

Abstract
Osteopontin (OPN) is a secreted, integrin‐binding matrix phosphorylated glycoprotein that is overexpressed in many advanced cancers. However, the functional mechanisms by which OPN contributes to the development of ovarian cancer are poorly understood. Here, we reveal that acquired expression of OPN by HO‐8910 ovarian cancer cells greatly promoted the progression of ovarian cancer. OPN expression dramatically increased the colony formation of ovarian cancer cells in vitro and tumor growth in vivo. Under the stress induced by serum depletion or curcumin treatment, OPN expression promoted the survival of ovarian cells through preventing stress‐induced apoptosis. At the molecular level, both endogenous and exogenous OPN expression activated the PI3‐K/Akt survival pathway and dramatically decreased p53 expression under serum depletion. In addition, HIF‐1α was induced in OPN‐producing cells under normoxia. Furthermore, we also found that inhibition of the PI3‐K/Akt pathway attenuated OPN‐mediated HIF‐1α up‐regulation in ovarian cancer cells. Taken together, these results indicate that OPN can increase the survival of ovarian cancer cells under stress conditions in vitro and promote the late progression of ovarian cancer in vivo, and the survival‐promoting functions of OPN are mediated through Akt activation and the induction of HIF‐1α expression. (Cancer Sci 2008; 99: 1901–1907)
Osteopontin (OPN), also known as early T‐cell activation‐1 (Eta‐1), is a secreted, integrin‐binding matrix phosphorylated glycoprotein that plays important roles in a wide range of biological processes, including tissue remodeling, inflammation, angiogenesis, tumor development, and immunity to infectious disease.( 1 , 2 , 3 ) These diverse biological functions reflect the ability of secreted OPN to bind to different cell surface integrin receptors and CD44 variants, and the ability of intracellular OPN to interact with other signal‐transduction molecules.( 4 , 5 , 6 , 7 ) OPN has been shown to inhibit apoptosis and contribute to cell survival by its phosphorylation,( 8 ) interaction with CD44,( 9 , 10 ) activation to nuclear factor–kappa B (NF‐κB),( 11 ) alternation of the expression of the proapoptotic proteins Bim, Bak, and Bax,( 11 ) and activation of the PI3‐K/Akt signaling pathway.( 10 ) It has been found that OPN expression is up‐regulated in several malignant tumor tissues; and it has been reported to be associated with late progression and metastasis of tumors.( 1 , 12 ) However, the mechanisms by which OPN functions in tumor development are still not well defined.
The development of a tumor consists of a series of complex sequential events.( 13 ) During the process of tumor development, cancer cells have to overcome a number of stresses such as hypoxia, nutrient depletion, acidosis, and loss of adhesion that may all induce cell death.( 14 ) Hypoxia is an inevitable stress that tumor cells have to face during the development of solid tumors. Hypoxia‐inducible factor‐1 (HIF‐1), which is a heterodimeric basic helix‐loop‐helix transcriptional activator possessing two subunits, HIF‐1α and HIF‐1β,( 15 ) is stabilized and activated to promote the transcription of several genes, whose products are required for critical aspects of tumor progression. HIF‐1β is constitutively expressed, while the expression and activity of the HIF‐1α subunit are induced by the exposure of cells to hypoxia or growth factors.( 16 ) Under normal oxygen tension (normoxia), HIF‐1α can be modified by prolyl hydroxylation, which permits binding of von Hippel‐Lindau protein, a recognition component of the E3 ligase complex. HIF‐1α undergoes rapid ubiquitination and degradation by proteasomes afterwards, so HIF‐1α is maintained at a low level in normoxic cells.( 17 ) Under hypoxic conditions or treatment with iron chelators, the praline hydroxylases remain inactivated, and HIF‐1α degradation is blocked and so the HIF‐1α activity is stable. Recent reports have shown that the increase of HIF‐1α activity is not only induced by intratumoral hypoxia but also by genetic alteration under normoxic conditions, such as von Hippel‐Lindau protein( 18 ) and PTEN.( 19 ) In addition, some activation of oncogenes can also increase HIF‐1α activity through activating the PI3‐K/Akt, mitogen‐activated protein kinase (MAPK) or other signal pathways.( 20 , 21 ) Although the PI3‐K/Akt and HIF pathways share many common features such as promoting cell survival, angiogenesis, tumor malignancy, and metastasis, how PI3‐K/Akt regulates HIF activity is still controversial.( 22 , 23 )
Ovarian cancer is one of the aggressive and fatal diseases which spreads in an early stage, occurs late, and it is difficult to cure because it is scarcely detected until its recurrence in the advanced stages. It tends to recur. According to the 2008 American Cancer Society report, ovarian cancer accounts for about 3% of all cancers among women and is ranked second among gynecologic cancers.( 24 ) Despite the improvements in surgical and chemotherapeutic treatment in the past few decades, the long‐term survival rates of ovarian carcinoma remain poor. The combination of chemotherapeutic agents is the major strategy against ovarian cancer treatment. As a result, using these agents at clinically effective doses is often accompanied by severe toxicity. Such a lack of specificity has stimulated the development of a new strategy that targets the molecular signaling transduction pathways in cancer cells and thus tends to be less toxic to normal cells than conventional chemotherapies.( 25 ) Previous reports have indicated that OPN was overexpressed in the tumors and serums of patients with ovarian cancer,( 26 , 27 , 28 ) but the contribution of OPN overexpression in the development of ovarian cancer remains unclear. Here, we examine the effects and mechanisms of OPN in the progression of ovarian cancer development. Our study demonstrates that OPN can increase the survival of ovarian cancer cells under stress conditions through PI3‐K/Akt activation and the induction of HIF‐1α expression. The elucidation of these intricate mechanisms of OPN indicates that OPN may be a useful molecular target for the therapeutics of ovarian cancer.
