

Abstract
Cholangiocarcinoma (CCA) is a fatal disease with high resistance to anticancer drugs. This is probably in part due to enhanced resistance to apoptosis. We have previously shown that galectin‐3 (Gal‐3), a β‐galactoside‐binding lectin, is highly expressed in CCA tissues. In this study, we demonstrated further that Gal‐3 plays a direct role in anti‐apoptosis regardless of the apoptotic insults. The anti‐apoptotic activity and chemoresistance of CCA cells were related to Gal‐3 expression level. Suppression of Gal‐3 expression with siRNA stimulated apoptosis. siGal‐3‐K626 transiently depleted Gal‐3 expression to the baseline and dramatically induced apoptosis, while siGal‐3‐K402 suppressed Gal‐3 expression by 50% and provoked cell apoptosis, but only under apoptotic insults (hypoxic conditions or short UV radiation). These actions were reversed in Gal‐3 overexpressing CCA cells. The correlation between the degree of anti‐apoptotic activity and the level of endogenous Gal‐3 was demonstrated. Suppression of Gal‐3 expression in CCA cells with siGal‐3‐K402 significantly enhanced apoptosis induced by cisplatin or 5‐fluorouracil by approximately 10 times, whereas overexpression of Gal‐3 led to an increased resistance to drugs. In summary, the present study showed that the cellular level of Gal‐3 might contribute to the anti‐apoptotic activity and chemoresistance of CCA cells. Hence, Gal‐3 expression level in cancer cells or tissues may be a marker for predicting chemotherapeutic response, and Gal‐3 may be a specific gene‐targeting therapy option for treating CCA. (Cancer Sci 2009; 00: 000–000)
Cholangiocarcinoma (CCA), bile duct cancer, occurs with a varying frequency in different geographic regions of the world. It is rare in Western countries; however, in Khon Kaen, a province in the north‐east of Thailand, the incidences of CCA in male and female residents are highest in the world.( 1 ) The risk factor for CCA in this region has been shown to be the infection of liver fluke, Opisthorchis viverrini,( 2 ) whereas primary sclerosing cholangitis, hepatolithiasis, and choledochal cysts are risk factors for CCA in Western countries.
Cholangiocarcinoma (CCA) has traditionally resulted in a high mortality rate and poor prognosis. Although surgery is potentially curative in certain patients, failure has usually occurred due to recurrence. Adjuvant or neo‐adjuvant therapy by chemotherapeutic drugs has been shown to improve local control, provide palliation, and prolong survival in various cancers; however, this is uncommon for CCA, owing to its poor response to therapy.( 3 ) Currently, adjuvant therapy, which enhances the cytotoxicity of chemotherapeutic drugs or sensitizes tumor cells to anticancer drugs, is showing encouraging results. This approach may increase the effectiveness of chemotherapy and improve prognosis in patients with CCA.
Recently, we have reported that all 53 CCA tissues expressed galectin 3 (Gal‐3) regardless of histological type. A lower intensity was found in poorly differentiated CCA and was related to lymphatic invasion.( 4 ) Even though there is substantial evidence regarding Gal‐3 expression in various cancers, the significance of Gal‐3 in the carcinogenesis and progression of CCA is yet to be determined.
Galectins are a family of proteins characterized by their affinity for β‐galactoside, and the sequence similarities in the carbohydrate‐recognition domain.( 5 ) To date, at least 15 galectin members have been identified and classified according to their structures into proto, chimera, and tandem‐repeat types.( 6 ) Galectin‐3 (Gal‐3), a multifunctional protein, participates in a variety of biological events, for example cell adhesion, differentiation, proliferation, and apoptosis.( 5 , 7 , 8 ) Gal‐3 has recently been shown to play a role in anti‐apoptosis in several cell types.( 9 , 10 , 11 , 12 , 13 ) Peritoneal macrophages from Gal‐3‐deficient mice were more sensitive to apoptotic stimuli than those from control mice.( 14 ) Gal‐3 could inhibit epithelial cell apoptosis induced by staurosporine, cisplatin, genistein, and anoikis.( 9 , 10 , 11 , 13 ) In addition, transfecting Gal‐3 into epithelial cells provided them with resistance to apoptotic insult.( 9 , 10 , 11 , 12 , 13 )
In this study, we examined the role of Gal‐3 in anti‐apoptosis in CCA cell lines by using RNA interference (RNAi) to knockdown endogenous Gal‐3 expression. The possibility of using RNAi‐mediated knockdown of Gal‐3, in combination with a chemotherapeutic agent, is explored as a more effective treatment for CCA.
Materials and Methods
Cell culture. Four CCA cell lines were established from different histological grading of primary CCA tumors, KKU‐OCA17 from a well‐differentiated type, KKU‐M055 and KKU‐M214 from moderately differentiated types, and KKU‐100 from a poorly differentiated type, as described by Sripa, et al. ( 15 ) CCA cells were cultured in Ham’s F‐12 (Life Technologies, Rockville, MD, USA) supplemented with 10% fetal calf serum, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C and 5% CO2.
Short interference RNA transfection. The coding sequence of the Gal‐3 gene in the human genome was submitted to Ambion siRNA Target Finder (http://www.ambion.com/techlib/misc/siRNA_finder.html). Two siRNAs specific for human galectin‐3, siGal‐3‐K402 and siGal‐3‐K626, derived from the mRNA sequences beginning at nt 402 (5′‐GGTGCCTCGCATGCTGATAAC‐3′) and nt 626 (5′‐AAGTACTGGTTGAACCTGACC‐3′), respectively, were purchased from JbioS (JbioS, Saitama, Japan). The lyophilized siRNAs were dissolved in annealing buffer, reheated to 95°C for 1 min, and incubated for 1 h at 37°C, by the protocol described previously.( 16 ) Transfection of both sequences was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. CCA cells (8 × 104 cells in 2 mL complete culture medium) were seeded into a six‐well plate for 24 h before transfection. The double‐stranded siRNAs (100 pm) were transiently transfected into CCA cell lines KKU‐100 and KKU‐M214 using (2 μg) Lipofectamine 2000 according to the manufacturer’s instructions. After 4 h, the medium was changed to complete culture medium (Ham’s F‐12 with 10% fetal calf serum), and siRNA treated cells were used in the in vitro study within 48–72 h after transfection. As a control, cells were treated with siRNA‐scramble (Ambion, Austin, TX, USA) or Lipofectamine 2000 under identical conditions.
