

Abstract
We recently cloned a variant form of erythropoietin‐producing hepatocyte (Eph)B6, a member of the Eph receptor tyrosine kinase family. In the present study, we examined the expression of the EphB6 variant (EphB6v) in a panel of brain tumor cell lines and glioblastoma tissues and we found that EphB6v was preferentially expressed in malignant brain tumors, such as glioblastomas and anaplastic astrocytomas. The EphB6v has a unique 54 amino acid sequence at the C‐terminal that is not found in normal EphB6. Therefore, we attempted to identify antigenic peptides unique to EphB6v for immunotherapy. The two EphB6v‐derived peptides exhibited the ability to bind to human leukocyte antigen (HLA)‐A0201 molecules, and each of them was able to induce cytotoxic T lymphocytes in vitro in the peripheral blood mononuclear cells of HLA‐A2+ glioma patients. The cytotoxicity was mediated by peptide‐specific CD8+ T cells in an HLA‐A2‐restricted manner. The expression of EphB6v was also observed in different types of cancer (e.g. lung, colon, stomach, liver and pancreatic) cells. Taken together, the two peptides derived from EphB6v might be appropriate targets for peptide‐based specific immunotherapy for HLA‐A2+ patients with various cancers. (Cancer Sci 2008; 99: 1656–1662)
The primary characteristics of malignant gliomas are their ability to infiltrate and invade the surrounding normal brain tissue, which results in a poor prognosis for patients with malignant glioma.( 1 , 2 ) Therefore, despite advances in neurosurgery, radiation and chemotherapy, the development of new treatment modalities is needed. One promising modality in the treatment of malignant tumors is specific immunotherapy, which induces tumor‐reactive cytotoxic T lymphocytes (CTL).( 3 ) Recent advances in tumor immunology have led to the discovery of numerous cancer antigen peptides that are recognized by cancer‐specific CTL and that have been applied in clinical trials as cancer vaccines.( 4 , 5 , 6 , 7 ) A personalized peptide vaccination strategy recently developed by our group has shown some clinical benefits in cases of malignant gliomas.( 8 ) However, the characteristics of cancer cells and immunological status with regard to cancer showed high clinical heterogeneity among patients, even in those with the same histological types of cancer and identical major histocompatibility complex (MHC) types. In this regard, a pool of vaccine peptides should be available from which suitable vaccine peptides can be chosen for individual patients with malignant gliomas. The identification of new antigen peptides related to malignant gliomas will be important for the development of specific immunotherapy for malignant glioma patients.
We have identified a new target molecule, erythropoietin‐producing hepatocyte (Eph)B6, of malignant gliomas using a cDNA microarray, which enabled us to comprehensively profile the gene expression, and to compare the genes in malignant glioma tissues with those in benign gliomas and normal brain tissue (Ryuya Yamanaka et al., unpublished data, 2008). EphB6 is a member of the Eph receptor tyrosine kinase family and is expressed in a variety of human tissues; EphB6 has been shown to lack protein kinase activity.( 9 , 10 ) In a previous study, we found an alternative splicing variant of EphB6.( 11 ) This variant form (EphB6v) lacks the transmembrane and cytoplasmic domains and has a unique 54 amino acid sequence encoded by intron 9; this sequence is not found in normal EphB6. Expression of EphB6v was detected in malignant glioma cell lines, but rarely found in the normal tissues; that is, only low level expression was detected in the testis, thymus and peripheral blood mononuclear cells (PBMC).( 11 ) In the same study, we identified two antigenic peptides unique to the 54 amino acid sequence of EphB6v that were recognized by CTL in a human leukocyte antigen (HLA)‐A24 restricted manner.( 11 )
In this study, we precisely analyzed the expression of EphB6v at the mRNA level in panels of brain tumor cell lines and malignant tumor tissues, as well as in a panel of various types of cancer cells from non‐brain tumors. We also attempted to identify epitope peptides unique to EphB6v recognized by CTL in a HLA‐A2, but not HLA‐A24, restricted manner, because HLA‐A2 is the most dominant allele of HLA worldwide and a larger number of cancer patients would therefore be expected to receive benefit from this approach.
Materials and Methods
Patients. PBMC were obtained from HLA‐A2+ malignant glioma patients and healthy donors after they had submitted their written informed consent. The institutional ethical review board of Kurume University approved study protocol. None of the participants was infected with HIV. The PBMC were prepared by Ficoll–Conray density gradient centrifugation. All of the samples were cryopreserved until used in the experiments. The HLA type (HLA‐A2+ or ‐A2−) was determined by flow cytometry using anti‐HLA‐A2 monoclonal antibody (mAb) (clone BB7.2) (One Lambda, Canoga, CA, USA).