Materials and Methods
Materials. Human recombinant OPN protein was purchased from R&D Systems Europe (Abingdon, UK). Akt‐specific inhibitor (1 L‐6‐Hydroxymethyl‐chiro‐inositol 2‐[R]‐2‐O‐methyl‐3‐O‐octadecylcarbonate), the PI3‐K inhibitor LY294002, and ubiquitin‐proteasome degradation inhibitor MG132 were bought from Calbiochem (San Diego, CA, USA). Soft agar was provided by Merck (Darmstadt, Germany). Mouse anti‐Flag, anti‐Akt, anti‐β‐actin, and goat anti‐OPN antibodies were purchased from Sigma Chemical Co. (St Louis, MO, USA). Mouse anti‐p53 antibody was purchased from Calbiochem. Rabbit anti‐phospho‐Akt (Ser 473) antibody was provided by R&D Systems Europe. Mouse anti‐HIF‐1α and anti‐rabbit IgG, anti‐mouse IgG, and anti‐goat IgG antibodies were bought from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Surgical specimens of gastric cancer and normal tissue were selected randomly from the Division of Tumor Pathology, Zhongshan Hospital, Xiamen University, China; tissue procurement was approved by the institutional review board of Xiamen University. In vivo animal experiments with female Balb/c nude mice were done in the Cancer Research Center Laboratory, Xiamen University, China.
Cell culture and generation of OPN‐producing ovarian cancer cells. The human ovarian cancer cell line HO‐8910 was obtained from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, Shanghai, and was cultured as previously described.( 25 ) The full open reading frame of human OPN cDNA (a kind gift from Dr Xiao‐Fan Wang's laboratory at Duke University Medical Center) was cloned into pcDNA3.1 mammalian expression vector (Invitrogen) with a Flag tag at the C‐terminus of OPN protein. Cell transfections were performed using FuGene 6 reagent (Roche Diagnostics) according to the manufacturer's instructions. The OPN/pcDNA3.1 plasmid or the vector alone was introduced into HO‐8910 ovarian cancer cells, and the stable cell clones were obtained by 200 µg/mL hygromycin B (Amresco) selection in the culture medium. Two OPN‐overexpressing clones (No. O28 and O40) and two control clones (No. V1 and V2) were chosen for the subsequent experiments.
Soft‐agar growth. Cells (1 × 103 cells/dish) were plated in 35‐mm‐diameter dishes with a top layer of 0.3% agar and a bottom layer of 0.6% agar in medium. 0.3 mL of medium was supplemented every 3 days and the dishes were examined under microscope. After 2 weeks, the number of cell clusters (≥50 cells per cluster) per dish was counted, and cell clusters were photographed. The results were quantified from three independent experiments. Colony formation efficiency was calculated as follows: Number of clusters/Number of plated cells × 100%. Assays were performed in triplicate for each group of cells and data were expressed as mean ± SD.
Cell survival assay under stress conditions. OPN‐producing HO‐8910 cells and the vector‐transfected HO‐8910 cells were grown in the normal medium until 70–80% confluency. Then, the cells were incubated with serum‐free medium, 200 µM CoCl2 or 40 µM curcumin (Sigma). After a definite time of exposure, the cells were observed under an inverted‐phase microscope (Leica DM IRB) and the cell numbers were determined after Trypan blue staining of viable cells in parallel plates. Triplicate assays were performed in each group of cells under both serum‐depleted and curcumin‐treated conditions and data were expressed as mean ± SD. The cell survival rate (%) was calculated as follows: Numbers of survival cells of the experimental group/Numbers of the control group × 100%.
Western blot analysis. Western blot analysis was performed as reported in our previous publications.( 29 , 30 ) After harvesting, the experimental cells were washed and lyzed in lysis buffer (20 mM Tris‐HCl, 100 mM NaCl, 20 mM KCl, 1.5 mM MgCl2, 50 mM β‐GPA, 10 mM NaF, 0.5% NP‐40, plus proteinase inhibitors and phosphatase inhibitors). The total protein, as determined by Bio‐Rad protein assay, was mixed with 4 × loading buffer, and preheated at 95°C for 10 min. The samples were then loaded on SDS‐polyacrylamine gel. The proteins were transferred onto PVDF membrane by semidry transfer system (Bio‐Rad). The membrane was blocked in 5% milk, and then incubated 1 h or overnight at 4°C with primary antibody. After hybridization with primary antibody, the membrane was washed and incubated with horseradish peroxidase–labeled secondary. Final detection was performed with enhanced chemiluminescence Western blotting reagents (Amersham Pharmacia Biotech). The blot was then stripped in buffer (62.5 mM Tris‐HCl [pH 6.8], 2% SDS, 200 mM 2‐mercaptoethanol) at 50°C for 30 min. After extensive washing, the blot was used again for probing the next molecule, beginning from the blocking.
