been reported by several groups. Although several molecules are found to inhibit both Aurora A and Aurora B protein kinases, the major cellular phenotypic response they produced is consistent with inhibition of Aurora B,( 10 , 12 , 13 ) suggesting that these Aurora kinase inhibitors mediate antitumor activity primarily through inhibition of Aurora B activity.( 16 ) To gain a clearer understanding of the function of Aurora A in cell growth and survival, Aurora kinase inhibitor, which selectively inhibits Aurora A is necessary. MLN8054 is the first oral selective Aurora A inhibitor to enter the clinic. It is based on a benzazepine scaffold with a fused aminopyrimidine ring.( 17 ) MLN8237 is a second‐generation of Aurora A selective inhibitor. Like its predecessor MLN8054, it is orally bioavailable and rapidly absorbed. It is a highly selective small molecule inhibitor of the serine/threonine protein kinase Aurora A kinase with potential antitumor activity.( 16 ) MLN8237 binds to and inhibits Aurora A kinase, which may result in disruption of the assembly of the mitotic spindle apparatus, disruption of chromosome segregation, and inhibition of cell proliferation. The potential benefits for MLN8237 over parent compound MLN8054 are an increased potency of inhibition accompanied by decreased benzodiazepine‐like central nervous system effects. MLN8237 shows preliminary evidence of antitumor activity in patients with advanced solid tumors and lymphomas.( 18 , 19 ) Recently, it has entered Phase II clinical investigation.
Human T‐cell leukemia virus type 1 (HTLV‐1) is a retrovirus that transforms human T lymphocytes.( 20 ) After a long latency period of 50–60 years, about 2–4% of individuals infected with HTLV‐1 succumb to adult T‐cell leukemia (ATL).( 20 ) Although the mechanisms involved in HTLV‐1‐mediated cell transformation remain unclear, Tax has been recognized as the major viral protein with oncogenic potential.( 20 ) We showed previously that Aurora A protein is highly expressed in HTLV‐1‐infected T‐cell lines and primary ATL cells.( 21 ) Knockdown of Aurora A expression by siRNA more effectively suppressed the growth of HTLV‐1‐infected T‐cell lines than that of ‐uninfected T‐cell lines. A pan‐Aurora kinase inhibitor, which inhibits both Aurora A and Aurora B kinases, suppresses the growth of HTLV‐1‐infected T‐cell lines and primary ATL cells by inducing apoptosis through inhibition of nuclear factor‐kappa B (NF‐κB) activity.( 21 ) Because Aurora A selective inhibitor was not available at that time, we could not evaluate the specific effects of Aurora A inhibition on the proliferation and survival of HTLV‐1‐infected T‐cells. In this study, we evaluated the effects of MLN8237 in vitro in HTLV‐1‐infected T‐cell lines compared to HTLV‐1‐uninfected T‐cell lines.
Materials and Methods
Reagents.
MLN8237, the Aurora A selective inhibitor, was provided by Milleniam Pharmaceuticals (Cambridge, MA, USA). It was dissolved in 100% DMSO (Nakalai Tesque, Kyoto, Japan) to a stock concentration of 1 mm and stored at −80°C. Nocodazole was purchased from Sigma‐Aldrich (St. Louis, MO, USA).
Cell lines.
The HTLV‐1‐uninfected T‐cell leukemia lines Jurkat and CCRF‐CEM, and the HTLV‐1‐transformed T‐cell line MT‐4 and HUT‐102, that is established from a patient with ATL with an unclear clonal origin, were maintained in RPMI‐1640 medium supplemented with 10% heat‐inactivated fetal bovine serum, 50 U/mL penicillin, and 50 μg/mL streptomycin (Sigma‐Aldrich) at 37°C in 5% CO2.
Antibodies.
Anti‐Caspase‐9, anti‐cleaved Caspase‐3 (Asp175), anti‐cleaved poly ADP ribose polymerase (PARP) (Asp214), anti‐phospho‐Aurora A (Thr288), anti‐histone H3, and anti‐phospho‐histone H3 (Ser10) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti‐p53, anti‐p21, and anti‐actin antibodies were obtained from Lab Vision (Fremont, CA, USA). Anti‐Aurora A antibody was purchased from BD Biosciences (San Jose, CA, USA). Horseradish peroxidase‐conjugated secondary antibodies were purchased from GE Healthcare (Waukesha, WI, USA).
Cell viability assay.
