

Abstract
Human papillomavirus (HPV) infection is a causative event for the development of uterine cervical carcinoma. Human papillomavirus (HPV) 16, 18, and 33 DNA has been also detected frequently in lung adenocarcinomas (AdCs) in East Asian countries; however, its prevalence in Japan remains unclear. We therefore screened for HPV 16/18/33 DNA in 297 lung AdCs in a Japanese population by multiplex PCR with type‐specific primers. As reported previously, HPV 16 DNA was detected in two cervical cancer cell lines, CaSki and SiHa, while HPV 18 DNA was detected in HeLa cells, and 0.1–1.0 copies of HPV‐DNA per cell were detectable by this method. However, with this method, none of the 297 lung AdCs showed positive signals for HPV 16/18/33 DNA, indicating that HPV‐DNA is not or is very rarely integrated in lung AdC genomes in the Japanese. Furthermore, none of the lung AdCs showed positive signals by nested PCR with HPV 16/18 type‐specific primers. Therefore, we further attempted to detect HPV 16/18/33 DNA in 91 lung cancer cell lines, including 40 AdC cell lines. Among them, 30 have been established in Japan and the remaining 61 in the USA. No HPV signals were obtained in any of the 91 cell lines by either multiplex or nested PCR, while the p53 gene was mutated in 81 of them including 35 of the 40 AdC cell lines. These results indicate that HPV 16/18/33 infection does not play a major role in the development of lung AdC in Japan nor in the USA. (Cancer Sci 2010)
Infection with human papillomavirus (HPV) is a critical event for the development of uterine cervical cancer.( 1 ) E6 protein, encoded by HPV, binds the host cellular tumor suppressor protein p53, and triggers its degradation through the ubiquitin pathway.( 2 , 3 ) Therefore, the biological significance of continuous p53 degradation by HPV‐E6 protein in cervical carcinoma is thought to be equivalent to that of p53 inactivation by genetic alterations in various other types of cancers in human carcinogenesis. The p53 gene is frequently inactivated in lung adenocarcinoma (AdC) by mutations and/or deletions of both alleles, and the prevalence of p53 mutations in lung AdC is approximately 50% with a higher incidence in smokers.( 4 , 5 ) However, p53 is not genetically altered in the other half of lung AdCs. Therefore, it is possible that p53 is inactivated by other mechanisms in lung AdC cells without p53 mutations. For this reason, there have been many reports investigating the involvement of HPV in lung AdC development. However, the prevalence of HPV infection in lung AdCs varies drastically among the reports.( 5 , 6 , 7 ) Recently, reasons for a wide variation in the prevalence of HPV infection in lung cancer were investigated by two systematic surveys of a large number of publications.( 6 , 7 ) A higher prevalence in Asia than in Europe was pointed out by these two investigations,( 6 , 7 ) and a higher prevalence in studies using HPV type‐specific primers than in those using consensus HPV primers was also pointed out in the latter investigation.( 7 ) In East Asia (Supplementary Table S1), a high incidence of HPV infection in lung AdC was reported from Taiwan (92.8%), China (46.9%), and Korea (55.1%).( 8 , 9 , 10 ) In particular, a high prevalence of HPV 16 and 18 infections was reported from Taiwan and China and of HPV 33 infection from Korea. In Japan, the incidence of HPV infection (0–19.4%) has been reported to be not as high as in other East Asian countries, but is still high enough to consider its involvement in lung AdC development.( 11 , 12 , 13 , 14 )
Taiwan, China, and Korea are geographically close to Japan and the people in these countries are ethnically also close to the Japanese. Therefore, in this study, we aimed to elucidate whether or not HPV 16, 18, and 33 are also involved in the development of lung AdC in Japan, as in Taiwan, China, and Korea. We applied a multiplex PCR method as well as a nested PCR method using type‐specific primers for detection of HPV 16, 18, and 33 DNA in 275 primary and 22 metastatic lung AdCs in Japanese, and also in 91 lung cancer cell lines established in either Japan or the USA. To validate the specificity and sensitivity of HPV detection methods, three cervical carcinoma cell lines were analyzed by the same methods. In 91 cell lines, the status of p53 mutations was comprehensively analyzed and the results were compared with several p53 databases to evaluate accurately the prevalence of p53 inactivation in lung cancers.
Materials and Methods
Patients and tissues. A total of 275 primary lung AdCs and 22 metastatic lung AdCs to the brain were obtained at surgery from patients treated at the National Cancer Center Hospital, Tokyo, and at Saitama Medical University Hospital. The tumors were pathologically diagnosed according to the tumor‐node‐metastasis classification of malignant tumors( 15 ) (Table 1). Tumor tissues were stored at –80°C until DNA extraction, and genomic DNA was extracted as previously described.( 16 ) This study was undertaken under the approval of the Institutional Review Board of the National Cancer Center.