Animals and treatments. In vivo tumorigenesis experiments were conducted in nude mice. Two OPN‐overexpressing clones (No. O28 and O40) and two vector control clones (No. V1 and V2) were used. Twelve female Balb/c nude mice, aged 8 weeks were randomly divided into four groups (three mice per group). Two groups were injected with OPN‐expressing HO‐8910 cells (3 × 106/animal) via subcutaneous injection. The control groups were injected with the same number of vector‐transfected HO‐8910 cells. Mice were sacrificed 5 weeks after injection and examined for the growth of subcutaneous tumors.
Results
Overexpression of OPN in ovarian cancer cells potently promotes tumor growth in vitro and in vivo. Previous studies have showed that OPN was overexpressed in human cancers, and the level of OPN in the serum of ovarian cancer patients was elevated.( 26 , 27 , 28 ) We also found that OPN was highly expressed in human ovarian cancer tissues by immunohistochemical staining with an antibody against human OPN, while OPN was undetectable in normal ovarian tissues (Suppl. Fig. 1). To further assess the functional significance of elevated OPN expression during the development of ovarian cancer, we investigated whether stable overexpression of OPN in a human ovarian cancer cell line could alter the tumor growth in vitro and in vivo. We selected a human ovarian cancer cell line, HO‐8910, which expresses a very low level of endogenous OPN (Fig. 1a) and displays low metastatic potential in vivo. An OPN construct tagged with Flag protein at the C‐terminus was transfected into the HO‐8910 cells. Two stable clones (No. O28 and O40) and two vector‐transfected controls (No. V1 and V2) were verified with specific antibody against Flag‐tag and OPN (Fig. 1a,b). Overexpression of OPN in HO‐8910 cells led to a morphological change with an appearance of fibroblast‐like cell shape (Fig. 1c), but did not cause an alteration in the rate of cell proliferation in normal serum conditions in vitro (Suppl. Fig. 2).
Figure 1.
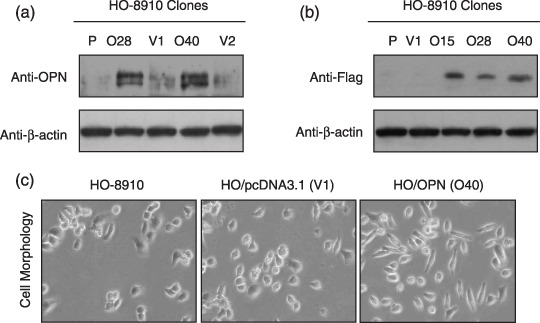
Stable overexpression of osteopontin (OPN) in HO‐8910 human ovarian cancer cells. (a,b) Characterization of OPN‐overexpressing HO‐8910 clones and vector control clones. Equivalent number of parental HO‐8910 cells (P) (2 × 106), two vector control HO‐8910 clones (V1, V2), and three OPN‐producing HO‐8910 clones (O15, O28, O40) were seeded in culture medium in 100 mm culture dishes. Twenty‐four hours later, cells were washed with phosphate‐buffered saline and underwent starvation in 4 mL serum‐free medium for another 48 h. Then the conditional media from the culture were subjected to Western blot assay using anti‐OPN or anti‐Flag antibody. The whole‐cell lysates from all samples were probed with anti‐β‐actin antibody for the loading control. (c) Overexpression of OPN in HO‐8910 cells resulted in cellular morphological change with an appearance of fibroblast‐like cell shape. (×100).
We next examined whether OPN overexpression could affect the colony formation efficiency in soft agar. The colony formation efficiency in soft agar measures the ability of cells to grow independently from anchorage and correlates with the degree of tumor malignancy.( 31 ) As shown in Fig. 2a,b, colony formation analysis indicated that the clone size of HO‐8910 cells transfected with empty vector was smaller than HO‐8910 cells transfected with OPN construct. The colony formation efficiency was notably different between HO/pcDNA3.1 and HO/OPN clones. To further investigate the impact of OPN overexpression on tumor growth in vivo, we injected the tumor cells into the nude mice to assess the potential differences in tumor growth between the OPN‐overexpression and the vector control clones. As shown in Fig. 2c,d, OPN overexpression appeared to significantly enhance the subcutaneous tumor growth over the control groups, and the tumor weight of OPN‐expressing clones was about two‐fold of that in vector clones, and the difference was significant (P < 0.05). These results indicated that OPN overexpression could enhance tumor growth in vivo.
Figure 2.
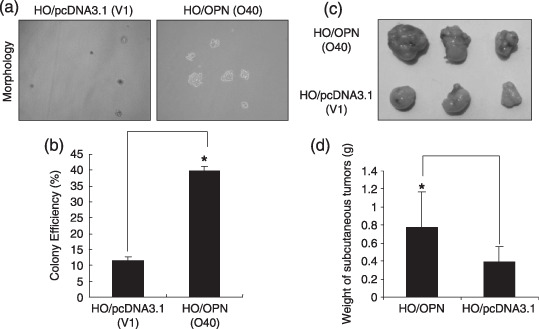
Overexpression of osteopontin (OPN) increased the colony formation in vitro and promoted ovarian tumor growth in vivo. (a) HO/pcDNA3.1 (V1) and HO/OPN cells (O40) were plated in triplicates with a top layer of 0.3% soft agar and a bottom layer of 0.6% soft agar. After 2 weeks, the representative micrographs of cell colonies were taken in random microscopic fields (×400). (b) Analysis of the clone formation efficiency. The frequency of clone formations was performed as described in ‘Materials and Methods’, and the results were obtained from three independent experiments. *P < 0.05 compared with vector control (SPSS 11.0). (c,d) Two OPN‐overexpressing clones (No. O28 and O40) and two vector control clones (No. V1 and V2) were injected into Balb/c nude mice via subcutaneous injection. After 35 days, the mice were sacrificed and analyzed for tumor formation in vivo. *P < 0.05 compared with vector control (SPSS 11.0).