Peptides. All of the peptides used in this study had more than 90% purity and were obtained from Hokkaido System Science (Sapporo, Japan) or GeneNet (Fukuoka, Japan). The names and sequences of the peptides are as follows: EphB6v‐3, SLLLISSLV; EphB6v‐4, SLAFRIPCL; EphB6v 472–486, GELFSLAFRIPCLRS; EBV‐A2 (an Epstein–Barr virus‐derived HLA‐A0201‐restricted peptide), GLCTLVAML; Flu‐A2 (an influenza virus‐derived HLA‐A0201‐restricted peptide), GILGFVFTL; HIV‐A2 (a HIV‐derived HLA‐A0201‐restricted peptide), SLYNTVATL; and EBV‐A24 (an Epstein–Barr virus‐derived HLA‐A2402‐restricted peptide), TYGPVFMCL. Peptides were dissolved in dimethylsulfoxide at 10 mg/mL and stored at –20°C.
Cell lines. The following brain tumor cell lines were used in this study: glioblastoma cell line T98G, KNS‐60, KNS‐81, KALS‐1, Becker, Marcus, U251MG (R596) and B2‐17; anaplastic astrocytoma Kings‐1, nos. 10 and 11; gliosarcoma KNS‐89, GI‐1; and astrocytoma SF‐126, SK‐MG‐1. These cell lines had previously been obtained from the Institute for Fermentation, Osaka, Japan, and were maintained by our laboratory. The following cell lines maintained in our cell laboratory were also used in this study: pancreas cancer Panc‐1, YPK‐1, MIA Paca‐2 and PK‐8; hepatocellular carcinoma HAK‐1B, HAK‐2, KIM‐1, HepG2 and KYN‐1; lung squamous cell carcinoma QG56, Sq‐1 and LC‐1‐Sq; lung adenocarcinoma LK87, PC‐9 and YM‐21; lung large cell carcinoma LC99A; lung small cell carcinoma LC65A; stomach cancer MKN‐7, SSTW‐9, NS‐8, MKN‐28 and MKN‐45; colon cancer COLO201, COLO205, COLO320, SW480 and KM12LM; and fibroblast cell line VA‐13. A TAP‐deficient HLA‐A2‐expressing lymphoblastic cell line, T2, was also used. RMA‐S‐A0201 and KNS‐81‐A0201 are stable transfectants expressing HLA‐A0201. The HLA‐A0201 gene, cloned into the expression vector pCR3.1,( 12 ) was cut by PvuI (Takara, Otsu, Japan) and transfected into RMA‐S cells by electroporation and into KNS‐81 cells using FuGENE6 transfection reagent (Roche, Mannheim, Germany). Stable cell lines were obtained by geneticin (1 mg/mL) selection, and the single clone was obtained by limiting dilution.
Reverse transcriptase polymerase chain reaction. Total RNA was purified from the cell lines using RNAzol B (Tel.Test, Friendswood, TX, USA). Template cDNA were prepared using the SuperScript Preamplification System for First‐Strand cDNA Synthesis (Invitrogen, Carlsbad, CA, USA). The primers were as follows: 5′‐TCAGCACTCATGCTACACTGG‐3′ (sense) and 5′‐TCTGCCTGGTCATAGTAGCG‐3′ (antisense) for EphB6, and 5′‐CCAAGCGTACCATCCAGTTT‐3′ (sense) and 5′‐CACGTTTGGCATACATCAGG‐3′ (antisense) for tubulin. Reverse transcription polymerase chain reaction (RT‐PCR) was performed using TaqDNA polymerase (ExTaq; Takara, Otsu, Japan) in a DNA thermal cycler (iCycler; Bio‐Rad Laboratories, Hercules, CA, USA) for 35 cycles of 98°C for 10 s, 55°C for 30 s and 72°C for 60 s, and the PCR products were separated by electrophoresis on 2% agarose gel. The sequences of the PCR products have been confirmed by DNA sequencing as previously reported.( 11 )
Western blot analysis. Rabbits were repeatedly immunized with EphB6v 472–486 peptide conjugated keyhole limpet hemocyanin (KLH) with TiterMax Gold adjuvant (Funakoshi, Tokyo, Japan). After six immunizations, sera were obtained from the rabbits and used for Western blot analysis. Cells were lysed with a buffer containing 10 mM Tris‐HCl, pH 7.4, 150 mM NaCl, 1% Triton X‐100, 0.2 mM phenylmethylsulfonyl fluoride, and 0.03 TIU/mL aprotinin for 30 min on ice, and centrifuged at 18000 g for 20 min at 4°C. The lysate was separated by 10% sodium dodecylsulfate polyacrylamide gel electrophoresis and transferred to a Hybond‐PVDF membrane (GE Healthcare, Buckinghamshire, UK) and incubated overnight at 4°C with 1:500 diluted anti‐EphB6v serum or pre‐immune control serum after blocking with BlockAce (Yukijirushi, Sapporo, Japan). Method of the subsequent detection of the proteins has been described previously.( 12 )
HLA stabilization assay. The ability of peptides to bind to HLA‐A0201 molecules was examined by a HLA stabilization assay using RMA‐S‐A0201 cells as previously reported( 12 , 13 ) with minor modifications. Briefly, the cells were cultured at 26°C for 20 h in RPMI‐1640 medium supplemented with 20% fetal bovine serum (FBS). Then, the cells were incubated in Opti‐MEM (Invitrogen) supplemented with human β2‐microglobulin (2 µg/mL) and various concentrations (1–100 µmol/L) of peptides at 26°C for 2 h, followed by incubation at 37°C for 3 h. After being washed with phosphate‐buffered saline, the cells were incubated with antihuman MHC class I mAb (PT85A; VMRD, Pullman, WA, USA) for 30 min on ice, followed by staining with Alexa Fluor 488 goat antimouse IgG (Invitrogen) for 30 min on ice. After washing, the fluorescence intensity of the cells was analyzed by EPICS‐XL (Beckman Coulter, Fullerton, CA, USA). Binding activity was evaluated by the mean fluorescence intensity of the HLA‐A0201 molecules. In this experiment, EBV‐A2 was used as a positive control, and EBV‐A24 was used as a negative control.