Table 1.
Clinicopathologic characteristics of lung adenocarcinomas
| PCR | Primary tumor | Brain metastasis | ||
|---|---|---|---|---|
| Multiplex (%) | Nested (%) | Both (%) | ||
| No. of cases | – | 275 | 138 | 22 |
| Gender | Male | 161 (59) | 81 (59) | 15 (68) |
| Female | 114 (41) | 57 (41) | 7 (32) | |
| Age (years) | Mean | 60.7 | 62.0 | 57.3 |
| Range | 30–84 | 30–84 | 48–74 | |
| Pathological stage | I | 201 (73) | 124 (90) | – |
| II | 27 (10) | 6 (4) | – | |
| III | 45 (16) | 8 (6) | – | |
| IV | 2 (1) | 0 (0) | – | |
| Smoking history | Smoker | 71 (55) | 69 (58) | 15 (68) |
| Nonsmoker | 57 (45) | 51 (43) | 7 (32) | |
| Unknown | 147 | 18 | 0 | |
| p53 mutation | + | 34 (32) | 34 (33) | 16 (73) |
| – | 72 (68) | 70 (67) | 6 (27) | |
| ND | 169 | 34 | 0 | |
ND, not determined.
Cell line DNA. DNA from 91 lung cancer cell lines( 17 , 18 ) was screened for HPV‐DNA in its genome. These cell lines consisted of 40 AdCs, 11 squamous cell carcinomas (SqCs), two adenosquamous carcinomas (ASCs), nine large‐cell carcinomas (LCCs), 27 small‐cell lung carcinomas (SCLCs), and two others (one carcinoid tumor and one neuroendocrine tumor), as listed in Table 2. Detailed information will be provided upon request. DNA from three cervical carcinoma cell lines, CaSki, SiHa, and HeLa, and HPV 33 containing plasmid DNA, was used as positive controls for detection of HPV‐DNA.
Table 2.
Status of the p53 gene in 91 lung cancer cell lines
| No. | Cell line | Hist. | Amino acid | Nucleotide |
|---|---|---|---|---|
| Point mutation | ||||
| 1 | ABC1 | AdC | p.P278S | c.832C>T |
| 2 | CALU‐3 | AdC | p.M237I | c.711G>T |
| 3 | HCC44 | AdC | p.S94X+p.R175L | c.281C>G+c.524G>T |
| 4 | HCC78 | AdC | p.S241F | c.722C>T |
| 5 | HCC193 | AdC | p.R248Q | c.743G>A |
| 6 | HCC515 | AdC | p.L194F | c.580C>T |
| 7 | Ma10 | AdC | p.G245V | c.734G>T |
| 8 | Ma17 | AdC | p.Y126C | c.377A>G |
| 9 | Ma24 | AdC | p.R337C | c.1009C>T |
| 10 | H23 | AdC | p.M246I | c.738G>C |
| 11 | H441 | AdC | p.R158L | c.473G>T |
| 12 | H820 | AdC | p.T284P | c.850A>C |
| 13 | H1437 | AdC | p.R267P | c.800G>C |
| 14 | H1975 | AdC | p.R273H | c.818G>A |
| 15 | H2009 | AdC | p.R273L | c.818G>T |
| 16 | H2087 | AdC | p.V157F | c.469G>T |
| 17 | H2122 | AdC | p.Q16L+p.C176F | c.527G>T+c.47A>T |
| 18 | H2126 | AdC | p.E62X | c.184G>T |
| 19 | PC3 | AdC | p.R282W | c.844C>T |
| 20 | PC7 | AdC | p.H214R | c.641A>G |
| 21 | PC9 | AdC | p.R248Q | c.743G>A |
| 22 | PC14 | AdC | p.R248W | c.742C>T |
| 23 | RERF‐LCMS | AdC | p.R248L | c.743G>T |
| 24 | RERF‐LC‐OK | AdC | p.F113C | c.338T>G |
| 25 | VMRC‐LCD | AdC | p.R175H | c.524G>A |
| 26 | II‐18 | AdC | p.K164X | c.490A>T |
| 27 | H322 | AdC | p.R248L | c.743G>T |
| 28 | EBC1 | SqC | p.E171X | c.511G>T |
| 29 | LC1/Sq | SqC | p.M237I | c.711G>T |