OPN promoted cellular survival of ovarian cancer cells under stress. To explore the functional mechanisms by which OPN promotes tumor growth, we examined the biological effects of OPN on ovarian cancer cells under in vitro conditions that mimic the cellular stress inside tumors, such as nutrient deprivation by serum depletion, hypoxia, and chemical reagent treatment. We have confirmed that 40 µM curcumin could induce significant apoptosis in HO‐8910 cells.( 29 ) As shown in Fig. 3a,b, results from three independent experiments with two clones (No. O28 and O40) revealed that OPN‐producing HO‐8910 cells were more resistant to serum starvation in comparison with the control cells (No. V1 and V2) (P < 0.01). Similarly, OPN‐producing HO‐8910 cells were shown to prevent curcumin‐induced apoptosis (Fig. 3c,d) (P < 0.01). The increase in cell number associated with OPN expression may be due to increased cellular proliferation or resistance to cell death. However, we found no relative difference in cell proliferation between OPN‐producing HO‐8910 cells and vector control cells (Suppl. Fig. 2). In addition, we also detected the cell viability under 200 µM CoCl2 treatment which mimicked the hypoxia condition and it was found that overexpression of OPN did not affect ovarian cancer cell viability under this stress condition (Suppl. Fig. 3a,b). These results indicated that OPN could act to promote cellular survival for ovarian cancer cells under a variety of stresses.
Figure 3.
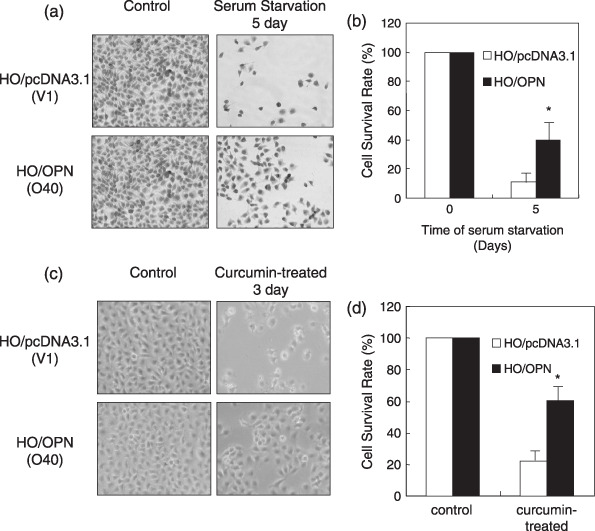
HO‐8910 ovarian cancer cells expressing osteopontin (OPN) displayed resistance to apoptosis. OPN‐overexpressing cells (O40) and vector control cells (V1) were incubated with serum‐free medium for starvation for 5 days or incubated with 40 µM curcumin for 3 days respectively. (a,b) Overexpression of OPN in HO‐8910 cells enhanced cellular survival under starvation. (c,d) Overexpression of OPN in HO‐8910 cells prevented HO‐8910 cells from curcumin‐induced apoptosis. Cells were observed and the representative cell micrographs were taken in random microscopic fields (×100), and the cell numbers were counted after Trypan blue staining of viable cells. Results in (b) and (d) were obtained from three independent experiments. *P < 0.01 compared with vector control (SPSS 11.0).
OPN activated the PI3‐K/Akt survival pathway and down‐regulated the cellular level of p53 protein under serum depletion. To explore the molecular mechanism by which OPN promotes cellular survival under serum depletion, we examined OPN expression or introduction of the exogenous human recombinant OPN on the activation of the PI3‐K/Akt cellular survival pathway. As shown in Fig. 4a, phosphorylation on Ser 473 that indicates the activation of Akt pathway was readily detected in the OPN‐producing HO‐8910 cells, but not in the vector‐transfected HO‐8910 cells under the serum‐free conditions. Similarly, treatment of HO‐8910 cells with 200 ng/mL OPN under the serum‐free conditions led to Akt phosphorylation on Ser 473 (Fig. 4a). PI3‐K and Akt have been strongly linked to cell survival and are resistant to apoptotic stimuli.( 32 ) We used PI3‐K inhibitor and Akt inhibitor to block the PI3‐K and Akt functions, and examined the effect on the OPN‐mediated enhancement with respect to cellular survival under serum‐free conditions. For this purpose, parental HO‐8910 cells were preincubated with the PI3‐K inhibitor, LY294002, or a specific Akt inhibitor, 1L‐6‐Hydroxymethyl‐chiro‐inositol 2‐[R]‐2‐O‐methyl‐3‐O‐octadecylcarbonate, and treated either with control conditions or exogenous OPN. As shown in Fig. 4b, both the PI3‐K inhibitor and Akt inhibitor could minimize the impact of OPN treatment on Akt phosphorylation. These data firmly established that PI3‐K and Akt activities were required for the contribution of OPN to promote cellular survival under serum‐free conditions. In addition, HO‐8910 cells under serum‐free conditions that constitutively overexpress OPN displayed a significantly lower level of p53 (Fig. 4c).
Figure 4.