Induction of peptide‐specific CTL from PBMC. The methods used for the induction and detection of peptide‐specific CTL have been reported previously.( 14 ) Flu‐A2 and EBV‐A2 peptides were used as positive controls, and HIV‐A2 peptides were used as a negative control. PBMC (1 × 105 cells/well) were incubated with 10 µg/mL of each peptide in quadruplicate in a U‐bottom 96‐well microculture plate in 200 µL of culture medium consisting of 45% RPMI‐1640, 45% AIM‐V medium (Life Technologies, Gaithersburg, MD, USA), 10% FBS, 100 units/mL interleukin‐2 and 0.1 mmol/L MEM Nonessential Amino Acid Solution (Life Technologies). On days 13–15 of the culture period, cells in each well were washed and aliquoted into four wells. The cells in two of the wells were cultured with the corresponding peptide‐pulsed T2 cells, and those in the remaining two wells were cultured with HIV‐A2 peptide‐pulsed T2 cells. After 24–30 h of incubation at 37°C, the supernatants were collected and the γ‐interferon (IFN‐γ) concentration was measured by enzyme‐linked immunosorbent assay (ELISA). The assay was carried out in eight wells, and the results were considered positive if the values of the test peptide were significantly higher than those of the HIV‐A2 peptide at P < 0.05.
Cytotoxicity assay. Peptide‐stimulated PBMC were tested for their cytotoxicity against the glioma cell lines KNS‐81 (HLA‐A2−) and KNS‐81‐A0201 (HLA‐A2+) by a standard 6‐h 51Cr‐release assay. PBMC from HLA‐A2+ healthy donors were cultured with 20 µg/mL of phytohemagglutinin (PHA‐P; DIFCO, Detroit, MI, USA) for 3 days and were used as phytohemagglutinin (PHA) blasts. The effector cells were incubated with 2 × 103 cells/well of 51Cr‐labeled target cells at various effector/target (E/T) ratios in a U‐bottom 96‐well microculture plate. The specific lysis was calculated according to the following formula: % specific lysis = (test sample release – spontaneous release)/(maximum release – spontaneous release) × 100(%). Spontaneous release was determined using the supernatant of the target cells incubated with no effecter cells, and the maximum release was determined by a supernatant of the target cells incubated with 1% Triton‐X100 (Wako Pure Chemical, Osaka, Japan).
CD8+ T cells purified from the peptide‐stimulated PBMC using a CD8 Positive Isolation Kit (Dynal, Oslo, Norway) were used as effector cells for the cold‐target inhibition assay. Either control HIV‐A24 peptide‐ or corresponding peptide‐pulsed radioisotope‐unlabeled C1R‐A2402 cells (2 × 103 cells/well) were added to the cytotoxicity assay culture.
For the inhibition of cytotoxicity by mAb, 10 µg of anti‐HLA class I (W6/32, mouse IgG2a) or an isotype‐matched control (anti‐CD14) mAb was added to the wells at the initiation of the assay.
Results
Expression of EphB6v and EphB6 in various glioma cell lines and tissues. We examined the gene expression of EphB6 and EphB6v in cell lines of glioblastomas, anaplastic astrocytomas, gliosarcomas and astrocytomas by RT‐PCR (Fig. 1a). The majority of the cell lines expressed EphB6v mRNA, particularly glioblastoma and anaplastic astrocytoma cell lines expressed relatively high levels of EphB6v mRNA. In contrast, expression of EphB6 was more rarely detected in the tested cell lines. These results suggest that EphB6v rather than EphB6 is a more suitable target molecule for immunotherapy against malignant brain tumors. Expression of EphB6 and EphB6v at mRNA levels in glioblastoma tissues was also examined (Fig. 1b). All (8/8) and seven of the eight glioblastoma specimens expressed EphB6v and EphB6, respectively. Next, we examined the expression of EphB6v in cultured non‐tumor cells. PHA‐activated PBMC after 7 days culture (PHA‐blast) and fibroblast cell line VA‐13, as well as PBMC, expressed EphB6v (Fig. 1c). The expression of EphB6v in the PBMC has been reported previously.( 11 ) Semiquantitative amplification of template cDNA in this system was confirmed by using serial dilution of cDNA samples from KNS‐60 cells (Fig. 1d). To confirm the expression of EphB6v at protein level, we made antiserum specific for a unique sequence of EphB6v (amino acid position at 472–486) and analyzed by Western blot. An estimated molecular mass of EphB6v is 56 KDa and a clear band at 56 KDa was observed in KNS‐81, that was EphB6v positive at mRNA level, but only a background level of band was detected in U251MG (R596), that was EphB6v negative at mRNA level (Fig. 1e). These results suggest that EphB6v mRNA is actually translated to protein in the cells.