| 30 | LK2 | SqC | p.V272M | c.814G>A |
| 31 | HCC15 | SqC | p.D259V | c.776A>T |
| 32 | H520 | SqC | p.W146X | c.438G>A |
| 33 | SK‐MES‐1 | SqC | p.E298X | c.892G>T |
| 34 | PC10 | SqC | p.G245C | c.733G>T |
| 35 | HCC366 | ASC | p.Y220C | c.659A>G |
| 36 | H596 | ASC | p.G245C | c.733G>T |
| 37 | Lu65 | LCC | p.E11Q | c.31G>C |
| 38 | Ma2 | LCC | p.R175H | c.524G>A |
| 39 | Ma25 | LCC | p.M237I | c.711G>T |
| 40 | H661 | LCC | p.R158L+p.S215I | c.473G>T+c.644G>T |
| 41 | H1155 | LCC | p.R273H | c.818G>A |
| 42 | PC13 | LCC | p.G334V | c.1001G>T |
| 43 | HCC33 | SCLC | p.C242Y | c.725G>A |
| 44 | Lu134 | SCLC | p.P278L | c.833C>T |
| 45 | Lu135 | SCLC | p.G244C | c.730G>T |
| 46 | Lu139 | SCLC | p.V157F | c.469G>T |
| 47 | N417 | SCLC | p.E298X | c.892G>T |
| 48 | H69 | SCLC | p.E171X | c.511G>T |
| 49 | H128 | SCLC | p.E62X | c.184G>T |
| 50 | H345 | SCLC | p.Y236C | c.707A>G |
| 51 | H446 | SCLC | p.Q154V | c.461G>T |
| 52 | H841 | SCLC | p.C242S | c.724A>T |
| 53 | H1184 | SCLC | p.G334V | c.1001G>T |
| 54 | H1450 | SCLC | p.L194R | c.581T>G |
| 55 | H1607 | SCLC | p.P151H | c.452C>A |
| 56 | H1963 | SCLC | p.V147D+p.H214R | c.440T>A+c.641A>G |
| 57 | H2107 | SCLC | p.K101X | c.301A>T |
| 58 | H2141 | SCLC | p.R209X | c.625A>T |
| 59 | H2171 | SCLC | p.Q144X | c.430C>T |
| 60 | H2195 | SCLC | p.V157F | c.469G>T |
| 61 | H1618 | SCLC | p.R248L | c.743G>T |
| 62 | H187 | SCLC | p.S241C | c.722C>G |
| 63 | H510 | SCLC | p.R282G | c. 844C>G |
| 64 | H1770 | Neuroendocrine | p.R248W | c.741‐742CC>TT |
| Small insertion/deletion (≤9 nucleotides) | ||||
| 1 | Ma29 | AdC | p.V121fs | c.363delT |
| 2 | H522 | AdC | p.P191fs | c.572delC |
| 3 | H1648 | AdC | p.L35fs | c.103‐104insT |
| 4 | HCC95 | SqC | p.G334fs | c.1000(‐1003) 1G del |
| 5 | H157 | SqC | p.L35fs+p.E298X | c.103‐104insT+c.892G>T |
| 6 | H727 | Carcinoid | p.Q165‐S166insYKQ | c.496‐497ins9 |
| Large deletion | ||||
| 1 | H358 | AdC | p? | Large deletion |
| 2 | H1299 | LCC | p? | Large deletion |
| Splicing‐site mutation | ||||
| 1 | H1703 | AdC | p.G262fs | g. Ivs8 +1g>t |
| 2 | H1819 | AdC | p.A307fs | g. Ivs9 +1g>t |
| 3 | H2347 | AdC | p.Y126fs | g.375G>A |
| 4 | H1650 | AdC | p.V225fs | g.Ivs6 ‐2a>g |
| 5 | Sq1 | SqC | p.Y126fs | g. Ivs4 +2t>c |
| 6 | H82 | SCLC | p.Y126fs | g.375G>T |
| 7 | H209 | SCLC | p.V225fs | g. Ivs6 ‐2a>t |
| 8 | H526 | SCLC | p.S33fs | g. Ivs3 ‐1g>c |
| 9 | H1339 | SCLC | p.I332fs | g.Ivs9 +1g>t |
| Wild type | ||||
| 1 | A427 | AdC | — | — |
| 2 | A549 | AdC | — | — |
| 3 | Ma12 | AdC | — | — |
| 4 | Ma26 | AdC | — | — |
| 5 | H1395 | AdC | — | — |
| 6 | H226 | SqC | — | — |
| 7 | Lu99A | LCC | — | — |
| 8 | H460 | LCC | — | — |
| 9 | Lu24 | SCLC | — | — |
| 10 | Ms18 | SCLC | — | — |
p, c, and g indicate protein, cDNA, and genomic DNA.