Western blot analysis. Endogenous Gal‐3 levels were determined using Western blotting. Before cell lysate preparation, apoptotic cells were washed out with PBS and then the adhered cells were extracted with lysis buffer (8 m Urea, 4% CHAPS) containing a complete protease inhibitor cocktail (Roche Molecular Biochemicals, Mannheim, Germany). The lysate was subjected to 12.5% SDS–polyacrylamide gel electrophoresis,( 17 ) and the protein expression of Gal‐3 in CCA cells was determined by immunoblotting.( 18 ) Gal‐3 was detected by 1:10 000 anti‐Gal‐3 antibody (Chemicon, Temecula, CA, USA), or 1:5000 anti‐β‐actin antibody (Sigma‐Aldrich, St. Louis, MO, USA) as an internal control. The membrane was probed with horseradish peroxidase–conjugated antibody at 1:10 000 dilution (GE Healthcare, Buckinghamshire, UK). The immunoreactive proteins were visualized by Western Lightning Chemiluminescence Reagents (PerkinElmer, Boston, MA, USA). Quantitative analysis of Gal‐3/β‐actin expression was performed using the Gel‐Pro analyzer (Media Cybernetics, Silver Spring, MD, USA).
Time lapse assay. After 4 h of siRNA transfection, cells cultured in complete medium were placed under a time‐lapse microscope and cultured further for 72 h, at 37°C and 5% CO2. The images were obtained using an ×20 UPlan Apo objective (Olympus, Tokyo, Japan). The camera, shutters, and filter wheel were controlled by MetaMorph imaging software (Universal Imaging, Dawningtown, PA, USA), and the images were collected at 5‐min intervals for 48 h. Analysis was performed using MetaMorph software (Universal Imaging).
Construction of GFP Gal‐3 expression vector. Human Gal3‐inserted pET vector (Novagen, Madison, WI, USA) was a gift from Dr Ryuji Nagai (Department of Biochemistry, Kumamoto University School of Medicine, Kumamoto, Japan). DNA coding for Gal‐3 was cut from the vector at the EcoRI site, followed by insertion into the multiple cloning site of the vector pEGFP‐C1 (Clontech, Mountain View, CA, USA). The orientation of the construct was confirmed by digestion with SmaI, and the DNA sequence of the insertion was confirmed with a DNA sequencer (Applied Biosystems, Framingham, MA, USA). The plasmid was then transfected into KKU‐M055 using Lipofectamine 2000 according to the manufacturer’s instructions. After 24 h, the medium was changed to Medium G (Ham’s F‐12 medium containing 10% fetal calf serum and 0.6 mg/mL of G418 [Gibco, Carlsbad, CA, USA]). The cells were cultured in medium G for 2 weeks, and colonies resistant to G418 were screened by immunoblotting using the anti‐Gal‐3 antibody (Chemicon). Cells transfected with pEGFP‐C1 vector without Gal‐3 cDNA were used as a control.
Cell proliferation assay and drug treatment. After 24 h of siRNA transfection, cells were treated with various concentrations of cisplatin and 5‐fluorouracil (5‐FU) then incubated at 37°C, 5% CO2 for another 48 h. The number of cells was determined using a sulforhodamine B assay (SRB; Sigma‐Aldrich).( 19 ) Briefly, the culture medium was removed, and 10% cold trichloroacetic acid was added for 1 h at 4°C, and subsequently washed five times with deionized water. The plates were then air‐dried and 0.4% SRB in 1% acetic acid was added for 30 min. Unbound dye was washed out five times with 1% acetic acid. After air‐drying, SRB dye within cells were solubilized with 200 μL of 10 mm Tris‐base solution, and plates were shaken for at least 5 min. Absorbance was measured at 540 nm using a microplate reader (Tecan Austria, Salburg, Austria).
Hypoxic conditions and UV irradiation. After 24 h of siRNA transfection, the cells were grown further at 37°C under hypoxic conditions (5% CO2, 10% H2, and 85% N2) for 48 h, or exposed directly to UV radiation (250 mJoule/m2) for 4 s and then incubated at 37°C, 5% CO2 for 48 h. The number of cells was determined using the sulforhodamine B assay.
Immunocytofluorescence staining. Cells were fixed in 4% paraformaldehyde for 15 min, permeabilized with 0.2% Triton X‐100 for 5 min, and blocked with 1% bovine serum albumin in PBS for 1 h. Then, cells were incubated in 1:8000 of mouse monoclonal anti‐Gal‐3 antibody (Chemicon) for 1 h, then 1:1000 FITC goat antimouse IgG (BioSource, Camarillo, CA, USA) and 1:200 Hoechst 33342 (Molecular Probes, Eugene, OR, USA) for 1 h. The stained cells were observed using immunofluorescent microscope (Olympus).
Detection of apoptotic cells by flow cytometry using annexin V assay. After 24 h of siRNA transfection, cells were gently trypsinized and washed with ice‐cold PBS. The cell pellet was resuspended in 100 μL of Annexin‐V‐FLUOS labeling solution (Roche, Penzberg, Germany) and incubated at room temperature for 15 min. At least 20 000 cells were counted by flow cytometry (FACSCalibur flow cytometer) and data were analyzed using Cell Quest software (Becton Dickinson, San Jose, CA, USA).
Statistical analysis. Statistical analyses were performed using Sigma Stat version 3.1 software (Systat Software UK, London, UK). Results from flow cytometry, drug treatment, hypoxic condition, and UV irradiation are presented as mean ± SD, and the significance of the differences were addressed by Student’s t‐test. A P‐value of <0.05 was considered statistically significant.