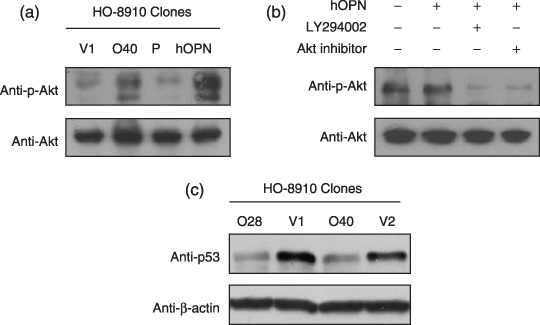
Osteopontin (OPN) activated the PI3‐K/Akt survival pathway and suppressed p53 expression induced by serum starvation. (a) OPN enhanced Akt phosphorylation on Ser 473 induced by serum starvation. The OPN‐overexpressing clone (O40) and the control clone (V1) were plated with serum overnight, followed by serum starvation for 3 days, and the total protein was lyzed for detection of the phosphorylation of Akt on Ser 473. The parental HO‐8910 cells were serum‐starved overnight, and treated with 200 ng/mL human OPN (hOPN) for 30 min before the detection of the phosphorylation of Akt on Ser 473. (b) PI3‐K activity is necessary for OPN‐mediated Akt phosphorylation on Ser 473. Serum‐starved parental HO‐8910 cells were pretreated with LY294002 (50 µM), Akt inhibitor (15 µM), respectively, for 3 h, followed with phosphate‐buffered saline or human OPN (200 ng/mL) treatment for 30 min, and the total protein was lyzed for detection of the phosphorylation of Akt on Ser 473. (c) OPN down‐regulated cellular level of p53 protein induction by serum starvation. The OPN‐overexpressing clones and control clones were incubated with serum overnight, followed by starvation for 3 days; the total protein was lyzed for p53 detection and analyzed by Western blot.
Inhibition of the PI3‐K/Akt signaling pathway attenuated OPN‐mediated HIF‐1α up‐regulation. To further explore the molecular mechanism by which OPN promotes cellular survival, we examined the potential effect of OPN on the activity of HIF‐1α that plays a key role during cancer development under hypoxia conditions. To this end, we found that OPN could increase the cellular level of HIF‐1α protein under normoxia. As shown in Fig. 5a, under normoxic conditions, HIF‐1α was readily detected in the OPN‐producing HO‐8910 cells, but not in the vector‐transfected cells. To define whether the impact of OPN on HIF‐1α up‐regulation is a primary or secondary effect to other cellular effects on long‐term OPN expression, we also examined the effect of exogenous human recombinant OPN on HIF‐1α of the parental HO‐8910 cells. Similarly, treatment of HO‐8910 cells with 400 ng/mL of OPN under the serum‐free conditions led to HIF‐1α up‐regulation (Fig. 5b).
Figure 5.
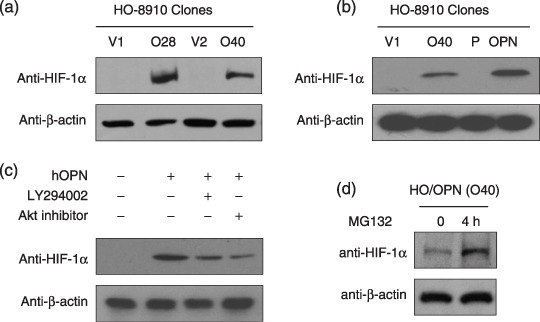
PI3‐K/Akt pathway inhibition attenuated osteopontin (OPN)–mediated hypoxia‐inducible factor‐1α (HIF‐1α) up‐regulation. (a) The OPN‐overexpressing clones (No. O28 and O40) and control clones (No. V1 and V2) were cultured until about 90% confluency under normoxic conditions, and the total protein was lyzed for HIF‐1α detection. (b) OPN up‐regulated the cellular level of HIF‐1α under normoxic conditions. The parental HO‐8910 cells were treated with 400 ng/mL human OPN (hOPN) for 24 h before HIF‐1α detection. (c) PI3‐K pathway inhibition attenuated OPN induction of HIF‐1α in HO‐8910 ovarian cancer cells. The parental HO‐8910 cells were pretreated with LY294002 (50 µM) or Akt inhibitor (15 µM) for 3 h and followed by either phosphate‐buffered saline or human OPN (400 ng/mL) treatment for 24 h, then lyzed for HIF‐1α detection. (d) OPN increased the level of HIF‐1α dramatically by inhibiting its ubiquitin‐proteasome degradation process. The OPN‐overexpressing cells (O40) were treated with MG132 (100 µM) for 4 h, then the total protein was lyzed for HIF‐1α detection. All blots were stripped and reprobed with anti‐β‐actin antibody for the loading control.
To elucidate the possible role of the PI3‐K/Akt pathway on OPN‐mediated HIF‐1α up‐regulation under normoxic conditions, we used PI3‐K inhibitor and Akt inhibitor to block the PI3‐K and Akt functions and examined the effect on the OPN‐mediated HIF‐1α up‐regulation. As shown in Fig. 5c, the chemical inhibitor of PI3‐K and Akt had a modest effect on the level of HIF‐1α. Recent reports indicated that some growth factors could increase the expression level of HIF‐1α protein either through inhibiting its degradation or stimulating its expression.( 16 ) To probe how OPN regulated the level of HIF‐1α, we pretreated the cells with MG132 that could specifically inhibit HIF‐1α degradation. As shown in Fig. 5d, after treatment with 100 µM MG132 for 4 h, the protein expression level of HIF‐1α increased dramatically. This result indicated that the mechanism by which OPN increased the cellular level of HIF‐1α is different from that of MG132 and CoCl2. Collectively, our data suggest that OPN protects ovarian cancer cells from stress‐induced death, which may be associated with the HIF‐1α up‐regulation.