AdC, adenocarcinoma; ASC, adenosquamous carcinoma; LCC, large‐cell carcinoma;
SCLC, small‐cell lung carcinoma; SqC, squamous cell carcinoma.
Multiplex PCR with HPV type‐specific primers. Sequences for the E1 and L2 regions of HPV 16 and for the E1 region of HPV 18 and 33, together with the aminolevulinate, delta‐, synthase 1 (ALAS1) gene segment as an internal positive control, were simultaneously amplified by multiplex PCR in a single tube, as reported.( 19 ) The primer sequences are shown in Supplementary Table S2. Multiplex PCR was performed with Takara Taq (Takara, Shiga, Japan) with a volume of 50 μL containing 1× PCR buffer, 2.5 mM MgCl2, 0.2 mM dNTPs, 0.025 U Taq polymerase, 3 nM primers, and 10 ng template DNA. Amplifications were performed with the following cycling profiling using a GeneAmp PCR system 9700 apparatus (Applied Biosystems, Foster City, CA, USA): Taq polymerase activation by incubation at 95°C for 1 min, followed by 40 cycles of denaturation at 94°C for 30 s, annealing at 70°C for 90 s, and elongation at 72°C for 60 s. Five micro liters of the amplicons were analyzed by electrophoresis on 3% agarose gels and ethidium bromide staining.
Nested PCR with HPV type‐specific primers. Sequences from the upstream regulatory region (URR) to the E7 region of HPV 16 and HPV 18 were first amplified by PCR with outer primers, and the HPV 16 E6/E7 and HPV 18 E6 regions were secondly amplified by nested PCR with inner primers, as reported previously.( 20 ) The primer sequences are shown in Supplementary Table S2. The first round of PCR was performed under the following conditions: Taq polymerase activation at 95°C for 1 min, followed by 35 cycles of denaturation at 95°C for 1 min, annealing at 60°C for 1 min, and elongation at 72°C for 1 min. The second round of PCR was performed as follows: 95°C for 1 min, followed by 20 cycles of denaturation for 1 min at 95°C, 1 min of annealing at 60°C, and 1 min of elongation at 72°C. Polymerase chain reaction (PCR) was performed with a Takara Taq with a volume of 20 μL containing 1× PCR buffer, 0.2 mM dNTPs, 0.05 U Taq polymerase, 2 nM of primers, and 10 ng of template DNA for the first round PCR and 1 μL of the first round PCR products for the second round PCR using a GeneAmp PCR system 9700 apparatus (Applied Biosystems).
Mutation analysis of the p53 gene. A total of 106 of the 275 primary lung AdCs and all of the 22 metastatic lung AdCs were previously examined for mutations in exons 4–8 of the p53 gene by genomic PCR and direct sequencing.( 21 , 22 ) All of the 91 lung cancer cell lines were examined for mutations in exons 2–11 covering all the coding sequences of the p53 gene by genomic PCR and direct sequencing as previously described.( 18 , 23 ) Sequence data for the cell lines obtained in this study were compared with those of the Catalogue of Somatic Mutations in Cancer (COSMIC) (http://www.sanger.ac.uk/cosmic/).( 24 )
Results
Detection of HPV 16, 18, and 33 DNA by multiplex PCR. Recently, Nishiwaki et al. developed a rapid and sensitive multiplex PCR‐based HPV genotyping method that allows the simultaneous amplification of 16 different HPV genotypes in a single tube reaction.( 19 ) This method is based on the amplification of multiple HPV‐DNA sequences with a set of HPV type‐specific primers, and the HPV types are visually distinguished by the size of amplified fragments after separation by gel electrophoresis. Since DNA for HPV 16, 18, and 33 types has been frequently detected in lung AdC cells in East Asia, four primer sets for these three HPV types, in addition to a primer set for a control genome sequence, were used in this study. Two sets of primers were prepared for the amplification of the HPV 16 DNA( 19 ) because of a possible high prevalence of HPV 16 DNA integration in lung AdC genomes.