The antiproliferative effects of MLN8237 against HTLV‐1‐infected and ‐uninfected T‐cell lines were measured by the WST‐8 method (Cell Counting Kit‐8; Wako Pure Chemical Industries, Osaka, Japan), based on the MTT assay, as described previously.( 22 ) Briefly, the 5 × 103 cells were incubated in triplicate in a 96‐well microculture plate in the presence of different concentrations of MLN8237 in a final volume of 0.1 mL at 37°C. Thereafter, 5 μL Cell Counting Kit‐8 solution (5 mm WST‐8, 0.2 mm 1‐methoxy 5‐methylphenazinium methylsulfate, and 150 mm NaCl) was added, and the cells were further incubated for 4 h. The number of surviving cells was measured by a 96‐well multiscanner autoreader at an optical density of 450 nm. Cell viability was determined as percentage of the control (absence of MLN8237). The 50% inhibitory concentration (IC50) was extrapolated from trend line data.
Cell cycle analysis.
Cell cycle analysis was performed with the CycleTEST PLUS DNA reagent kit (Becton Dickinson, San Jose, CA, USA). Briefly, cells were washed with a buffer solution containing sodium citrate, sucrose, and DMSO, suspended in a solution containing RNase A, and stained with 125 μg/mL propidium iodide (PI) for 10 min. Cell suspensions were analyzed on a Epics XL‐MCL flow cytometer (Beckman Coulter, Fullerton, CA, USA) using EXPO32 software. The first peak of DNA content in the histogram was defined as 2C, and the following peaks were defined as 4C, 8C, and 16C.
Apoptosis assay.
Apoptosis was quantified by staining with Annexin‐V‐Fluos (Roche Diagnostics, Mannheim, Germany) and PI, using the instructions provided by the manufacturer, followed by analysis on an Epics XL‐MCL flow cytometer using EXPO32 software.
Western blotting.
Western blot analysis was performed as described previously.( 23 ) In brief, whole cell lysates were subjected to SDS‐PAGE and electroblotted onto polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA), and then analyzed for immunoreactivity with the appropriate primary and secondary antibodies as indicated in the Figures. Reaction products were visualized using enhanced chemiluminescence reagent, according to the instructions provided by the manufacturer (GE Healthcare).
Immunocytochemistry.
Cells were collected by centrifugation; the pellet was washed with PBS, resuspended in 4% paraformaldehyde, and fixed for 20 min at room temperature. After fixation, the cells were centrifuged to remove the paraformaldehyde, permeabilized in a solution of 1% bovine serum albumin and 0.1% saponin, then incubated in 100× diluted anti‐phospho‐histone H3 (Ser10) antibody for 1 h. Cells were washed and incubated with Alexa 546‐conjugated secondary antibody (Invitrogen/Molecular Probes, Eugene, OR, USA) for 1 h in the dark. After incubation, cells were washed and applied onto a microscope slide and counterstained with DAPI stain. Images of the cells positive for phospho‐histone H3 (Ser10) were acquired using a Leica TCS‐SP5 confocal microscope (Leica, Mannheim, Germany).
Results
MLN8237 inhibits the viability of HTLV‐1‐infected T‐cell lines.
First, we examined the effects of MLN8237 on the cell viability of HTLV‐1‐infected T‐cell lines. MLN8237 markedly reduced the viability of HTLV‐1‐infected T‐cell lines (MT‐4 and HUT‐102) 24 h after exposure to MLN8237, but the effects on HTLV‐1‐uninfected T‐cell lines (Jurkat and CCRF‐CEM) were mild at this time point (Fig. 1, left panel). Although after 48 or 72 h exposure to 50 nm MLN8237, viability of Jurkat and CCRF‐CEM cells was decreased to 40–60% and 60–80%, respectively, that of MT‐4 and HUT‐102 cells at those time points was still lower than that of Jurkat and CCRF‐CEM cells (Fig. 1, middle and right panels). The concentrations of MLN8237 required to inhibit growth of the cells by 50% (IC50) are shown in Table 1. Viability of normal PBMCs from healthy donors was not affected by MLN8237 treatment (IC50 > 50 nm). These results suggested that MLN8237 more effectively inhibited the cell viability of HTLV‐1‐infected T‐cell lines than ‐uninfected T‐cell lines.
Figure 1.
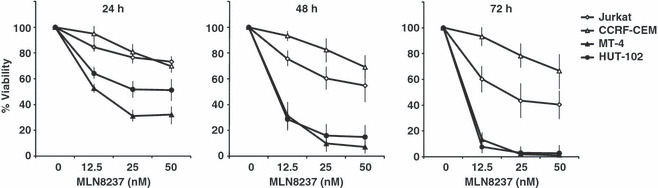
MLN8237 inhibits the viability of human T‐cell leukemia virus type 1 (HTLV‐1)‐infected T‐cell lines. HLTV‐1‐infected (MT‐4 and HUT‐102) and ‐uninfected (Jurkat and CCRF‐CEM) T‐cell lines were treated with different concentrations of MLN8237 (0, 12.5, 25, or 50 nm) for 24, 48, or 72 h. Cell viability was measured by WST‐8 assay. Data are mean ± SD of triplicate experiments.
Table 1.