Discussion
Cancer cells commonly develop mechanisms by which they resist cell death either through disruption of apoptotic processes or activation of survival signals. Within a growing tumor mass, the sequential acquisition of a number of genetic and epigenetic alterations during tumor progression also enable cancer cells to gain the ability to escape from apoptosis, induce angiogenesis, and metastasize to distant organs.( 14 , 33 ) We have previously demonstrated that a secreted protein called periostin which is overexpressed in colon cancers can activate the Akt signaling pathway through the αvβ3 integrins to augment the survival of both cancer cells and endothelial cells, and dramatically enhance metastatic growth of colon cancers.( 14 ) OPN is another secreted protein that plays an important role in the progression of tumor development.( 1 , 2 , 3 ) In this report, we demonstrated that OPN overexpression displayed a striking phenotype of greatly accelerated colony formation of the cells in soft agar in vitro and tumor growth in nude mice in vivo, which indicates that OPN overexpression may be associated with the tumorigenicity and late progression of ovarian cancer. Several reports suggested that MAPK,( 34 ) NF‐κB,( 11 ) and PI3‐K/Akt( 10 ) pathways may play roles in modulating OPN‐mediated cell survival, migration, and angiogenesis. Here we revealed that both endogenous and exogenous OPN could activate the PI3‐K/Akt survival pathway in human ovarian cancer cells. Our results suggest that the underlying mechanism of OPN‐mediated promotion of tumor development is largely associated with the ability of Akt activation to enhance cell survival under stresses. In this regard, acquired expression of OPN and similar types of proteins may enable tumor cells to thrive in the hypoxic microenvironment. The discovery of a link between OPN and the PI3‐K/Akt signaling pathway provides a molecular explanation for the prosurvival activity of OPN in the context of tumor progression.
The inner microenvironment of solid tumors is commonly hypoxic. How cancer cells adapt to the hypoxic condition in a solid tumor is critical to the tumor progression. Currently, intratumoral hypoxia has been increasingly considered as a critical microenvironmental factor that promotes tumor aggressiveness and metastasis. Stabilization of the HIF‐1α transcription complex can promote tumor progression and metastasis, leading to treatment failure and mortality in various human cancers.( 17 , 35 , 36 ) Here we found that OPN also could increase the cellular level of HIF‐1α protein. This may be another mechanism by which OPN stimulates the growth of ovarian tumor in vivo, since OPN did not show any effect on the proliferation of ovarian cancer cells in vitro. Furthermore, we also demonstrated that MG132 that specifically inhibits the process of HIF‐1α degradation could greatly increase the level of HIF‐1α in OPN‐producing cells. Therefore, the discovery of OPN‐mediated HIF‐1α up‐regulation may provide a molecular explanation for the prosurvival activity of OPN.
In addition to the induction of HIF‐1α expression by OPN, we had also observed that OPN could suppress p53 induction by serum starvation. Previous studies have suggested that HIF‐1α might promote or protect against apoptosis induced by serum deprivation or by anticancer agents in a cell type– and/or stimulus‐dependent manner. For example, HIF‐1α promoted cell death through an increase in p53 or other proapoptotic proteins such as BNIP3 or BNIP3L (NIX).( 37 , 38 ) On the contary, HIF‐1α exerted its antiapoptotic function by transcriptional activation of antiapoptotic proteins or transcriptional suppression of proapoptotic proteins, such as the Bcl‐2 family, p53, or by alterations in cellular energy metabolism through increased glucose uptake and glycolysis.( 39 , 40 ) Here, we found the proapoptotic protein p53 was down‐regulated by the induction of OPN. These data point to the prosurvival fuctions of OPN potentially through regulating the PI3‐K/Akt/HIF‐1α/p53 pathway. However, further investigations should be done to elucidate the molecular interplay between the HIF‐1α and p53 pathways under OPN induction.
In conclusion, the present study has demonstrated that acquired expression of OPN may enable ovarian cancer cells to thrive under stress conditions by up‐regulating the cellular level of HIF‐1α protein and activating the PI3‐K/Akt survival pathway. These findings indicate that OPN is closely associated with the progression of ovarian cancer, and OPN may be considered as a useful molecular target for the therapeutic development of ovarian cancer.
Supporting information
Fig. S1. Overexpression of osteopontin (OPN) in human ovarian cancer tissues. OPN expression in ovarian tissues by immunohistochemical analysis (×400). The tissue sections of the primary ovarian cancers and matched normal ovarian tissues were immunostained with an affinity‐purified anti‐OPN polyclonal antibody. The arrow indicates positive staining for OPN protein which was colored brown. All sections were counterstained with hematoxylin which was colored blue.
Fig. S2. Overexpression of osteopontin (OPN) did not alter the rate of cell proliferation in vitro. HO/pcDNA3.1 and HO/OPN cells were cultured in six‐well plates. Cells were trypsinized and counted after Trypan blue staining of viable cells for 1, 3, and 6 days.