The sensitivity and specificity of this method was validated using genomic DNA from three cervical cancer cell lines, CaSki, SiHa, and HeLa, and a lung cancer cell line, A549. Human papillomavirus (HPV) 16 has been shown to be integrated into chromosomal DNA in the CaSki and SiHa cell lines, while HPV 18 is integrated in the HeLa cell line.( 25 , 26 , 27 ) A cell line with integration of HPV 33 was not available; therefore, HPV 33 containing plasmid DNA was mixed with A549 cell DNA as a ratio of one copy of HPV 33 DNA per diploid human genome. Specific DNA fragments for HPV 16, 18, and 33 of different sizes from each other were successfully amplified with the control genomic DNA fragment (Genome in Fig. 1) in CaSki, SiHa, and HeLa cells, as well as A549 cells mixed with HPV 33 DNA (Fig. 1). Two bands for HPV 16 DNA (HPV16‐U and HPV16‐L) were detected in CaSKi and SiHa cell DNA, while a band for HPV 18 DNA was detected in HeLa cell DNA. Human papillomavirus (HPV) 33‐specific DNA was amplified from the mixture of plasmid DNA and A549 cell DNA, while no HPV‐specific DNA was amplified from A549 cell DNA. Therefore, by this method, three different HPV types were successfully identified and distinguished by the difference in the sizes of amplified DNA. To determine the sensitivity of this method, each sample was serially diluted and mixed with A549 cell DNA to obtain genomic DNA with 0.1–1.0 copies of each HPV‐DNA. Approximately 600 copies of HPV 16 DNA are integrated in CaSki cells, one to two copies of HPV 16 DNA are integrated in SiHa cells, and 20–50 copies of HPV 18 DNA are integrated in HeLa cells.( 25 , 26 , 27 ) As shown in Figure 1, 0.1–1.0 copies of the HPV‐DNA sequence per cell were detected by this method. Therefore, this method allowed us to detect one copy of HPV 16, 18, and/or 33 DNA integrated in chromosomal DNA of human cells. Further validation of this method was performed using DNA isolated from 18 primary cervical cancers because the presence of the HPV 16/18 DNA in these tumors was previously determined by Southern blot analysis.( 28 , 29 ) Human papillomavirus (HPV) types detected by multiplex PCR analysis were completely the same as those by Southern blot analysis, and the sensitivity of multiplex PCR analysis for detection of HPV 16 DNA was higher than that of Southern blot analysis. Four cases negative for HPV 16 DNA by Southern blot analysis were positive by multiplex PCR analysis (data not shown). Therefore, we concluded that the sensitivity of the multiplex PCR analysis is higher than that of Southern blot analysis for detection of HPV 16 DNA in cancer cells.
Figure 1.
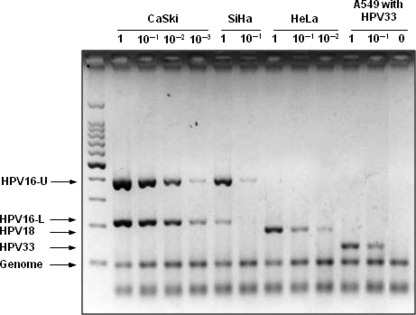
Detection of human papillomavirus (HPV) 16, 18, and 33 DNA in cervical cancer cell lines by multiplex PCR analysis. Specificity and sensitivity for detection of HPV 16, 18, and 33 DNA. Polymerase chain reaction (PCR) was performed using DNA from CaSki (∼600 copies of HPV 16 integrated), SiHa (1–2 copies of HPV 16 integrated), HeLa (20–50 copies of HPV 18 integrated), and A549 cells with/without HPV 33 containing plasmid DNA. Each sample was serially diluted with A549 cell DNA up to the copy number of 0.1–1.0 per cell for HPV‐DNA. Five micro liters of the amplicons were analyzed by electrophoresis on 3% agarose gels and ethidium bromide staining. 100 bp DNA Ladder (Takara, Shiga, Japan) was used as a size marker.
We then applied this method for detection of HPV 16/18/33 DNA in 275 primary lung AdCs and 22 metastatic lung AdCs to the brain (Table 1). However, HPV‐specific DNA was not amplified in any of these 297 lung AdCs. Thus, it was strongly suggested that HPV 16/18/33 DNA is not integrated in the chromosomal DNA of these lung AdCs.