Results
Suppression of Gal‐3 induced apoptosis. To address the functional importance of Gal‐3, we employed RNAi to transiently deplete the expression of Gal‐3 in two CCA cell lines, KKU‐100 and KKU‐M214, which had high expression levels of Gal‐3. Two siRNAs of 21‐mer oligoribonucleotides targeting Gal‐3, siGal‐3‐K402 and siGal‐3‐K626, were designed to suppress Gal‐3 expression. The efficiency of the siGal‐3s in suppression of endogenous Gal‐3 is shown in Figure 1. As shown by Western blot analysis, transfection of KKU‐100 CCA cells with siGal‐3‐K626 dramatically reduced Gal‐3 protein >90% within 24 h, whereas cells transfected with siGal‐3‐K402 showed partial suppression of Gal‐3 expression (∼50%) (Fig. 1A). The efficiency of the siRNA was observed as early as 6 h after siGal‐3 transfection and before apoptotic effect was observed (Fig. S1). Moreover, siGal‐3 treatment effectively suppressed the level of Gal‐3 in a time‐dependent manner, and the suppression effects were detected until 72 h after transfection (Fig. 1B). Similar results were observed for KKU‐M214 cells (data not shown). Neither siRNA‐scramble, nor Lipofectamine 2000, affected the level of Gal‐3 expression.
Figure 1.
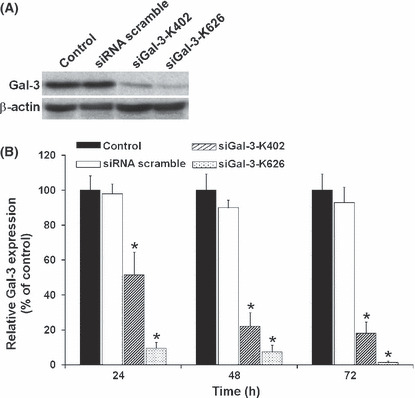
Galectin‐3 (Gal‐3) suppression by siGal‐3. Cholangiocarcinoma (CCA) cell line KKU‐100 cells were transfected with siGal‐3‐K626 or siGal‐3‐K402 or siRNA‐scramble using Lipofectamine 2000. (A) Expression of Gal‐3 protein by immunoblotting with anti‐Gal‐3 after siGal‐3 transfection for 72 h. (B) Relative Gal‐3 expression levels (% of control) after siGal‐3 transfection for 24 h, 48 h, and 72 h. Results are the mean ± SE (bars) of three independent experiments. *P < 0.05 compared with control cells.
Within 24 h of siGal‐3treatment, apoptotic nuclei were readily observed in the cells treated with siGal‐3‐K626, but not in those treated with siGal‐3‐K402 or in control cells treated with Lipofectamine 2000 (Fig. 2A). To determine the anti‐apoptotic activity of Gal‐3, KKU‐100 CCA cells transfected with siGal‐3‐K626 for 24 h were analyzed by flow cytometry using Annexin V and propidium iodide staining for early and late apoptotic cells. As shown in Figure 2(B), the number of Annexin V positive cells was much higher in KKU‐100 cells treated with siGal‐3‐K626 compared to the control. Gal‐3 suppressed cells underwent 70% apoptotic cell death, whereas only 5% of apoptotic cells were detected in the Gal‐3 expressing cells (P < 0.05) (Fig. 2C).
Figure 2.
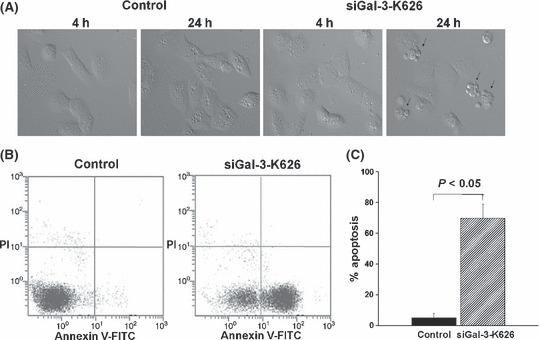
Induction of apoptosis in cholangiocarcinoma (CCA) cell lines treated with siGal‐3‐K626. (A) Cell morphology of control and siGal‐3‐K626‐treated cells was observed by time‐lapse microscopy at 4 h and 24 h after transfection. (B) Apoptotic cells were detected in siGal‐3‐K626‐treated cells using Annexin V and PI (propidium iodide) staining followed by flow cytometry. (C) The percentages of apoptotic cells were compared between siGal‐3‐K626‐treated cells and controls. Results are the mean ± SE of three independent experiments.
Suppression of Gal‐3 enhanced hypoxia and UV induced apoptosis. To demonstrate the anti‐apoptotic role of Gal‐3, CCA cell lines KKU‐100 and KKU‐M214, with or without transfection of siGal‐3‐K402, were induced to apoptosis by hypoxic conditions or UV irradiation. Compared to the parental control and siRNA‐scramble‐treated cells, the exposure of siGal‐3‐K402‐treated cells to hypoxic conditions for 48 h resulted in significantly more apoptotic cells: 82.1 ± 0.58% and 65.5 ± 1.2% for KKU‐100 and KKU‐M214, respectively (P < 0.05) (Fig. 3A). Similarly, a short UV irradiation induced apoptosis in siGal‐3‐treated cells more than it did in the controls (P < 0.05). Exposure of CCA cells to UV caused an increase of 29.4 ± 2.6% and 13.9 ± 1.2% in apoptotic cells in siGal‐3‐K402‐treated KKU‐100 and KKU‐M214, respectively (Fig. 3B). These results suggest a common role for Gal‐3 in the anti‐apoptotic process, regardless of the apoptotic inducer.
Figure 3.