IC50 for inhibition of cell growth of MLN8237
| Cells | HTLV‐1 | IC50 for inhibition of cell growth (nm) | ||
|---|---|---|---|---|
| 24 h | 48 h | 72 h | ||
| Jurkat | − | >50 | >50 | >50 |
| CCRF‐CEM | − | >50 | >50 | 18.9 |
| MT‐4 | + | 13.3 | 8.8 | 7.2 |
| HUT‐102 | + | 25.4 | 8.7 | 6.8 |
MLN8237 induces polyploidy in HTLV‐1‐uninfected T‐cell lines, but not in HTLV‐1‐infected T‐cell lines.
As Aurora A is a key regulator of mitosis, we examined the effects of MLN8237 on the cell cycle profile by flow cytometry‐based assays. Exposure of HTLV‐1‐uninfected Jurkat (Fig. 2a) and CCRF‐CEM (Fig. 2b) cells to MLN8237 prominently increased in polyploid (≥8C DNA content) cells. After 72 h exposure to MLN8237, cells with 16C DNA content increased in these cell lines. In contrast, exposure of HTLV‐1‐infected MT‐4 (Fig. 2c) and HUT‐102 (Fig. 2d) cells to MLN8237 increased in few polyploid cells even after 72 h treatment.
Figure 2.
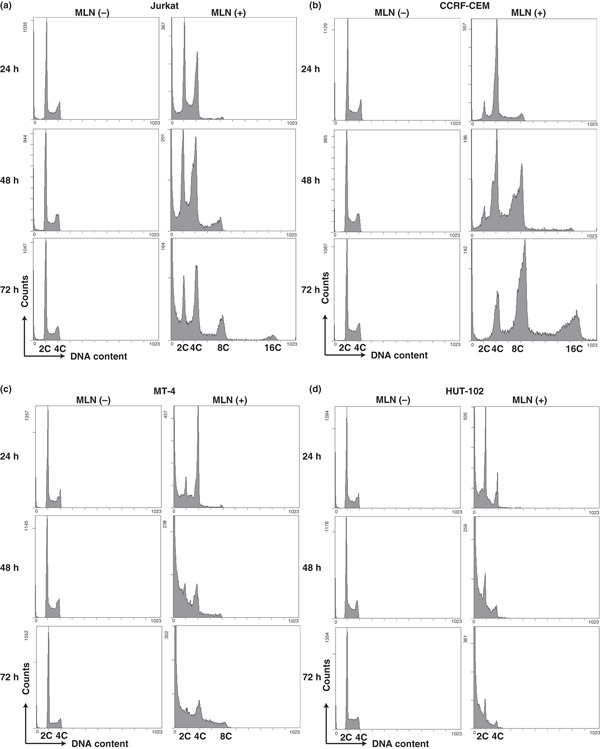
MLN8237 induces polyploidy in human T‐cell leukemia virus type 1 (HTLV‐1)‐uninfected T‐cell lines (a,b), but not in HTLV‐1‐infected T‐cell lines (c,d). Cells were exposed to 12.5 nm MLN8237 (MLN [+]) or vehicle (MLN [−]) for 24, 48, or 72 h. The cell cycle was analyzed by flow cytometry. Similar results were observed in triplicate experiments.
HTLV‐1‐infected T‐cell lines increase in the apoptotic fractions after exposure to MLN8237.
Dual staining with fluorescent Annexin V antibody and PI followed by flow cytometry demonstrated the induction of apoptosis by treatment with MLN8237 in MT‐4 cells (Fig. 3b) but not in CCRF‐CEM cells (Fig. 3a). A percentage of Annexin V‐positive and PI‐negative cells, indicating early apoptosis, were analyzed in the cells treated with MLN8237 (Fig. 3c). Marked increase of apoptosis in MT‐4 and HUT‐102 cells but not in Jurkat and CCRF‐CEM cells was observed. We also confirmed MLN8237‐induced HTLV‐1‐infected T‐cell specific apoptosis using western blotting. The cleaved form of caspase‐9, caspase‐3, and PARP, markers of apoptosis, were increased in MT‐4 and HUT‐102 cells but not in Jurkat and CCRF‐CEM cells treated with MLN8237 for 24 h (Fig. 4a) or 48 h (Fig. 4b). The expression of Aurora A was not changed by treatment of MLN8237 (Fig. 4a). Moreover, MLN8237 increased p53 and p21 protein expression in HTLV‐1‐infected T‐cell lines but not in ‐uninfected T‐cell lines (Fig. 4a,b). Previous study showed that polyploidy induced by Aurora kinase inhibitors are associated with compromised p53‐dependent postmitotic checkpoint function.( 24 ) Our findings suggest that MLN8237‐induced apoptosis without polyploidy in HTLV‐1‐infected T‐cell lines might be due to the integrity of the p53‐dependent postmitotic checkpoint. Then, the morphology of nuclei was examined in MLN8237 treated cells. MLN8237 increased the cells with unusual large nuclei, which were multinucleated cells, in Jurkat and CCRF‐CEM cells (Fig. 5a,c). In contrast, nuclei of MLN8237‐treated MT‐4 and HUT‐102 cells were fragmented, indicating induction of apoptosis (Fig. 5e,g).