Detection of HPV 16 and 18 DNA by nested PCR. It was reported that only a part of HPV‐DNA, from the URR to the E6/E7 region, is commonly integrated in chromosomal DNA of cervical cancer cells, and that deletions of other regions occur in the course of viral DNA integration into host cell DNA.( 25 , 26 , 30 ) The primers for HPV 16, 18, and 33 in the above multiplex PCR analysis were designed to amplify the E1 or L2 region (Supplementary Table S2).( 19 ) Therefore, it was possible that multiplex PCR analysis failed to detect the HPV‐DNA sequences because of integration of truncated HPV genomes without the E1 and L2 regions into host cell DNA. To pursue the possible integration of HPV 16 and 18 DNA in lung AdC cells, we performed a nested PCR analysis for the E6 and E7 regions of HPV 16 and 18. The URR to the E7 region of both HPV 16 and 18 genomes was first amplified using outer primers, then, the E6 to E7 region of the HPV 16 DNA and the E6 region of the HPV 18 DNA were amplified using inner primers (Supplementary Table S2), respectively, according to the method previously described.( 20 ) As in the multiplex PCR analysis, HPV 16‐ and 18‐specific DNA fragments were successfully amplified from the CaSki, SiHa, and HeLa cell lines, but not from A549. Next, 138 of the 275 primary AdCs and all of the 22 metastatic AdCs used for multiplex PCR analysis were subjected to nested PCR analysis (Table 1). However, none of them showed positive signals for the E6/E7 regions of the HPV 16 or 18. The results of multiplex PCR analysis as well as those of nested PCR analysis strongly indicated that HPV 16 and 18 are not integrated in lung AdCs developed in Japan, at least in the Tokyo area.
Absence of HPV 16, 18, and 33 DNA sequences in lung cancer cell lines. We next attempted to detect HPV 16, 18, and 33 DNA in a panel of 91 human lung cancer cell lines established in either Japan or the USA. Among the 91 cell lines, 30 originated from Japanese, 42 from Caucasians, and five from African‐Americans. Detailed information was not available for the remaining 14 cell lines. Forty cell lines were derived from AdC and the remaining 51 were from other histological types. Both multiplex PCR and nested PCR were performed on all of these cell lines. However, no HPV‐specific signals were obtained in any of these cell lines. Therefore, HPV 16, 18, and 33 DNA is not integrated in the 91 lung cancer cell lines established in Japan and the USA. Eleven cell lines were derived from SqC, and 27 cell lines were derived from SCLC; therefore, HPV 16/18/33 integration was not evident in any major histological types of lung cancer.
Status of p53 mutations in lung cancer cell lines. We previously examined for p53 mutations in 106 of the 275 primary tumors and all 22 brain metastases,( 21 , 22 ) and the mutations were detected in 34 of the 106 primary tumors (32%) and 16 of the 22 brain metastases (73%) (Table 1). We recently reported the status of p53 mutations in 87 of the 91 cell lines analyzed in the present study.( 18 ) In that study, mutation data of several cell lines were obtained not only by direct sequencing of the p53 coding regions but also from the COSMIC database, and the mutations were detected in 70 of the 87 cell lines (80%). However, during this study, we noticed that data for p53 mutations are not the same among three major databases, COSMIC, UMD_TP53 database (http://p53.free.fr), and IARC p53 database (http://www‐p53.iarc.fr).( 24 , 31 , 32 ) Absence of HPV 16/18/33 integration as well as p53 mutations in 17 lung cancer cell lines prompted us to re‐investigate the status of p53 mutations in these cell lines. Therefore, the p53 mutation status in all the 91 cell lines was determined by direct sequencing of all the coding exons, from exon 2 to exon 11, together with exon–intron boundaries of these exons (Table 2). If mutations were detected in the exon–intron boundaries, a possible occurrence of splicing abnormalities due to the mutations was examined by direct sequencing of p53 cDNA products from the corresponding cell lines. Point mutations were detected in 64 of the 91 cell lines, small insertions/deletions in six of them, and large deletions in two of them. Splice‐site mutations were detected in nine cell lines, in which shifts of open reading frames due to either exon skipping or intron retention were confirmed. Accordingly, only 10 cell lines were shown to carry the wild‐type p53 gene and express normal p53 protein, including five of the 40 AdC cell lines.
The status of 36 cell lines was not available in COSMIC and thus was defined by our studies (Supplementary Table S3‐1) ( 18 , 21 , 22 , 23 , this study). The status of 45 cell lines was concordant between our data and COSMIC data (Supplementary Table S3‐2), whereas that of the remaining 10 cell lines was discordant (Supplementary Table S3‐3). Therefore, although 10 of the 91 lung cancer cell lines carry the wild‐type p53 gene, HPV 16, 18, or 33 are not integrated in these cell lines.