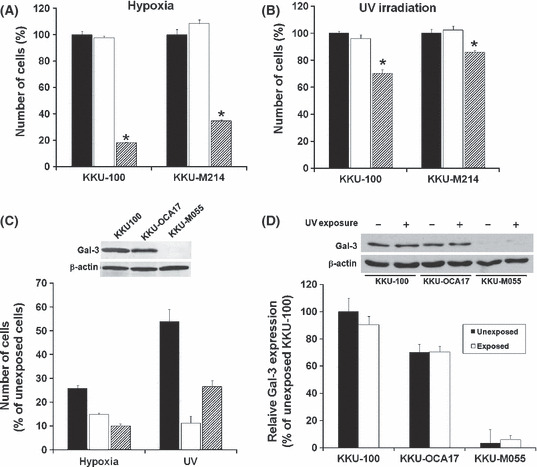
siGal‐3‐K402 enhanced apoptosis in cholangiocarcinoma (CCA) cells under hypoxia and UV‐induced apoptosis. CCA cell lines KKU‐100 or KKU‐M214 were treated with siRNA‐scramble or siGal‐3‐K402 for 24 h, then grown (A) under hypoxic conditions for 48 h or (B) briefly exposed to UV radiation, then incubated at 37°C and 5% CO2 for 48 h. The cell numbers were determined by sulforhodamine‐B (SRB) assay thereafter: siGal‐3‐K402‐treated cells (slash bar), parental cells (black bar), and siRNA‐scramble cells (white bar). (C) Effects of hypoxia and UV‐induced apoptosis on CCA cell lines with different endogenous galectin‐3 (Gal‐3) expressions: KKU‐100 (high expression, black bar), KKU‐OCA17 (moderate expression, white bar), and KKU‐M055 (low expression, slash bar). (D) Endogenous Gal‐3 expression of CCA cell lines after a short UV irradiation and culture for a further 48 h. Expressions of Gal‐3 in KKU‐100, KKU‐OCA17, and KKU‐M055 were determined by immunobloting (upper inset) and relative Gal‐3 expression level (% of unexposed KKU‐100) between UV‐unexposed cells (black bar) and UV‐exposed cells (white bar) were compared. Results are the mean ± SE of three independent experiments. *P < 0.05.
To investigate whether constitutive Gal‐3 expression in CCA cells affected resistance to apoptotic stress, the anti‐apoptotic activity of three CCA cell lines with different levels of Gal‐3 expression, KKU‐100 (high), KKU‐OCA17 (moderate), and KKU‐M055 (low), were examined under hypoxic conditions or UV irradiation. As shown in Figure 3(C), when comparing the corresponding control cells without apoptotic induction, the exposure of CCA cells to hypoxic conditions resulted in apoptosis of 74.3 ± 1.4% in KKU‐100, 85.3 ± 0.8% in OCA17, and 89.9 ± 0.8% in KKU‐M055. Similarly, the exposure of CCA cells to UV radiation induced apoptosis of 46.2 ± 5.2% in KKU‐100, 88.9 ± 3.9% in OCA17, and 73.4 ± 2.5% in KKU‐M055. These data reveal an important role of Gal‐3 in regulating cell responses to apoptotic insults. The anti‐apoptotic activity corresponded to the level of endogenous Gal‐3 expression: the higher the level of endogenous Gal‐3 expression, the higher the anti‐apoptotic activity.
To ensure that UV irradiation did not alter Gal‐3 expression, endogenous Gal‐3 protein levels in KKU‐100, KKU‐OCA17, and KKU‐M055 cells were determined 48 h after UV exposure. As shown in Figure 3(D), the levels of Gal‐3 expression in all cell lines were not varied after UV irradiation; hence the anti‐apoptotic response was based on the endogenous Gal‐3.
Overexpression of Gal‐3 rescued cells from apoptosis. To reveal the contribution of Gal‐3 to anti‐apoptosis, we next constructed an overexpressed Gal‐3 CCA cell line using GFP expression vector pEGFP‐C1‐hGal3. KKU‐M055, which had a low level of endogenous Gal‐3, was transfected with pEGFP‐C1‐hGal3 or the control vector (pEGFP‐C1), as controls. GFP‐expressing cells observed by phase‐contrast and fluorescence microscopy are shown in Figure 4(Aa,b). Transfection of KKU‐M055 cells with pEGFP‐C1‐hGal3 GFP vector resulted in 50–60% of GFP‐labeled cells. The efficiency of transfection was monitored via GFP expressing cells. Approximately 50–60% of KKU‐M055 cells were transfected with pEGFP‐C1‐hGal3. Moreover, cytofluorescence staining indicated that GFP‐Gal‐3 expressed in pEGFP‐C1‐hGal3‐transfected cells could function in a similar manner to endogenous Gal‐3 observed in KKU‐100 cells. GFP‐Gal‐3 was found predominantly in the nucleus (Fig. 4Ac,d,g–h), and it translocated from the nucleus to the cytoplasm under apoptotic insult (Fig. 4Ai,j), as it did in KKU‐100 cells (Fig. 4 Ae,f). The overexpression of Gal‐3 protein was confirmed by Western blot analysis (Fig. 4B). Gal‐3 expression in these cells was approximately 2.5‐fold higher than the parental cells or pEGFP‐C1‐treated cells (Fig. 4C).
Figure 4.
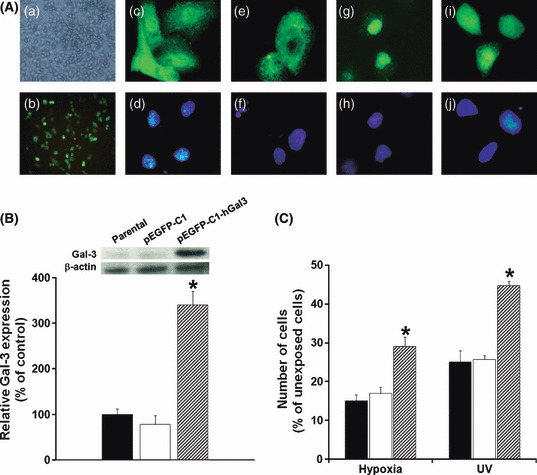
The anti‐apoptotic activity of galectin‐3 (Gal‐3) was demonstrated in Gal‐3 overexpressing cells. The CCA cell line, KKU‐M055, was transfected with the pEGFP‐C1‐hGal3 vector. The efficiency of the transfection was determined by (Aa) phase contrast, (Ab) GFP‐fluorescence, and (B) relative expression of Gal‐3 parental and vector pEGFP‐C1‐transfected cells determined by Western blotting of Gal‐3. The function of GFP‐Gal‐3 was shown to be similar to the native Gal‐3 of KKU‐100; (Ac,e) immunofluorescence of Gal‐3 in KKU‐100; (Ag,i) immunofluorescence of GFP‐Gal‐3 in KKU‐M055 transfected with pEGFP‐C1‐hGal3. (Ad,f,h,j) Hoechst 33342 staining for the nucleus. Endogenous Gal‐3 was prominent in the nucleus (Ac,g) and translocated to the cytoplasm under siGal‐3 treatment (Ad) or short UV exposure (Ai). (C) Effects of hypoxia and UV‐induced apoptosis on KKU‐M055 cells, as shown by comparing Gal‐3 overexpressing cells with controls. Results are the mean ± SE of three independent experiments. *P < 0.05.