Figure 1.
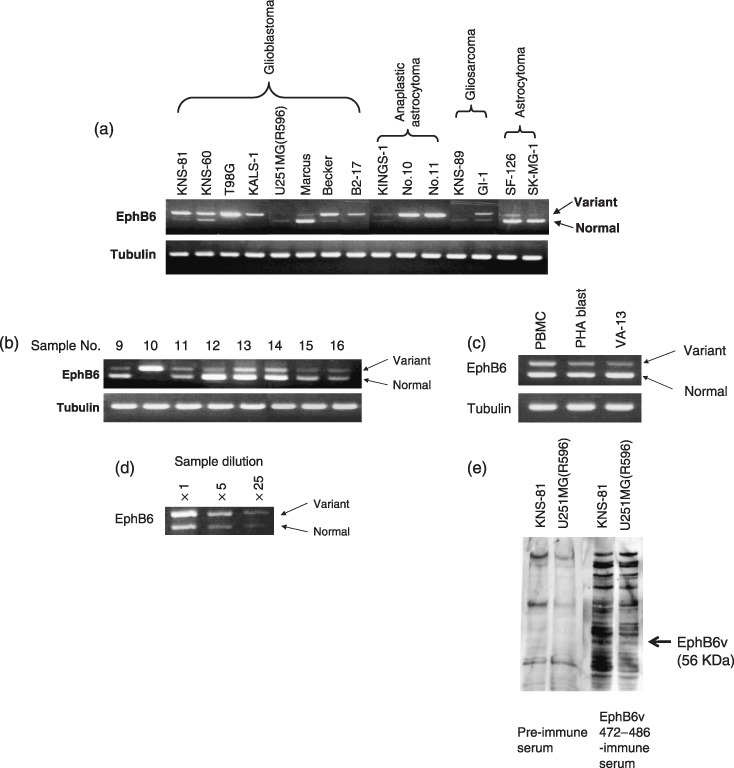
Comparison of erythropoietin‐producing hepatocyte (Eph)B6 expression in various cell lines and malignant glioma tissues. The mRNA levels of EphB6 and the EphB6 variant (EphB6v) in (a) brain tumor cell lines, (b) glioblastoma tissues and (c) non‐tumor cells were detected by reverse transcription polymerase chain reaction as described in Materials and Methods. Tubulin was used as an internal control. (d) Semiquantitative amplification of template cDNA in this system was confirmed by using serial dilution of cDNA samples from KNS‐60 cells. (e) The protein expression of EphB6v in brain tumor cell lines was confirmed by Western blot analysis using rabbit antiserum to EphB6v 472–486 peptide. PBMC, peripheral blood mononuclear cell; PHA, phytohemagglutinin.
Determination of CTL epitope peptides. The putative HLA‐A0201‐binding peptides derived from the unique 54 amino acid sequence of EphB6v were analyzed using BIMAS software (Bioinformatics and Molecular Analysis Section, National Institutes of Health; http://wye.cit.nih.gov/molbio/hla_bind/). Two 9‐mer peptides (EphB6v‐3 at positions 491–499 and EphB6v‐4 at positions 476–484) with the highest two binding scores were selected for further analyses. The BIMAS‐binding scores of the half‐life of the peptide‐MHC dissociation of EphB6v‐3 and ‐4 were 257 and 49, respectively. To confirm the binding of these two peptides to HLA‐A0201 molecules, we performed a HLA stabilization assay. RMA‐S‐A0201 cells incubated at 26°C exhibited a much higher HLA surface expression level, which rapidly decreased after incubation at 37°C due to the instability of HLA expression in the absence of the binding peptides.( 13 ) This assay system was used to evaluate the binding capacities of EphB6v peptides to HLA‐A2 molecules. As shown in Fig. 2, the EphB6v‐3 and EphB6v‐4 peptides, as well as the positive‐control EBV‐A2 peptides, led to the stabilization of HLA‐A0201 expression on RMA‐S‐A0201 cells, when compared to the effects of the negative‐control peptide (EBV‐A24).
Figure 2.
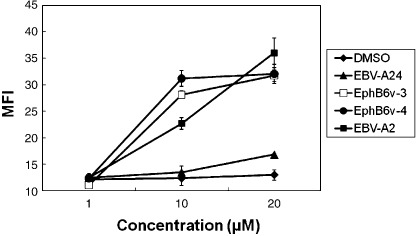
Binding capacity of erythropoietin‐producing hepatocyte B6 variants (EphB6v)‐3 and ‐4 peptides to human leukocyte antigen (HLA)‐A0201 molecules was examined by an HLA stabilizing assay using RMA‐S‐A0201 cells. Each point indicates the mean fluorescence intensity (MFI) of HLA expression from duplicate assays. DMSO, dimethylsulfoxide solvent alone; EBV, Epstein–Barr virus.