Discussion
To detect HPV‐DNA in lung cancer cells, we applied two different PCR methods with HPV type‐specific primers, one‐step multiplex PCR( 19 ) and nested PCR,( 20 ) because PCR with type‐specific primers was reported to be more sensitive than PCR with consensus primers to detect HPV‐DNA sequences in human cell DNA.( 7 ) The prevalence and genotype distribution of HPV in cervical cancer precursor lesions defined by one‐step multiplex PCR was reported to be compatible with several previous data.( 19 ) In addition, by using these methods, HPV 16 and 18 DNA was distinguishably and efficiently amplified from three cervical cancer cell lines. Therefore, the lack of HPV 16, 18, and 33 DNA in primary lung AdC as well as in lung cancer cell lines would not be due to the low sensitivity of this method for HPV detection. Accordingly, it was concluded from this study that HPV 16, 18, and 33 are not (or are rarely) integrated in lung AdC genomes in the Japanese, particularly those living in the Tokyo area. Lung cancer cell lines analyzed in this study have been established in either Japan or the USA, and consist of all major histological types of lung cancer. Absence of HPV 16/18/33 infection in primary lung AdCs in the US population and lung cancer cell lines established in the USA was previously reported.( 33 , 34 , 35 ) Therefore, the results in the cell lines are consistent with the results in primary AdCs in both Japan and the USA. Indeed, we further attempted to detect HPV‐associated DNA sequences in these cell lines by PCR under several low stringent conditions using a set of consensus primers for HPV 16, 18, and 33. However, no HPV‐specific signals were detected in any of the 91 lung cancer cell lines examined (data not shown). Therefore, we concluded that no HPV 16/18/33 DNA is integrated in these cell lines. Accordingly, HPV infection seems not to play an important role in the development of lung cancer in Japan nor in the USA, although it is still possible that other HPV types play some role in its development.
A Taiwanese study reported that female never‐smokers with lung cancer who were older than 60 years of age had a significantly higher prevalence of HPV 16/18 infections.( 8 ) However, in Korean lung cancer patients, HPV 16/18/33 infections were not associated with gender, smoking status, and histological type.( 10 ) In a study in China, HPV 16/18 infections were not correlated with any clinicopathological parameter, including age, gender, smoking status, and histological type, either.( 9 ) In this study, 41% (121/297) and 43% (64/150) of AdC patients were female and non‐smokers, respectively (Table 1). Therefore, the etiological role of HPV 16/18 in lung carcinogenesis in non‐smokers seems to be restricted to certain geographic areas, and in Japan, HPV 16/18 infection does not play a causative role in the development of lung AdC in female non‐smokers.
An inverse correlation of HPV 16/18 E6 protein expression with p53 expression was also reported in Taiwanese lung tumors.( 36 ) However, in a study in China, there was a relationship between the presence of HPV 16/18 DNA and abnormal p53 protein accumulation.( 37 ) Therefore, association of HPV infection with p53 inactivation is still unclear in lung cancer. We previously examined for p53 mutations in 128 of 297 lung AdCs analyzed in this study, and the mutations were detected in 50 cases (39%) (Table 1); therefore, it was possible that HPV is infected in another 78 cases. However, none of the 78 lung AdCs carried HPV 16/18/33 DNA in their genomes. Accordingly, HPV 16/18/33 infections appear to play a limited role in the development of lung AdC in Japan. These results prompted us to analyze comprehensively the status of the p53 gene in a large panel of lung cancer cell lines. The p53 gene is inactivated not only by mutations in the coding regions, but also by splicing abnormalities caused by mutations in the exon–intron boundaries and homozygous deletions, and the incidence of p53 genetic alterations in total was 89% (81/91). Therefore, although 10 of the 91 cell lines were shown to carry the wild‐type p53 gene, no HPV 16/18/33 DNA was detected in these cell lines. Since the status of the p53 gene in these cell lines was not consistent among several databases and reports, the results provided here will be highly informative to diverse scientists using these cell lines for molecular and biological studies.
In Japan, HPV‐DNA has been detected in <10% of lung AdC in Chiba and Hokkaido, and ∼20% in Okinawa (Supplementary Table S1). Therefore, we cannot totally rule out the involvement of HPV infection in the etiology of lung AdC in Japan. However, the present results strongly indicate that HPV infection plays only a limited role, if any, in the development of lung AdC in Japan.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Table S1. Prevalence of human papillomavirus (HPV) 16, 18, and 33 in lung adenocarcinomas in East Asia.
Table S2. Primer sequences for detection of human papillomavirus (HPV) DNA in cancer cell DNA.
Table S3‐1. p53 status defined in our studies but not registered in the COSMIC database.
Table S3‐2. Concordance of p53 status defined in our studies and registered in the COSMIC database.