Figure 3.
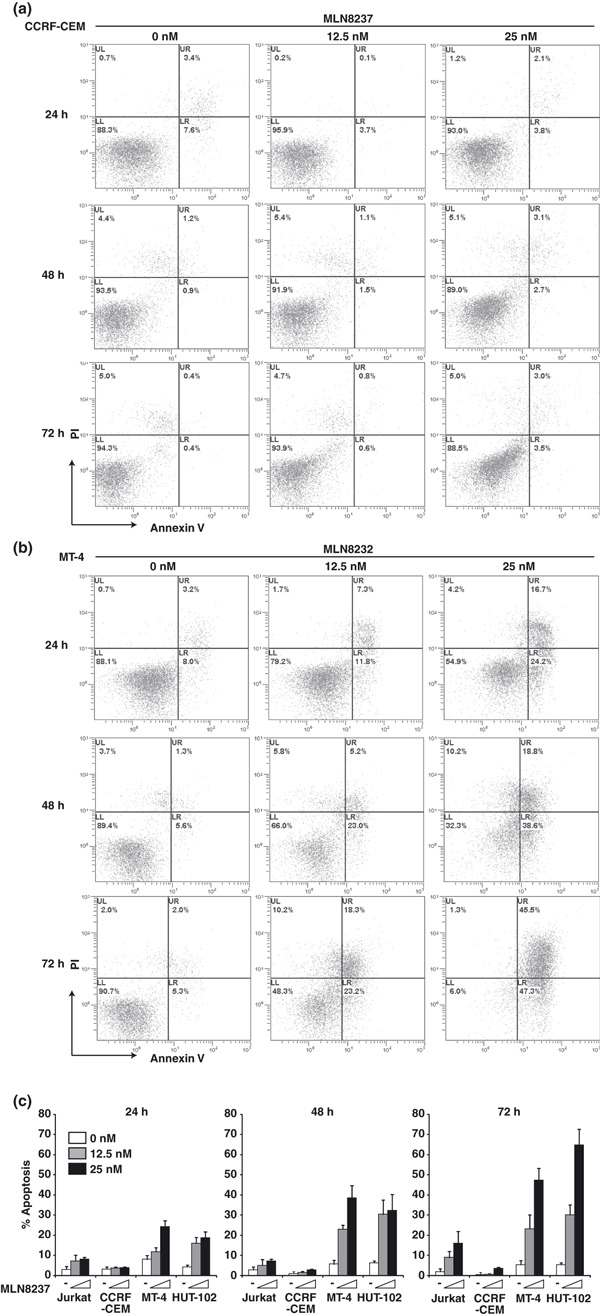
ensp;Human T‐cell leukemia virus type 1 (HTLV‐1)‐infected T‐cell lines increase in the apoptotic fractions after exposure to MLN8237. CCRF‐CEM (a) and MT‐4 (b) cells were exposed to 0, 12.5, or 25 nm MLN8237 for 24, 48, or 72 h. Cells were harvested and stained with Annexin V and propidium iodide (PI). Apoptosis was analyzed by flow cytometry. Similar results were observed in triplicate experiments. (c) The percentage of early apoptotic cells (Annexin V‐positive, PI‐negative cells) was determined by flow cytometry. Data are mean ± SD of three separate experiments.
Figure 4.
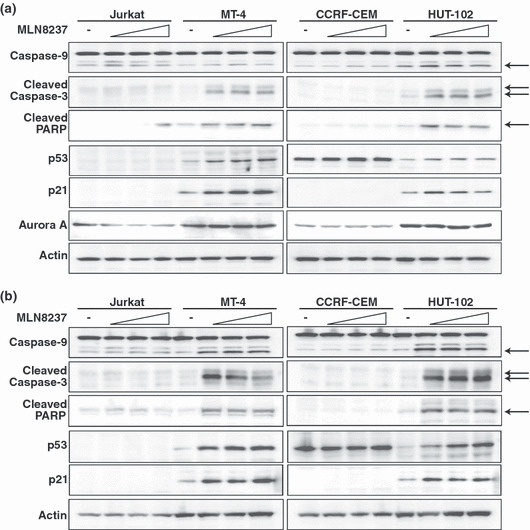
MLN8237 induces cleavage of caspase‐9, caspase‐3, and PARP. Western blotting shows the caspase‐9, cleaved caspase‐3, cleaved poly ADP ribose polymerase (PARP), p53, p21, and Aurora A expression at 24 h (a) or 48 h (b) after MLN8237 (0, 12.5, 25, or 50 nm) exposure in human T‐cell leukemia virus type 1 (HTLV‐1)‐infected and ‐uninfected T‐cell lines. Arrows indicate cleaved caspase‐9, caspase‐3, or PARP. Actin was evaluated as a loading control. Similar results were observed in triplicate experiments.