The anti‐apoptotic role of Gal‐3 in pEGFP‐C1‐hGal‐3‐transfected cells was compared with the parental (KKU‐M055) cells and the control pEGFP‐C1 vector–transfected cells, by culturing cells in hypoxic conditions or short term exposure to UV irradiation. As shown in Figure 4(E), hypoxic conditions or short UV exposure dramatically induced cell apoptosis of parental and control cells to <20% and 25% of the unexposed cells, respectively. Resistance to these apoptotic insults, however, was demonstrated in the Gal‐3 overexpressing KKU‐M055 cells. The numbers of pEGFP‐C1‐hGal3‐transfected cells that remained in both apoptotic‐induced conditions were significantly higher than those of the control cells (P < 0.05) (Fig. 4E).
Suppression of Gal‐3 expression enhanced anticancer drug–induced apoptosis. To determine whether drug‐induced apoptosis was enhanced by suppression of Gal‐3 expression in CCA cell lines, siGal‐3‐K402 treated cells were cultured in the absence or presence of an anticancer drug for 48 h, and cell numbers were determined by the sulforhodamine‐B (SRB) assay. Different concentrations of cisplatin at 4, 40, 400 μg/mL, and 5‐FU at 1‐1000 μg/mL for KKU‐100 and 0.3–30 μg/mL for KKU‐M214, were tested.
The chemotherapeutic drugs cisplatin and 5‐FU induced apoptosis of CCA cells in a dose‐dependent manner (Fig. 5A–D). Suppression of Gal‐3 expression significantly enhanced drug‐induced apoptosis in comparison with that of the control. Knockdown of Gal‐3 expression in CCA cells significantly enhanced apoptosis induced by cisplatin at all concentrations tested (P < 0.05). Suppression of Gal‐3 improved the action of cisplatin by approximately 10 times. Cells treated with siGal‐3 or with a combination of 4 μm or 40 μm cisplatin reduced cell numbers to the same levels as cells treated with 40 μm or 400 μm cisplatin (Fig. 5A,C). Similar observations were obtained for cells treated with 5‐FU (Fig. 5B,D). These results indicated that the RNAi‐mediated knockdown of Gal‐3 increased cell sensitivity to cisplatin‐ and 5‐FU‐induced apoptosis.
Figure 5.
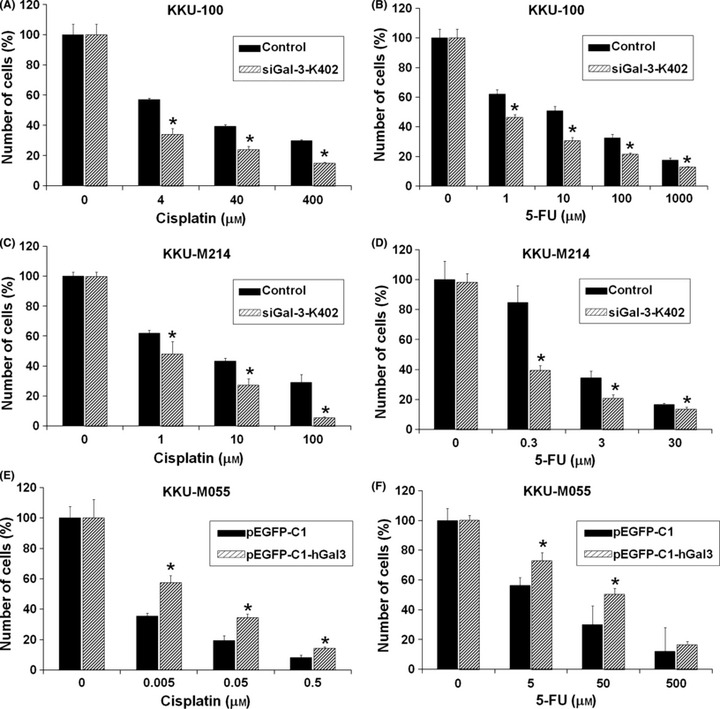
Galectin‐3 (Gal‐3) depletion enhances anticancer drug sensitivity whereas overexpressing Gal‐3 increases drug resistance. Cholangiocarcinoma (CCA) cell lines KKU‐100 or KKU‐M214 were treated with either Lipofectamine (black bar) or siGal‐3‐K402 (slash bar) for 24 h, followed by treatments with various concentrations of cisplatin (A,C) or 5‐fluorouracil (5‐FU) (B,D) for a further 48 h. KKU‐M055 cells were treated with pEGFP‐C1 (control, black bar) or pEGFP‐C1‐hGal3 (Gal‐3 overexpressing cells, slash bar) for 24 h, followed by treatments with various concentrations of cisplatin (E) or 5‐FU (F) for a further 48 h as above. The number of cells was determined by sulforhodamine‐B (SRB) assay. Results are the mean ± SE (bars) of three independent experiments. * P < 0.05.
To examine whether or not the enhancement of apoptosis shown in anticancer‐drug‐treated Gal‐3 knockdown cells was associated with Gal‐3, Gal‐3 overexpressing cells (pEGFP‐C1‐hGal3‐transfected KKU‐M055 cells) were treated with different concentrations of cisplatin (0.005, 0.05, 0.5 μg/mL) (Fig. 5E) and 5‐FU (5, 50, 500 μg/mL) (Fig. 5F). Overexpression of Gal‐3 significantly diminished the cisplatin‐ or 5‐FU‐induced apoptosis in KKU‐M055 cells (P < 0.05).