Fig. S3. No differences in cell growth between HO/pcDNA3.1 and HO/osteopontin (OPN) cells under hypoxic conditions. Cells were cultured in six‐well plates and treated for 5 days with 200 µM CoCl2 that mimicked hypoxia conditions. (a) Cells were observed under microscope (×100); and (b) cell numbers were counted after Trypan blue staining of viable cells.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 30370307, 30400239, 30570935) and the Program for New Century Excellent Talents in Xiamen University.
References
- 1. Wai PY, Kuo PC. Osteopontin: regulation in tumor metastasis. Cancer Metastasis Rev 2008; 27: 103–18. [DOI] [PubMed] [Google Scholar]
- 2. Weber GF. The metastasis gene osteopontin: a candidate target for cancer therapy. Biochim Biophys Acta 2001; 1552: 61–85. [DOI] [PubMed] [Google Scholar]
- 3. Rangaswami H, Bulbule A, Kundu GC. Osteopontin: role in cell signaling and cancer progression. Trends Cell Biol 2006; 16: 79–87. [DOI] [PubMed] [Google Scholar]
- 4. Shinohara ML, Lu L, Bu J et al . Osteopontin expression is essential for interferon‐alpha production by plasmacytoid dendritic cells. Nat Immunol 2006; 7: 498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhu B, Suzuki K, Goldberg HA et al . Osteopontin modulates CD44‐dependent chemotaxis of peritoneal macrophages through G‐protein‐coupled receptors: evidence of a role for an intracellular form of osteopontin. J Cell Physiol 2004; 198: 155–67. [DOI] [PubMed] [Google Scholar]
- 6. Miyauchi A, Alvarez J, Greenfield EM et al . Recognition of osteopontin and related peptides by an alpha v beta 3 integrin stimulates immediate cell signals in osteoclasts. J Biol Chem 1991; 266: 20 369–74. [PubMed] [Google Scholar]
- 7. Weber GF, Ashkar S, Glimcher MJ, Cantor H. Receptor–ligand interaction between CD44 and osteopontin (Eta‐1). Science 1996; 271: 509–12. [DOI] [PubMed] [Google Scholar]
- 8. Kaartinen MT, Linnala KA, Maenpaa PH. Cross‐linking of osteopontin by tissue transglutaminase increases its collagen binding properties. J Biol Chem 1999; 274: 1729–35. [DOI] [PubMed] [Google Scholar]
- 9. Lee JL, Wang MJ, Sudhir PR, Chen GD, Chi CW, Chen JY. Osteopontin promotes integrin activation through outside‐in and inside‐out mechanisms: OPN–CD44V interaction enhances survival in gastrointestinal cancer cells. Cancer Res 2007; 67: 2089–97. [DOI] [PubMed] [Google Scholar]
- 10. Lin YH, Yang‐Yen HF. The osteopontin‐CD44 survival signal involves activation of the phosphatidylinositol 3‐kinase/Akt Signaling pathway. J Biol Chem 2001; 276: 46 024–30. [DOI] [PubMed] [Google Scholar]
- 11. Hur EM, Youssef S, Haws ME, Zhang SY, Sobel RA, Steinman L. Osteopontin‐induced relapse and progression of autoimmune brain disease through enhanced survival of activated T cells. Nat Immunol 2007; 8: 74–83. [DOI] [PubMed] [Google Scholar]
- 12. Rittling SR, Chambers AF. Role of osteopontin in tumor progression. Br J Cancer 2004; 90: 1877–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 14. Bao S, Ouyang GL, Bai XF et al . Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the Akt/PKB pathway. Cancer Cell 2004; 5: 329–39. [DOI] [PubMed] [Google Scholar]
- 15. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia‐inducible factor 1 is a basic‐helix‐loop‐helix‐PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci 1995; 92: 5510–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Déry MAC, Michaud MD, Richard DE. Hypoxia‐inducible factor 1: regulation by hypoxic and non‐hypoxic activators. Int J Biochem Cell Biol 2005; 37: 535–40. [DOI] [PubMed] [Google Scholar]
- 17. Harris AL. Hypoxia: a key regulatory factor in tumour growth. Nat Rev Cancer 2002; 2: 38–47. [DOI] [PubMed] [Google Scholar]
- 18. Maxwell PH, Wiesener MS, Chang GW et al . The tumor suppressor protein VHL targets hypoxia‐inducible factors for oxygen‐dependent proteolysis. Nature 1999; 399: 271–5. [DOI] [PubMed] [Google Scholar]
- 19. Zundel W, Schindler C, Haas‐Kogan D et al . Loss of PTEN facilitates HIF‐1‐mediated gene expression. Gene Dev 2000; 14: 391–6. [PMC free article] [PubMed] [Google Scholar]
- 20. Dimova EY, Kietzmann T. The MAPK pathway and HIF‐1 are involved in the induction of the human PAI‐1 gene expression by insulin in the human hepatoma cell line HepG2. Ann NY Acad Sci 2006; 1090: 355–67. [DOI] [PubMed] [Google Scholar]
- 21. Zhong H, Chiles D, Feldser D et al . Modulation of hypoxia‐inducible factor 1α expression by the epidermal growth factor/phosphatidylinositol‐3 kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res 2000; 60: 1541–5. [PubMed] [Google Scholar]