Figure 5.
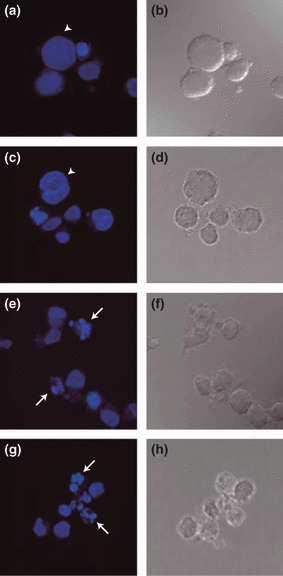
MLN8237 induces multinucleated cells in human T‐cell leukemia virus type 1 (HTLV‐1)‐uninfected T‐cell lines and nuclear fragmentation in HTLV‐1‐infected T‐cell lines. Jurkat (a,b), CCRF‐CEM (c,d), MT‐4 (e,f), and HUT‐102 (g,h) cells were treated with 12.5 nm MLN8237 for 24 h. Nuclei were stained with DAPI (blue) (a,c,e,g). DIC: differential interference contrast images (b,d,f,h). Arrows indicate fragmentation of the nuclei. Arrowheads indicate multinucleated cells. Similar images were observed in triplicate experiments.
MLN8237 selectively inhibits Aurora A over Aurora B in both HLTV‐1‐infected and ‐uninfected T‐cell lines.
The effect of MLN8237 on Aurora A activity was evaluated by western blotting of Aurora A autophosphorylation on Thr288 (p‐Aurora A). Aurora A kinase activity depends on autophosphorylation of Thr288 in the activation loop.( 25 , 26 ) Thus, detection of Aurora A autophosphorylation on Thr288 reflects the activity of the kinase in cells. Treatment of the nocodazole‐arrested cells at mitosis with MLN8237 (12.5, 25, or 50 nm) for 24 h inhibited Aurora A autophosphorylation on Thr288 in both HTLV‐1‐infected and ‐uninfected T‐cell lines (Fig. 6a), demonstrating that MLN8237 inhibits Aurora A activity in these cells. The selectivity of MLN8237 for Aurora A over the structurally related Aurora B kinase was evaluated by immunofluorescent detection of phosphorylated histone H3 on Ser10, an Aurora B‐specific substrate in cells.( 27 , 28 ) Although MLN8237 at 12.5 nm suppressed Aurora A activity (Fig. 6a), 50 nm MLN8237 was not sufficient to inhibit Aurora B activity, because mitotic cells remained phosphorylated histone H3‐immunopositive in both HTLV‐1‐infected and ‐uninfected T‐cell lines (Fig. 6b). MLN8237 treatment resulted in increase of phosphorylated histone H3 which is the marker of mitotic cells (Fig. 6c), consistent with a phenotype of Aurora A inhibition.( 17 ) Together, these results verify that MLN8237 was indeed well suited to discriminate between Aurora A and Aurora B in our experimental cell system.
Figure 6.
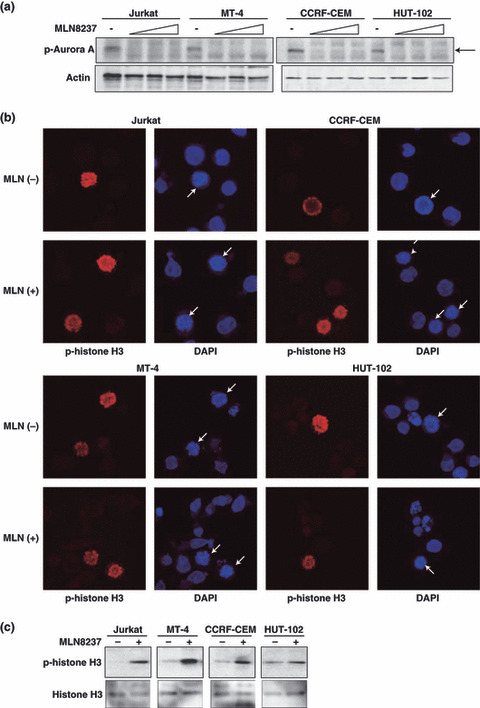
MLN8237 selectively inhibits Aurora A over Aurora B in both human T‐cell leukemia virus type 1 (HTLV‐1)‐ infected and ‐uninfected T‐cell lines. (a) The cells were pretreated for 16 h with 10 nm nocodazole to arrest the cell cycle at mitosis followed by treatment with various concentrations of MLN8237 (0, 12.5, 25, or 50 nm) for 2 h. Western blot analysis was used to detect phospho‐Aurora A (Thr288) and Actin. Similar results were observed in triplicate experiments. (b) Representative immunofluorescent images of the cells treated with 50 nm MLN8237 (MLN [+]) or vehicle (MLN [−]) for 24 h. Cells were stained for anti‐phospho‐histone H3 (Ser10) antibody (p‐histone H3; red) and DNA (DAPI; blue). Arrows indicated mitotic cells. (c) The cells were treated with 12.5 nm MLN8237 (+) or vehicle (−) for 24 h. Phosphrylation of histone H3 (Ser10) and whole histone H3 were analyzed by western blotting. Similar results were observed in triplicate experiments.