Discussion
Cholangiocarcinoma (CCA) is a fatal disease with poor prognosis and high recurrence. There is no effective treatment for CCA at present, probably due to its enhanced resistance to apoptosis. Accumulated evidence indicates that Gal‐3 has anti‐apoptotic effects in a variety of cells.( 9 , 10 , 11 , 12 , 13 ) In this study, it was demonstrated that Gal‐3 played a role in anti‐apoptosis; reduction of Gal‐3 expression significantly induced apoptosis in CCA cells and enhanced the responsiveness of CCA cells to chemotherapeutic agents.
The present study successfully established the RNA interference (RNAi)–mediated knockdown of Gal‐3 and an overexpression system for Gal‐3 in CCA cell lines. The anti‐apoptotic activity of Gal‐3 is dependent on the expression level of endogenous Gal‐3. Apoptosis was dramatically induced in CCA cells in which Gal‐3 expression was diminished to baseline levels with siGal‐3‐K626, whereas cells treated with siGal‐3‐K402 which suppressed Gal‐3 expression to 50%, did not induce apoptosis. Further experiment is needed to explore the mechanism that induces apoptosis in our model. However, there are a few reports related to the apoptotic pathway induced by Gal‐3 depletion. Inhibition of the phosphatidylinositol‐3‐kinase (PI3K)/Akt signaling pathway was shown to be involved in siGal‐3‐treated human papillary thyroid cancer cells.( 20 ) Gal‐3 also plays role in the Wnt/β‐catenin pathway. siRNA silencing of Gal‐3 expression inhibited T‐cell factor (TCF)‐receptor activity, decreased cytosolic β‐catenin level, and induced apoptosis in human colorectal cancer cells.( 21 )
A reduction in the anti‐apoptotic activity of Gal‐3 in siGal‐3‐K402‐treated cells, however, was readily observable when the cells were exposed to hypoxia or UV‐induced apoptotic conditions. Conversely, cells with overexpression of endogenous Gal‐3 resisted the above two apoptotic insults. This observation indicated that the GFP‐Gal‐3 protein produced in Gal‐3‐overexpressing cells was effectively active.
The correlation of anti‐apoptotic activity to the level of Gal‐3 expression was also demonstrated in this study. CCA cell line KKU‐100, with high endogenous Gal‐3, showed higher resistance to apoptotic inductions than did KKU‐OCA17 and KKU‐M055, which both had lower endogenous Gal‐3. These findings suggest the possibility of using expression level of Gal‐3 to predict the anti‐apoptotic potential of cancer cells or tissues toward chemotherapeutic drugs. This suggestion is supported by the evidence that CCA with high expression of Gal‐3 exhibited high chemoresistance whereas those with low expression of Gal‐3 were chemosensitive. Among the five human intrahepatic CCA cell lines (KKU‐100, KKU‐M055, KKU‐M156, KKU‐M214, and KKU‐OCA17), CCA cell line KKU‐100, with the highest Gal‐3 expression, was highly resistant to all the chemotherapeutic drugs tested, whereas KKU‐M055, which does not express Gal‐3 constitutively, was the most sensitive.( 22 ) However, this suggestion is a correlative observation and further experimental studies would be needed for a definitive conclusion.
Galectin‐3 (Gal‐3) seems to be a common key for the anti‐apoptotic phenomenon in biological systems. As shown in this study, despite the apoptotic inducers, for example hypoxia and UV irradiation, reduction of Gal‐3 expression significantly enhances apoptosis cell death of CCA cells in both conditions. In addition to our study, the anti‐apoptotic action of Gal‐3 in response to different apoptotic stimulations, anti‐Fas‐induced cell death and anoikis induced apoptosis, have been reported in B‐cell lymphocytes( 23 ) and human breast epithelial cells,( 10 ) respectively.
The finding that Gal‐3 plays a role in the anti‐apoptosis of CCA cells led to further evaluation as to whether reduction of Gal‐3 expression in CCA cells affected their response to the pro‐apoptotic action of chemotherapeutic agents. The combined effects of siGal‐3 and two chemotherapeutic agents, cisplatin and 5‐FU, widely used anticancer agents frequently provided to CCA patients, were evaluated in two CCA cell lines. As expected, the RNAi‐mediated knockdown of Gal‐3 synergistically enhanced the cytotoxicity of cisplatin and 5‐FU in both CCA cell lines. Gal‐3 knockdown by RNAi might reduce the anti‐apoptotic activity and hence enhance the cytotoxicity of these anticancer drugs. The direct association of Gal‐3 suppression with the enhancement of antitumor efficacy was shown in cells overexpressing Gal‐3. Increasing Gal‐3 expression in KKU‐M055 reduced the apoptotic response to anticancer drugs. The nuclear export of phosphorylated galectin‐3,( 24 ) translocation of Gal‐3 to the perinuclear membrane, and enrichment of Gal‐3 in mitochondria( 25 ) have been shown to be the mechanism by which galectin‐3 regulates its anti‐apoptotic activity in response to cisplatin in the BT‐549 human breast carcinoma cell line. In the present study, the translocation of Gal‐3 from the nucleus to cytoplasm was also observed under apoptotic insult. More experimental data are needed to indicate the definite mechanism by which Gal‐3 exerted its anti‐apoptotic activity in our study.
Galectin‐3 (Gal‐3) contains the NWGR (N, asparagine; W, tryptophan; G, glycine; R, arginine) anti‐death motif of the Bcl‐2 family. One of the mechanisms by which Gal‐3 acts on anti‐apoptosis in response to anticancer drugs has been shown to be the stimulation of Bcl‐2 associated death (Bad) protein phosphorylation and decreasing Bad expression, which resulted in the stabilization of mitochondrial membrane integrity, and subsequently the inhibition of cytochrome c release and caspase‐3 activation, which finally suppressed apoptosis.( 26 ) Additionally, an anti‐apoptotic effect of Gal‐3, which is meditated by the activation of the extracellular signal‐regulated kinases (ERK) and c‐Jun NH2‐terminal kinase (JNK) pathways, is also suggested.( 27 ) The overexpression of Gal‐3 probably protects cells from apoptotic death by inhibiting the caspase pathway,( 9 ) or by maintaining mitochondrial homeostasis.( 28 )
Resistance to chemotherapeutic agents is the major cause of failure for anticancer drug treatment in many cancers, including CCA. Thymidilate synthase, multidrug resistant protein (MRP1), multidrug resistant‐associated proteins (MDR3), and glutathione‐S‐transferase 1 (GSTP1) are highly expressed in several CCA cell lines, including KKU‐100 and KKU‐M214.( 22 ) The actions of these genes may underline the chemo‐resistant properties of CCA cells. The finding that the suppression of Gal‐3 expression enhances the sensitivity of antitumor drugs suggests the possibility that Gal‐3 could be a new target molecule for improving the response of CCA cells to chemotherapeutic drugs.