Induction of EphB6v peptide‐specific CTL from the PBMC of HLA‐A2+ donors. We next attempted to test the ability of the EphB6v‐3 and ‐4 peptides to induce peptide‐specific CTL from the PBMC of HLA‐A2+ patients with malignant gliomas. The PBMC were stimulated in vitro with each of the peptides or control peptides, and then the cultures were examined in terms of their IFN‐γ production in response to the corresponding peptide‐pulsed T2 cells. The results revealed that EphB6v‐3 and ‐4 successfully induced peptide‐specific CTL in five and seven of eight malignant glioma patients, respectively (Table 1). EBV‐A2 and Flu‐A2 peptides were used as positive controls. We also examined whether the EphB6v‐3 and ‐4 peptides had the potential to generate peptide‐specific CTL from HLA‐A2+ healthy donors, and we detected IFN‐γ production in seven and seven of nine healthy donors, respectively (Table 1). The number of positive wells per tested four‐well culture, each containing 1 × 105 cells/well, correlates with the approximate frequency of the CTL‐precursor. The mean numbers of positive wells per tested four‐well culture in the positive cases in the patients and healthy donors is 1.2 and 1.4, for EphB6v‐3, and 1.4 and 2.3 for EphB6v‐4, respectively. These results indicate that the EphB6v‐3 and ‐4 peptides possessed the ability to induce peptide‐specific CTL from both malignant glioma patients as well as healthy donors.
Table 1.
Cytotoxic T lymphocytes induction from human leukocyte antigen A2+ malignant glioma patients and healthy donors with erythropoietin‐producing hepatocyte B6 variants 3 and 4 peptides
| Donor | IFN‐γ production (pg/mL) † | |||
|---|---|---|---|---|
| EphB6v‐3 | EphB6v‐4 | EBV‐A2 | Flu‐A2 | |
| Glioma patients | ||||
| Pt 1 | 234 | 57, 78, 148, 513 | 137 | 76, 83 |
| Pt 2 | 49 | 89 | 445 | n.s. |
| Pt 3 | 42 | 290 | 55 | n.s. |
| Pt 4 | 17 | 65 | 1429 | 34, 101 |
| Pt 5 | n.s. | n.s. | 40 | n.s. |
| Pt 6 | n.s. | 83 | n.s. | n.s. |
| Pt 7 | n.s. | 46 | 97 | n.s. |
| Pt 8 | 78, 157 | >5000 | – | 129 |
| Positive cases/total cases | 5/8 | 7/8 | 6/8 | 3/8 |
| Mean number of positive wells in the positive cases ‡ | 1.2 | 1.4 | 1.0 | 2.0 |
| Healthy donors | ||||
| HD 1 | 366 | n.s. | n.s. | n.s. |
| HD 2 | n.s. | 342, 752, 1452 | n.s. | n.s. |
| HD 3 | n.s. | 437, 757, 861 | n.s. | 136 |
| HD 4 | 42 | 2356, >5000 | 44 | n.s. |
| HD 5 | 497 | n.s. | 123 | 179 |
| HD 6 | 261 | 716, >5000 | 31 | 55 |
| HD 7 | 24, 197 | 67, 137, 310 | – | 26 |
| HD 8 | 55 | 15, 64 | 33 | 47 |
| HD 9 | 55, 95, 172 | 727 | n.s. | 84, 88 |
| Positive cases/total cases | 7/9 | 7/9 | 4/9 | 6/9 |
| Mean number of positive wells in the positive cases ‡ | 1.4 | 2.3 | 1.0 | 1.1 |
Only statistically significant values are shown in this Table.
Mean number of positive wells per tested 4‐wells, each contained 1 × 105 cells/well, in the positive cases. EBV, Epstein–Barr virus; EphB6v, erythropoietin‐producing hepatocyte B6 variants; HD, healthy donor; IFN‐γ, γ‐interferon; n.s., not significant; Pt, patient.
Cytotoxicity of EphB6v peptide‐specific CTL from the PBMC of HLA‐A2+ donors. The cytotoxicity of EphB6v peptide‐induced CTL from the PBMC of HLA‐A2+ patients with malignant gliomas and healthy donors was further examined by 51Cr‐release assay. PBMC from the donors were repeatedly stimulated in vitro with each of the two peptides, and their cytotoxicity against KNS‐81 (EphB6v+HLA‐A2−) glioma cells, KNS‐81‐A0201 (EphB6v+HLA‐A2+) cells, U251MG (R596) (EphB6v−HLA‐A2+) and PHA blasts (EphB6v+HLA‐A2+) was measured. The PHA blasts from HLA‐A2+ healthy donors were used as a non‐tumorous control of HLA‐A2+ cells. As shown in Fig. 3, CTL induced from PBMC from both malignant glioma patients and healthy donors exhibited higher levels of cytotoxicity against KNS‐81‐A0201 cells than against parental KNS‐81 cells, U251MG (R596) and PHA blasts. The cytotoxicity of the CD8+ T cells purified from EphB6v‐3 or ‐4 peptide‐induced effector cells from both malignant glioma patients and healthy donors was significantly inhibited by the addition of cold targets; namely, isotope unlabeled T2 cells (HLA‐A2+) pulsed with the corresponding peptide, but not those pulsed with HIV peptide, inhibited cytotoxicity (Fig. 4). The cytotoxicity of the peptide‐induced CD8+ T cells from a patient against KNS‐81‐A0201 cells was also significantly inhibited by the addition of anti‐HLA class I mAb (W6/32), but not by the addition of isotype‐matched anti‐CD14 control mAb (Fig. 5). These results indicated that EphB6v‐3 or ‐4 peptide‐induced CTL do possess cytotoxic activity against malignant glioma cells in a HLA‐A2‐restricted and antigen‐specific manner.