Discussion
Aurora kinase inhibitors target the mitotic functions of the Aurora kinases and induce apoptosis in culture cells as well as in human xenografts.( 10 , 11 , 12 , 13 , 14 , 15 , 16 , 29 , 30 ) However, the molecular mechanisms leading to the induction of tumor cell death are poorly understood, although a detailed knowledge about these mechanisms is most important to improve therapeutic strategies and drug combinations and to explain resistance on a molecular level. We showed previously that a pan‐Aurora kinase inhibitor induces apoptosis in HTLV‐1‐infected T‐cells at earlier time point compared to uninfected T‐cells.( 21 ) In this study, to analyze the specific function of Aurora A inhibition on the growth and survival of HTLV‐1‐infected T‐cells, we used MLN8237, a selective inhibitor of Aurora A. We demonstrate that MLN8237 exerts its antiproliferative and antisurvival activity against HTLV‐1‐infected T‐cell lines more effectively than ‐uninfected T‐cell lines through specific inhibition of Aurora A kinase.
It is unknown whether Aurora A or Aurora B is the better target for oncology therapy. In fact, the validity for targeting Aurora A as an anticancer therapeutic approach has been questioned, ( 16 ) largely because the major phenotype with pan‐Aurora kinase inhibitors is Aurora B inhibition. However, a recent more detailed study demonstrated that pan‐Aurora inhibitors also elicit a phenotype consistent with Aurora A inhibition.( 31 , 32 ) We reported previously that a pan‐Aurora kinase inhibitor suppressed cell viability and induced apoptosis by inhibiting NF‐κB signaling in HTLV‐1‐infected T‐cell lines and primary ATL cells.( 21 ) It has been well known that HTLV‐1 transforming protein Tax activates NF‐κB signaling.( 33 ) Primary ATL cells, which do not express Tax, showed constitutively activated NF‐κB in a Tax‐independent manner.( 34 ) Therefore, NF‐κB signaling is a good therapeutic target of ATL. However, we found that MLN8237 did not affect NF‐κB activity in HTLV‐1‐infected T‐cell lines (data not shown), suggesting that inhibition of Aurora B but not that of Aurora A might be responsible for pan‐Aurora kinase inhibitor‐induced NF‐κB inhibition in these cells. It is possible that inhibition of both Aurora kinases is necessary to suppress NF‐κB activity. Future analysis will be needed to elucidate the effect of Aurora B selective inhibitor on NF‐κB activity in HTLV‐1‐infected T‐cells.
Previous studies have shown that the phenotypic response to Aurora kinase inhibition appears to be dependent on the genetic background of the cells. For example, the status of the tumor suppressor protein p53 appears to be critical in determining the extent of endoreduprication and kinetics of cell death.( 35 ) Whether cells arrest with 4C DNA content in pseudo‐G1 or endoreduplicate with the accumulation of >4C DNA content likely depends on the integrity of the p53‐dependent postmitotic checkpoint.( 35 ) Previous study has indicated that Aurora kinase inhibitor VX‐680 induces not only endoredupulication but also apoptosis in cells with compromised p53‐dependent postmitotic checkpoint function.( 24 ) In contrast, we found that MLN8237 up‐regulated p53 and its transcriptional target p21 expression in HTLV‐1‐infected T‐cell lines but not in uninfected T‐cell lines at an early time point. The HTLV‐1‐infected T‐cell lines used in this study (MT‐4 and HUT‐102) have wild type p53. On the other hand, the status of the p53 gene in HTLV‐1‐uninfected T‐cell lines (Jurkat and CCRF‐CEM) was mutant. Thus, it seems that lack of polyploidy in HTLV‐1‐infected T‐cell lines treated with MLN8237 may be due to the functional p53‐dependent G1 postmitotic checkpoint. However, the viability of HTLV‐1‐negative PBMCs with wild type p53 was not affected by MLN8237 treatment (data not shown). Therefore, the exact mechanisms by which MLN8237 induces apoptosis in HTLV‐1‐infected T‐cell lines and the contribution of HTLV‐1 viral proteins are still unclear.