In conclusion, these results suggest that the expression of Gal‐3 provides tumor cells with an anti‐apoptotic advantage. Cells with high expression of Gal‐3 tend to resist chemotherapeutic treatment, whereas cells with low Gal‐3 expression are prone to apoptosis, and hence are sensitive to drug treatment. Based on this observation, Gal‐3 expression levels in cancer tissue may be a predictor of chemotherapeutic response, which could help clinicians select an appropriate treatment for CCA patients. Several approaches have been developed to reduce cellular Gal‐3 expression. For instance, siGal‐3 may be delivered into solid tumors by an efficient delivery system of siRNA( 29 ) that is becoming a conventional application for in vivo cancer therapy.( 30 , 31 ) Synthetic lactulose amines, for example lactulosyl‐L‐leucine and modified citrus pectin, have been designed and shown to be specific Gal‐3 inhibitors for therapeutic purposes.( 32 ) The treatment of cells with modified citrus pectin and lactulosyl‐L‐leucine was able to abrogate resistance to doxorubicin in angiosarcoma cells.( 33 ) Moreover, the inhibition of Gal‐3 anti‐apoptotic function by modified citrus pectin was sufficient to reverse multiple myeloma cells’ resistance to bortezomib, and enhance their response to apoptosis induced by dexamethasone.( 34 ) Since Gal‐3 is frequently expressed in CCA tissues, it is therefore proposed that interfering with Gal‐3 action, either by using RNAi‐mediated knockdown of Gal‐3, or specific Gal‐3 inhibitors, can either be developed as a specific gene‐targeting therapy to treat CCA, or used in combination with a chemotherapeutic agent to enhance apoptosis and chemosensitivity in CCA.
Supporting information
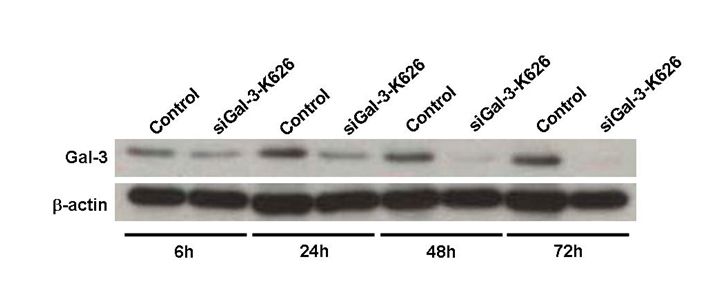
Fig. S1 Cholangiocarcinoma (CCA) cell line KKU‐100 cells were transfected with siGal‐3‐K626 and the endogenous galectin‐3 (Gal‐3) was determined by immunoblotting after siRNA transfection for 6, 24, 48, and 72 h. Expression of Gal‐3 was gradually suppressed and the effect of siRNA was observed at 6 h before apoptotic induction.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
{kind=link}
Acknowledgments
We are grateful for financial support from the Royal Golden Jubilee PhD Program (PHD/0140/2546) to M.J. and S.W.; Grant‐in‐Aid for Scientific Research on Priority Areas for Cancer Research from The Ministry of Education, Culture, Sports, Science and Technology, Japan (20014021) to Araki N; and the JSPS–Asia Africa Science Platform Program (Khon Kaen University and Kumamoto University). We also thank Professor James Will and Mr. Anthony W. Wilson for assistance with the English‐language presentation of the manuscript.
References
- 1. Vatanasapt V, Tangvoraphonkchai V, Titapant V, Pipitgool V, Viriyapap D, Sriamporn S. A high incidence of liver cancer in Khon Kaen Province, Thailand. Southeast Asian J Trop Med Public Health 1990; 21: 489–94. [PubMed] [Google Scholar]
- 2. Haswell‐Elkins MR, Mairiang E, Mairiang P et al. Cross‐sectional study of Opisthorchis viverrini infection and cholangiocarcinoma in communities within a high‐risk area in northeast Thailand. Int J Cancer 1994; 59: 505–9. [DOI] [PubMed] [Google Scholar]
- 3. Thongprasert S. The role of chemotherapy in cholangiocarcinoma. Ann Oncol 2005; 16 (Suppl 2): ii, 93–6. [DOI] [PubMed] [Google Scholar]
- 4. Junking M, Wongkham C, Sripa B, Sawanyawisuth K, Araki N, Wongkham S. Decreased expression of galectin‐3 is associated with metastatic potential of liver fluke‐associated cholangiocarcinoma. Eur J Cancer 2008; 44: 619–26. [DOI] [PubMed] [Google Scholar]
- 5. Barondes SH, Cooper DN, Gitt MA, Leffler H. Galectins. Structure and function of a large family of animal lectins. J Biol Chem 1994; 269: 20807–10. [PubMed] [Google Scholar]
- 6. Hirabayashi J, Kasai K. The family of metazoan metal‐independent beta‐galactoside‐binding lectins: structure, function and molecular evolution. Glycobiology 1993; 3 (4): 297–304. [DOI] [PubMed] [Google Scholar]
- 7. Hughes RC. Galectins as modulators of cell adhesion. Biochimie 2001; 83: 667–76. [DOI] [PubMed] [Google Scholar]
- 8. Yang RY, Liu FT. Galectins in cell growth and apoptosis. Cell Mol Life Sci 2003; 60: 267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Akahani S, Nangia‐Makker P, Inohara H, Kim HR, Raz A. Galectin‐3: a novel antiapoptotic molecule with a functional BH1 (NWGR) domain of Bcl‐2 family. Cancer Res 1997; 57 (23): 5272–6. [PubMed] [Google Scholar]
- 10. Kim HR, Lin HM, Biliran H, Raz A. Cell cycle arrest and inhibition of anoikis by galectin‐3 in human breast epithelial cells. Cancer Res 1999; 59 (16): 4148–54. [PubMed] [Google Scholar]
- 11. Lin HM, Moon BK, Yu F, Kim HR. Galectin‐3 mediates genistein‐induced G(2)/M arrest and inhibits apoptosis. Carcinogenesis 2000; 21: 1941–5. [DOI] [PubMed] [Google Scholar]