Figure 3.
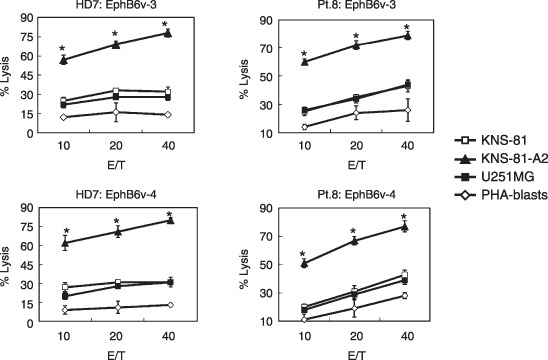
The cytotoxicity of erythropoietin‐producing hepatocyte B6 variants (EphB6v)‐3 (upper panels) and ‐4 (lower panels) peptide‐induced cytotoxic T lymphocytes from human leukocyte antigen (HLA)‐A2+ patients (right panels) and healthy donors (left panels) against KNS‐81 (EphB6v+HLA‐A2−), KNS‐81‐A0201 (EphB6v+HLA‐A2+), U251MG (R596) (EphB6v−HLA‐A2+), and phytohemagglutinin (PHA) blasts (EphB6v+HLA‐A2+) was examined by standard 6‐h 51Cr‐release assay. Representative results are shown. E/T, effector/target.
Figure 4.
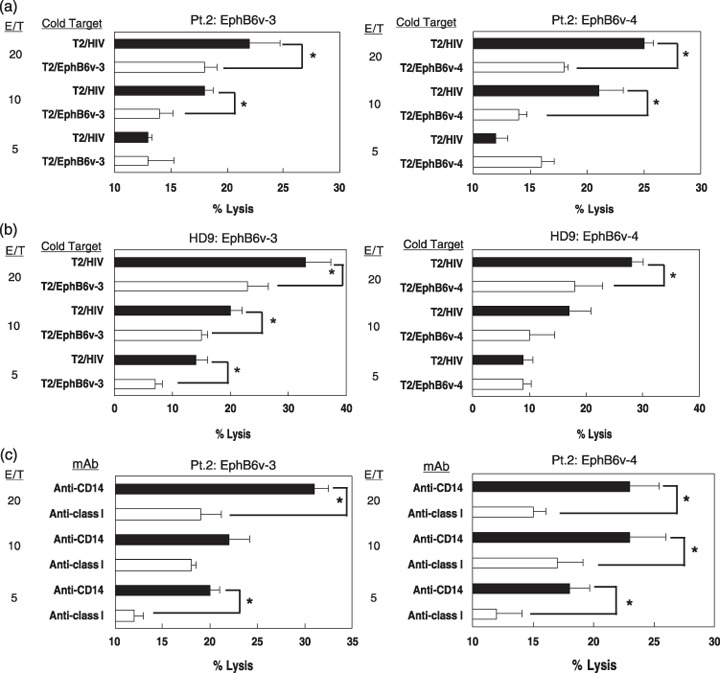
Cold‐target inhibition and antibody blocking of the cytotoxicity of erythropoietin‐producing hepatocyte B6 variant (EphB6v) peptide‐induced cytotoxic T lymphocytes (CTL) from malignant glioma patients and healthy donors. The purified CD8+ T‐cell fraction of peptide‐induced CTL from (a,c) human leukocyte antigen (HLA)‐A2+ malignant glioma patients or (b) healthy donors were tested for cytotoxicity against KNS‐81‐A0201 cells in the presence of cold T2 cells preloaded with either the corresponding peptide or the HIV peptide (a, b) or anti‐class I HLA (W6/32) mAb (c). Anti‐CD14 monoclonal antibody (mAb) was also used as an isotype‐matched control mAb. Cytotoxicity was significantly inhibited by the addition of either cold‐target cells or W6/32. *Values are statistically significant from that of corresponding controls as determined by Student's t‐test, P < 0.05. E/T, effector/target.
Figure 5.