- 22. Alvarez‐Tajado M, Alfranca A, Aragones J, Vara A, Landazuri MQ, Del Peso L. Lake of evidence for the involvement of the phosphoinositide 3‐kinase/Akt pathway in the activation of hypoxia‐inducible factors by low oxygen tension. J Biol Chem 2002; 277: 13 508–17. [DOI] [PubMed] [Google Scholar]
- 23. Arsham AM, Plas DR, Thompson CB, Simon MC. phosphoinositide 3‐kinase/Akt signaling is neither required for hypoxic stablilization of HIF‐1α nor sufficient for HIF‐1‐dependent target transcription. J Biol Chem 2002; 277: 15 162–70. [DOI] [PubMed] [Google Scholar]
- 24. American Cancer Society . Cancer Facts and Figures 2008. Atlanta, GA: American Cancer Society, 2008, 1–70. Available from URL: http://www.cancer.org. [Google Scholar]
- 25. Dann RB, Kelley JL, Zorn KK. Strategies for ovarian cancer prevention. Obstet Gynecol Clin North Am 2007; 34: 667–86. [DOI] [PubMed] [Google Scholar]
- 26. Wong KK, Cheng RS, Mok SC. Identification of differentially expressed genes from ovarian cancer cells by MICROMAX cDNA microarray system. Biotechniques 2001; 30: 670–5. [DOI] [PubMed] [Google Scholar]
- 27. Schorge JO, Drake RD, Lee H et al . Osteopontin as an adjunct to CA125 in detecting recurrent ovarian cancer. Clin Cancer Res 2004; 10: 3474–8. [DOI] [PubMed] [Google Scholar]
- 28. Visintin I, Feng Z, Longton G et al . Diagnostic markers for early detection of ovarian cancer. Clin Cancer Res 2008; 14: 1065–72. [DOI] [PubMed] [Google Scholar]
- 29. Shi M, Cai Q, Yao L, Mao Y, Ming Y, Ouyang G. Antiproliferation and apoptosis induced by curcumin in human ovarian cancer cells. Cell Biol Int 2006; 30: 221–6. [DOI] [PubMed] [Google Scholar]
- 30. Song G, Mao YB, Cai QF, Yao LM, Ouyang GL, Bao SD. Curcumin induces human HT‐29 colon adenocarcinoma cell apoptosis via activating p53 and regulating apoptosis‐related proteins. Braz J Med Biol Res 2005; 38: 1791–8. [DOI] [PubMed] [Google Scholar]
- 31. Zhang GX, He B, Weber GF. Growth factor signaling induces metastasis gene in transformed cells: molecular connection between Akt kinase and osteopontin in breast cancer. Mol Cell Biol 2003; 23: 6507–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med 2005; 9: 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cavallaro U, Christofori G. Molecular mechanisms of tumor angiogenesis and tumor progression. J Neurooncol 2000; 50: 63–70. [DOI] [PubMed] [Google Scholar]
- 34. Rangaswami H, Bulbule A, Kundu GC. Nuclear factor‐inducing kinase plays a crucial role in osteopontin‐induced MAPK/IkappaBalpha kinase‐dependent nuclear factor kappaB‐mediated promatrix metalloproteinase‐9 activation. J Biol Chem 2004; 279: 38 921–35. [DOI] [PubMed] [Google Scholar]
- 35. Weidemann A, Johnson RS. Biology of HIF‐1alpha. Cell Death Differ 2008; 15: 621–7. [DOI] [PubMed] [Google Scholar]
- 36. Yang MH, Wu MZ, Chiou SH et al . Direct regulation of TWIST by HIF‐1alpha promotes metastasis. Nat Cell Biol 2008; 10: 295–305. [DOI] [PubMed] [Google Scholar]
- 37. Carmeliet P, Dor Y, Herbert JM et al . Role of HIF‐1alpha in hypoxia‐mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998; 394: 485–90. [DOI] [PubMed] [Google Scholar]
- 38. Sowter HM, Raval RR, Moore JW, Ratcliffe PJ, Harris AL. HIF‐1‐dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res 2003; 63: 6130–42. [PubMed] [Google Scholar]
- 39. Kim JY, Ahn HJ, Ryu JH, Suk K, Park JH. BH3‐only protein Noxa is a mediator of hypoxic cell death induced by hypoxia‐inducible factor 1alpha. J Exp Med 2004; 199: 113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kilic M, Kasperczyk H, Fulda S, Debatin KM. Role of hypoxia inducible factor‐1 alpha in modulation of apoptosis resistance. Oncogene 2007; 26: 2027–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Overexpression of osteopontin (OPN) in human ovarian cancer tissues. OPN expression in ovarian tissues by immunohistochemical analysis (×400). The tissue sections of the primary ovarian cancers and matched normal ovarian tissues were immunostained with an affinity‐purified anti‐OPN polyclonal antibody. The arrow indicates positive staining for OPN protein which was colored brown. All sections were counterstained with hematoxylin which was colored blue.
Fig. S2. Overexpression of osteopontin (OPN) did not alter the rate of cell proliferation in vitro. HO/pcDNA3.1 and HO/OPN cells were cultured in six‐well plates. Cells were trypsinized and counted after Trypan blue staining of viable cells for 1, 3, and 6 days.
Fig. S3. No differences in cell growth between HO/pcDNA3.1 and HO/osteopontin (OPN) cells under hypoxic conditions. Cells were cultured in six‐well plates and treated for 5 days with 200 µM CoCl2 that mimicked hypoxia conditions. (a) Cells were observed under microscope (×100); and (b) cell numbers were counted after Trypan blue staining of viable cells.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item