Are the Aurora kinase inhibitors cancer specific? Given that they are key regulators of mitosis, they are not, in the strictest sense. Toxicity of MLN8237 to normal bone marrow is not unexpected given the mechanism of action of the drug and its known tendency to cause leukopenia in early clinical trials.( 18 ) As with other myelosuppressive anticancer agents, effective application of MLN8237 in the clinic will require due consideration of this issue. Aurora kinase inhibitors induce polyploidy in normal epithelial cells,( 10 ) raising the issue of long‐term clinical side effects such as mucositis or diarrhea. As we have shown here, HTLV‐1‐infected T‐cells are exceptionally sensitive to MLN8237, enabling delivery of minimally toxic doses that have significant anti‐ATL effects.
In conclusion, we described the antiproliferative and cytotoxic effects of MLN8237 on HTLV‐1‐infected T‐cell lines. These effects occurred at an early time point (within 24 h) after exposure to MLN8237, which affects the growth and survival of uninfected T‐cell lines less effectively at this time point. Our findings from current study support the further evaluation of MLN8237 in the treatment of ATL.
Acknowledgments
We thank Millennium Pharmaceuticals for the gift of MLN8237 and the Fujisaki Cell Center, Hayashibara Biomedical Laboratories (Okayama, Japan) for providing the HUT‐102. We also acknowledge the contribution of all members of our laboratory for the helpful comments and collaborations. This work was supported in part by a grant‐in‐aid (JSPS KAKENHI 21591212) from the Japan Society for the Promotion of Science, a grant (no. 07‐23905) from the Princes Takamatsu Cancer Research Fund, and grants from the Ichiro Kanehara Foundation, Mitsubishi Pharma Research Foundation, and the Yasuda Medical Foundation.
References
- 1. Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol 2001; 2: 21–32. [DOI] [PubMed] [Google Scholar]
- 2. Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol 2003; 4: 842–54. [DOI] [PubMed] [Google Scholar]
- 3. Tang CJ, Lin CY, Tang TK. Dynamic localization and functional implications of Aurora‐C kinase during male mouse meiosis. Dev Biol 2006; 290: 398–410. [DOI] [PubMed] [Google Scholar]
- 4. Marumoto T, Zhang D, Saya H. Aurora‐A ‐ a guardian of poles. Nat Rev Cancer 2005; 5: 42–50. [DOI] [PubMed] [Google Scholar]
- 5. Zhou H, Kuang J, Zhong L et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet 1998; 20: 189–93. [DOI] [PubMed] [Google Scholar]
- 6. Goepfert TM, Adigun YE, Zhong L, Gay J, Medina D, Brinkley WR. Centrosome amplification and overexpression of aurora A are early events in rat mammary carcinogenesis. Cancer Res 2002; 62: 4115–22. [PubMed] [Google Scholar]
- 7. Wang X, Zhou YX, Qiao W et al. Overexpression of aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene 2006; 25: 7148–58. [DOI] [PubMed] [Google Scholar]
- 8. Zhang D, Hirota T, Marumoto T et al. Cre‐loxP‐controlled periodic Aurora‐A overexpression induces mitotic abnormalities and hyperplasia in mammary glands of mouse models. Oncogene 2004; 23: 8720–30. [DOI] [PubMed] [Google Scholar]
- 9. Andrews PD. Aurora kinases: shining lights on the therapeutic horizon? Oncogene 2005; 24: 5005–15. [DOI] [PubMed] [Google Scholar]
- 10. Ditchfield C, Johnson VL, Tighe A et al. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp‐E to kinetochores. J Cell Biol 2003; 161: 267–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Long ZJ, Xu J, Yan M et al. ZM 447439 inhibition of aurora kinase induces Hep2 cancer cell apoptosis in three‐dimensional culture. Cell Cycle 2008; 7: 1473–9. [DOI] [PubMed] [Google Scholar]
- 12. Hauf S, Cole RW, LaTerra S et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore‐microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol 2003; 161: 281–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harrington EA, Bebbington D, Moore J et al. VX‐680, a potent and selective small‐molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med 2004; 10: 262–7. [DOI] [PubMed] [Google Scholar]
- 14. Soncini C, Carpinelli P, Gianellini L et al. PHA‐680632, a novel Aurora kinase inhibitor with potent antitumoral activity. Clin Cancer Res 2006; 12: 4080–9. [DOI] [PubMed] [Google Scholar]
- 15. Wilkinson RW, Odedra R, Heaton SP et al. AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin Cancer Res 2007; 13: 3682–8. [DOI] [PubMed] [Google Scholar]