- 12. Yang RY, Hsu DK, Liu FT. Expression of galectin‐3 modulates T‐cell growth and apoptosis. Proc Natl Acad Sci U S A 1996; 93 (13): 6737–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoshii T, Fukumori T, Honjo Y, Inohara H, Kim HR, Raz A. Galectin‐3 phosphorylation is required for its anti‐apoptotic function and cell cycle arrest. J Biol Chem 2002; 277 (9): 6852–7. [DOI] [PubMed] [Google Scholar]
- 14. Hsu DK, Yang RY, Pan Z et al. Targeted disruption of the galectin‐3 gene results in attenuated peritoneal inflammatory responses. Am J Pathol 2000; 156 (3): 1073–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sripa B, Leungwattanawanit S, Nitta T et al. Establishment and characterization of an opisthorchiasis‐associated cholangiocarcinoma cell line (KKU‐100). World J Gastroenterol 2005; 11 (22): 3392–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Elbashir SM, Harborth J, Weber K, Tuschl T. Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods 2002; 26: 199–213. [DOI] [PubMed] [Google Scholar]
- 17. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227 (5259): 680–5. [DOI] [PubMed] [Google Scholar]
- 18. Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A 1979; 76 (9): 4350–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Skehan P, Storeng R, Scudiero D et al. New colorimetric cytotoxicity assay for anticancer‐drug screening. J Natl Cancer Inst 1990; 82 (13): 1107–12. [DOI] [PubMed] [Google Scholar]
- 20. Lin CI, Whang EE, Abramson MA et al. Galectin‐3 regulates apoptosis and doxorubicin chemoresistance in papillary thyroid cancer cells. Biochem Biophys Res Commun 2009; 379: 626–31. [DOI] [PubMed] [Google Scholar]
- 21. Shi Y, He B, Kuchenbecker KM et al. Inhibition of Wnt‐2 and galectin‐3 synergistically destabilizes beta‐catenin and induces apoptosis in human colorectal cancer cells. Int J Cancer 2007; 121: 1175–81. [DOI] [PubMed] [Google Scholar]
- 22. Tepsiri N, Chaturat L, Sripa B et al. Drug sensitivity and drug resistance profiles of human intrahepatic cholangiocarcinoma cell lines. World J Gastroenterol 2005; 11 (18): 2748–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hoyer KK, Pang M, Gui D et al. An anti‐apoptotic role for galectin‐3 in diffuse large B‐cell lymphomas. Am J Pathol 2004; 164 (3): 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takenaka Y, Fukumori T, Yoshii T et al. Nuclear export of phosphorylated galectin‐3 regulates its antiapoptotic activity in response to chemotherapeutic drugs. Mol Cell Biol 2004; 24: 4395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yu F, Finley RL Jr, Raz A, Kim HR. Galectin‐3 translocates to the perinuclear membranes and inhibits cytochrome c release from the mitochondria. A role for synexin in galectin‐3 translocation. J Biol Chem 2002; 277: 15819–27. [DOI] [PubMed] [Google Scholar]
- 26. Fukumori T, Oka N, Takenaka Y et al. Galectin‐3 regulates mitochondrial stability and antiapoptotic function in response to anticancer drug in prostate cancer. Cancer Res 2006; 66 (6): 3114–9. [DOI] [PubMed] [Google Scholar]
- 27. Fukumori T, Takenaka Y, Yoshii T et al. CD29 and CD7 mediate galectin‐3‐induced type II T‐cell apoptosis. Cancer Res 2003; 63 (23): 8302–11. [PubMed] [Google Scholar]
- 28. Matarrese P, Tinari N, Semeraro ML, Natoli C, Iacobelli S, Malorni W. Galectin‐3 overexpression protects from cell damage and death by influencing mitochondrial homeostasis. FEBS Lett 2000; 473: 311–5. [DOI] [PubMed] [Google Scholar]
- 29. Takei Y, Kadomatsu K, Yuzawa Y, Matsuo S, Muramatsu T. A small interfering RNA targeting vascular endothelial growth factor as cancer therapeutics. Cancer Res 2004; 64 (10): 3365–70. [DOI] [PubMed] [Google Scholar]
- 30. Hingorani SR, Jacobetz MA, Robertson GP, Herlyn M, Tuveson DA. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res 2003; 63 (17): 5198–202. [PubMed] [Google Scholar]
- 31. Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus‐mediated RNA interference. Cancer Cell 2002; 2: 243–7. [DOI] [PubMed] [Google Scholar]
- 32. Rabinovich GA, Cumashi A, Bianco GA et al. Synthetic lactulose amines: novel class of anticancer agents that induce tumor‐cell apoptosis and inhibit galectin‐mediated homotypic cell aggregation and endothelial cell morphogenesis. Glycobiology 2006; 16 (3): 210–20. [DOI] [PubMed] [Google Scholar]
- 33. Johnson KD, Glinskii OV, Mossine VV et al. Galectin‐3 as a potential therapeutic target in tumors arising from malignant endothelia. Neoplasia (New York, NY) 2007; 9 (8): 662–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chauhan D, Li G, Podar K et al. A novel carbohydrate‐based therapeutic GCS‐100 overcomes bortezomib resistance and enhances dexamethasone‐induced apoptosis in multiple myeloma cells. Cancer Res 2005; 65 (18): 8350–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Cholangiocarcinoma (CCA) cell line KKU‐100 cells were transfected with siGal‐3‐K626 and the endogenous galectin‐3 (Gal‐3) was determined by immunoblotting after siRNA transfection for 6, 24, 48, and 72 h. Expression of Gal‐3 was gradually suppressed and the effect of siRNA was observed at 6 h before apoptotic induction.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item