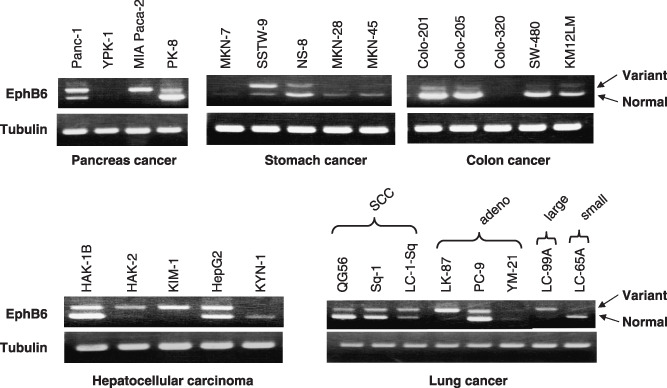
The expression of erythropoietin‐producing hepatocyte B6 (EphB6) was examined in pancreatic cancer, stomach cancer, colon cancer, liver cancer and lung cancer cell lines, as described in Materials and Methods. EphB6 expression was detected in all tested cancer cell lines; in particular, the EphB6 variant was preferentially expressed in hepatocellular carcinoma and in lung squamous and adenocarcinoma cell lines. Tubulin was used as an internal control. SCC, squamous cell carcinoma.
Expression of EphB6 genes in other cancer cell lines. RT‐PCR was also carried out to examine the gene expression of EphB6 and EphB6v in various types of cancer cells other than brain tumors, because the expression of EphB6v in brain tumors (Fig. 1) differs entirely from that of EphB6, and to date, no information has been obtained about the expression of EphB6v in other types of cancer cells. As shown in Fig. 6, EphB6v was frequently expressed in the other types of cancer cells studied; that is, pancreatic cancer (3/4), stomach cancer (4/5), colon cancer (4/5), hepatocellular carcinoma (4/5) and lung cancer (8/8, including three squamous cell carcinomas, three adenocarcinomas, one large cell carcinoma, and one small cell carcinoma). In particular, cell lines of pancreatic cancers, hepatocellular carcinomas and lung cancers expressed relatively high levels of EphB6v. The following expression frequencies for EphB6 were observed: pancreatic cancer (2/4), stomach cancer (4/5), colon cancer (4/5), hepatocellular carcinoma (3/5) and lung cancer (6/8).
Discussion
Among the 14 members of the Eph receptor tyrosine kinase family, EphB6 is the only kinase‐defective Eph receptor; this defect arises due to an alteration in the intracellular kinase domain.( 9 , 10 ) Generally, most kinase‐defective growth factor receptor proteins are associated with pathogenic conditions causative of human and other mammalian disorders.( 9 , 15 ) Nevertheless, EphB6 has been reported to be expressed in a variety of normal human tissues, such as the brain, heart, retina and thymus, as well as in blood cells.( 9 , 15 ) Expression of EphB6 has also been reported in human small cell lung carcinomas( 16 ) and neuroblastomas( 17 , 18 ) whereas no or low expression of EphB6 is observed in malignant melanomas and metastatic cancer.( 19 , 20 , 21 ) Furthermore, high levels of expression of EphB6 are predictive of a significantly favorable prognosis in human neuroblastomas.( 17 , 18 ) Therefore, EphB6 is considered to be an inhibitory factor with respect to the development of metastatic cancers. In this study, we investigated the expression of EphB6v in various types of brain tumors, and we found that the EphB6v was more preferentially expressed in malignant brain tumors, including glioblastomas and anaplastic astrocytomas. These results suggest that EphB6v might be a promotion‐related factor, but not an inhibitory factor, with respect to the progression of malignant tumors. Possible mechanisms of EphB6v on the progression of malignant tumors are as follows: (i) the secreted form of EphB6v competes with EphB6 for ephlin‐B ligand binding on the cell surface; and (ii) EphB6v makes a complex with unknown molecules on the cell surface through a C‐terminal unique sequence and provides antagonistic signals to the effects of EphB6.
Expression of EphB6v is not limited to malignant brain tumors, but is also found in other types of cancer cells, such as pancreatic cancers, hepatocellular carcinomas and lung cancers. These results suggest that EphB6v is a more suitable target molecule than EphB6 for immunotherapies against various types of malignant tumors. Therefore, we attempted to determine the antigenic peptides unique to EphB6v for immunotherapy. Two peptides unique to the 54 amino acid sequence of EphB6v exhibited the ability to bind to HLA‐A0201 molecules, and each of these peptides was able to induce CTL in vitro in a HLA‐A2‐restricted manner. We previously reported two EphB6v‐derived epitope peptides recognized by CTL in an HLA‐A24‐restricted manner.( 11 ) The approximate frequencies of the HLA‐A2 and ‐A24 populations are as follows: 40% and 60% in Japanese; 50% and 20% in Caucasians; and 30% and 12% in Africans, respectively.( 22 ) Therefore, the EphB6v‐derived peptides would be appropriate vaccine candidates for worldwide use in specific immunotherapy for the majority of patients with malignant tumors.
In conclusion, we identified two EphB6v‐derived peptides recognized by CTL in a HLA‐A2‐restricted manner that would be suitable for use as peptide vaccine candidates in patients with various types of malignant tumors. Present results also provide new insights into the functional roles of EphB6v not only in brain cancers, but also in other types of cancer.