- 16. Keen N, Taylor S. Aurora‐kinase inhibitors as anticancer agents. Nat Rev Cancer 2004; 4: 927–36. [DOI] [PubMed] [Google Scholar]
- 17. Pollard JR, Mortimore M. Discovery and development of aurora kinase inhibitors as anticancer agents. J Med Chem 2009; 52: 2629–51. [DOI] [PubMed] [Google Scholar]
- 18. Cervantes‐Ruiperez A, Elez ME, Rosello S et al. Phase I pharmacokinetic (PK) and pharmacodynamic (PD) study of MLN8237, a novel selective aurora A kinase (AAK) inhibitor, in patients (pts) with advanced solid tumors. J Clin Oncol (Meeting Abstract) 2009; 27: 2565. [Google Scholar]
- 19. Zhang M, Huck J, Hyer M, Ecsedy J, Manfredi M. Effect of Aurora A kinase inhibitor MLN8237 combined with rituximab on antitumor activity in preclinical B‐cell non‐Hodgkin’s lymphoma models. J Clin Oncol (Meeting Abstract) 2009; 27: 8553. [Google Scholar]
- 20. Matsuoka M, Jeang KT. Human T‐cell leukaemia virus type 1 (HTLV‐1) infectivity and cellular transformation. Nat Rev 2007; 7: 270–80. [DOI] [PubMed] [Google Scholar]
- 21. Tomita M, Toyota M, Ishikawa C et al. Overexpression of Aurora A by loss of CHFR gene expression increases the growth and survival of HTLV‐1‐infected T cells through enhanced NF‐κB activity. Int J Cancer 2009; 124: 2607–15. [DOI] [PubMed] [Google Scholar]
- 22. Ishiyama M, Tominaga H, Shiga M, Sasamoto K, Ohkura Y, Ueno K. A combined assay of cell viability and in vitro cytotoxicity with a highly water‐soluble tetrazolium salt, neutral red and crystal violet. Biol Pharm Bull 1996; 19: 1518–20. [DOI] [PubMed] [Google Scholar]
- 23. Tomita M, Choe J, Tsukazaki T, Mori N. The Kaposi’s sarcoma‐associated herpesvirus K‐bZIP protein represses transforming growth factor β signaling through interaction with CREB‐binding protein. Oncogene 2004; 23: 8272–81. [DOI] [PubMed] [Google Scholar]
- 24. Gizatullin F, Yao Y, Kung V, Harding MW, Loda M, Shapiro GI. The Aurora kinase inhibitor VX‐680 induces endoreduplication and apoptosis preferentially in cells with compromised p53‐dependent postmitotic checkpoint function. Cancer Res 2006; 66: 7668–77. [DOI] [PubMed] [Google Scholar]
- 25. Littlepage LE, Wu H, Andresson T, Deanehan JK, Amundadottir LT, Ruderman JV. Identification of phosphorylated residues that affect the activity of the mitotic kinase Aurora‐A. Proc Natl Acad Sci U S A 2002; 99: 15440–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Satinover DL, Leach CA, Stukenberg PT, Brautigan DL. Activation of Aurora‐A kinase by protein phosphatase inhibitor‐2, a bifunctional signaling protein. Proc Natl Acad Sci U S A 2004; 101: 8625–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Giet R, Glover DM. Drosophila aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J Cell Biol 2001; 152: 669–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goto H, Yasui Y, Nigg EA, Inagaki M. Aurora‐B phosphorylates Histone H3 at serine28 with regard to the mitotic chromosome condensation. Genes Cells 2002; 7: 11–7. [DOI] [PubMed] [Google Scholar]
- 29. Carpinelli P, Ceruti R, Giorgini ML et al. PHA‐739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Mol Cancer Ther 2007; 6: 3158–68. [DOI] [PubMed] [Google Scholar]
- 30. Shi Y, Reiman T, Li W et al. Targeting aurora kinases as therapy in multiple myeloma. Blood 2007; 109: 3915–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gadea BB, Ruderman JV. Aurora kinase inhibitor ZM447439 blocks chromosome‐induced spindle assembly, the completion of chromosome condensation, and the establishment of the spindle integrity checkpoint in Xenopus egg extracts. Mol Biol Cell 2005; 16: 1305–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Girdler F, Gascoigne KE, Eyers PA et al. Validating Aurora B as an anti‐cancer drug target. J Cell Sci 2006; 119: 3664–75. [DOI] [PubMed] [Google Scholar]
- 33. Sun SC, Yamaoka S. Activation of NF‐κB by HTLV‐I and implications for cell transformation. Oncogene 2005; 24: 5952–64. [DOI] [PubMed] [Google Scholar]
- 34. Mori N, Fujii M, Ikeda S et al. Constitutive activation of NF‐κB in primary adult T‐cell leukemia cells. Blood 1999; 93: 2360–8. [PubMed] [Google Scholar]
- 35. Margolis RL, Lohez OD, Andreassen PR. G1 tetraploidy checkpoint and the suppression of tumorigenesis. J Cell Biochem 2003; 88: 673–83. [DOI] [PubMed] [Google Scholar]