References
- 1. Stupp R, Mason WP, Weller M et al . Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352: 987–96. [DOI] [PubMed] [Google Scholar]
- 2. Yamanaka R. Novel immunotherapeutic approaches to glioma. Curr Opin Mol Ther 2006; 8: 46–51. [PubMed] [Google Scholar]
- 3. Dranoff G, Jaffee E, Lazenby A et al . Vaccination with irradiated tumor cells engineered to secrete murine granulocyte‐macrophage colony‐stimulating factor stimulates potent, specific, and long‐lasting anti‐tumor immunity. Proc Natl Acad Sci USA 1993; 90: 3539–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004; 10: 909–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Itoh K, Yamada A. Personalized peptide vaccines: a new therapeutic modality for cancer. Cancer Sci 2006; 97: 970–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Srivastava PK. Therapeutic cancer vaccines. Curr Opin Immunol 2006; 18: 201–5. [DOI] [PubMed] [Google Scholar]
- 7. Lewis JJ. Therapeutic cancer vaccines: using unique antigens. Proc Natl Acad Sci USA 2004; 101: 14 653–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yajima N, Yamanaka R, Mine T et al . Immunologic evaluation of personalized peptide vaccination for patients with advanced malignant glioma. Clin Cancer Res 2005; 11: 5900–11. [DOI] [PubMed] [Google Scholar]
- 9. Matsuoka H, Iwata N, Ito M et al . Expression of a kinase‐defective Eph‐like receptor in the normal human brain. Biochem Biophys Res Commun 1997; 235: 487–92. [DOI] [PubMed] [Google Scholar]
- 10. Gurniak CB, Berg LJ. A new member of the Eph family of receptors that lacks protein tyrosine kinase activity. Oncogene 1996; 13: 777–86. [PubMed] [Google Scholar]
- 11. Jin M, Komohara Y, Shichijo S et al . Identification of EphB6 variant‐derived epitope peptides recognized by cytotoxic T‐lymphocytes from HLA‐A24+ malignant glioma patients. Oncol Rep 2008; 19: 1277–83. [PubMed] [Google Scholar]
- 12. Nakao M, Shichijo S, Imaizumi T et al . Identification of a gene coding for a new squamous cell carcinoma antigen recognized by the CTL. J Immunol 2000; 164: 2565–74. [DOI] [PubMed] [Google Scholar]
- 13. Takiguchi M, Matsuda T, Tomiyama H. Polarity of the P1 anchor residue determines peptide binding specificity between HLA‐A*3101 and HLA‐A*3303. Tissue Antigens 2000; 56: 501–6. [DOI] [PubMed] [Google Scholar]
- 14. Hida N, Maeda Y, Katagiri K, Takasu H, Harada M, Itoh K. A simple culture protocol to detect peptide‐specific cytotoxic T lymphocyte precursors in the circulation. Cancer Immunol Immunother 2002; 51: 219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shimoyama M, Matsuoka H, Tamekane A et al . T‐cell‐specific expression of kinase‐defective Eph‐afmily receptor protein, EphB6 in normal as well as transformed hematopoietic cells. Growth Factors 2000; 18: 63–78. [DOI] [PubMed] [Google Scholar]
- 16. Tang XX, Brodeur GM, Campling BG, Ikegaki N. Coexpression of transcripts encoding EphB receptor protein tyrosine kinases and their ephrin‐B ligands in human small cell lung carcinoma. Clin Cancer Res 1999; 5: 455–60. [PubMed] [Google Scholar]
- 17. Tang XX, Evans AE, Zhao H et al . High‐level expression of EPHB6, EFNB2, and EFNB3 is associated with low tumor stage and high TrkA expression in human neuroblastomas. Clin Cancer Res 1999; 5: 1491–6. [PubMed] [Google Scholar]
- 18. Tang XX, Zhao H, Robinson ME, Cohen B, Cnaan A, London W. Implications of EPHB6, EFNB2, and EFNB3 expressions in human neuroblastoma. Proc Natl Acad Sci USA 2000; 97: 10 936–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hafner C, Bataille F, Meyer S et al . Loss of EphB6 expression in metastatic melanoma. Int J Oncol 2003; 23: 1553–9. [PubMed] [Google Scholar]
- 20. Fox BP, Kandpal RP. Transcriptional silencing of EphB6 receptor tyrosine kinase in invasive breast carcinoma cells and detection of methylated promoter by methylation specific PCR. Biochem Biophy Res Co 2006; 304: 268–76. [DOI] [PubMed] [Google Scholar]
- 21. Fox BP, Kandpal RP. Invasiveness of breast carcinoma cells and transcript profile: Eph receptors and ephrin ligands as molecular markers of potential diagnostic and prognostic application. Biochem Biophys Res Co 2004; 318: 882–92. [DOI] [PubMed] [Google Scholar]
- 22. Imanishi T, Akazawa T, Kimura A, Tokunaga K, Gojobori T. Allele and haplotype frequencies for HLA and complement loci in various ethnic groups. In: Tsuji K, Aizawa M, Sasazuki T, eds. HLA 1991, vol. 1. Oxford: Oxford Scientific Publications, 1992; 1065–220. [Google Scholar]