Table S3‐3. Discordance of p53 status defined in our studies and registered in the COSMIC database.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
This work was supported by Grants‐in‐Aid from the Ministry of Health, Labor and Welfare for the 3rd‐Term Comprehensive 10‐year Strategy for Cancer Control and for Cancer Research (16‐1) and a Grant‐in‐Aid for the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NiBio). We are grateful to Drs R. Nishikawa and K. Mishima of the Saitama Medical University Hospital for preparation of metastatic lung adenocarcinoma specimens.
References
- 1. Zur Hausen H. Papillomaviruses in the causation of human cancers ‐ a brief historical account. Virology 2009; 384: 260–5. [DOI] [PubMed] [Google Scholar]
- 2. Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990; 248: 76–9. [DOI] [PubMed] [Google Scholar]
- 3. Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990; 63: 1129–36. [DOI] [PubMed] [Google Scholar]
- 4. Nakanishi H, Matsumoto S, Iwakawa R et al. Whole genome comparison of allelic imbalance between noninvasive and invasive small‐sized lung adenocarcinomas. Cancer Res 2009; 69: 1615–23. [DOI] [PubMed] [Google Scholar]
- 5. Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers‐‐a different disease. Nat Rev Cancer 2007; 7: 778–90. [DOI] [PubMed] [Google Scholar]
- 6. Klein F, Kotb WF, Petersen I. Incidence of human papilloma virus in lung cancer. Lung Cancer 2009; 65: 13–8. [DOI] [PubMed] [Google Scholar]
- 7. Srinivasan M, Taioli E, Ragin CC. Human papillomavirus type 16 and 18 in primary lung cancers‐‐a meta‐analysis. Carcinogenesis 2009; 30: 1722–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheng YW, Chiou HL, Sheu GT et al. The association of human papillomavirus 16/18 infection with lung cancer among nonsmoking Taiwanese women. Cancer Res 2001; 61: 2799–803. [PubMed] [Google Scholar]
- 9. Fei Y, Yang J, Hsieh WC et al. Different human papillomavirus 16/18 infection in Chinese non‐small cell lung cancer patients living in Wuhan, China. Jpn J Clin Oncol 2006; 36: 274–9. [DOI] [PubMed] [Google Scholar]
- 10. Park MS, Chang YS, Shin JH et al. The prevalence of human papillomavirus infection in Korean non‐small cell lung cancer patients. Yonsei Med J 2007; 48: 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hirayasu T, Iwamasa T, Kamada Y, Koyanagi Y, Usuda H, Genka K. Human papillomavirus DNA in squamous cell carcinoma of the lung. J Clin Pathol 1996; 49: 810–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miyagi J, Kinjo T, Tsuhako K et al. Extremely high Langerhans cell infiltration contributes to the favourable prognosis of HPV‐infected squamous cell carcinoma and adenocarcinoma of the lung. Histopathology 2001; 38: 355–67. [DOI] [PubMed] [Google Scholar]
- 13. Kinoshita I, Dosaka‐Akita H, Shindoh M et al. Human papillomavirus type 18 DNA and E6‐E7 mRNA are detected in squamous cell carcinoma and adenocarcinoma of the lung. Br J Cancer 1995; 71: 344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hiroshima K, Toyozaki T, Iyoda A et al. Ultrastructural study of intranuclear inclusion bodies of pulmonary adenocarcinoma. Ultrastruct Pathol 1999; 23: 383–9. [DOI] [PubMed] [Google Scholar]
- 15. Sobin LH, Wittekind CH, eds. TNM classification of malignant tumours, 6th ed. New York: Wiley‐Liss; 2002: p. 99–103. [Google Scholar]
- 16. Sakamoto H, Mori M, Taira M et al. Transforming gene from human stomach cancers and a noncancerous portion of stomach mucosa. Proc Natl Acad Sci U S A 1986; 83: 3997–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kohno T, Otsuka A, Girard L et al. A catalog of genes homozygously deleted in human lung cancer and the candidacy of PTPRD as a tumor suppressor gene. Genes Chromosomes Cancer 2010; 49: 342–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Blanco R, Iwakawa R, Tang M et al. A gene‐alteration profile of human lung cancer cell lines. Hum Mutat 2009; 30: 1199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nishiwaki M, Yamamoto T, Tone S et al. Genotyping of Human Papillomaviruses by a Novel One‐Step Typing Method with Multiplex PCR and Clinical Applications. J Clin Microbiol 2008; 46: 1161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu EQ, Zhang GN, Yu XH et al. Evaluation of high‐risk human papillomaviruses type distribution in cervical cancer in Sichuan province of China. BMC Cancer 2008; 8: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tomizawa Y, Kohno T, Fujita T et al. Correlation between the status of the p53 gene and survival in patients with stage I non‐small cell lung carcinoma. Oncogene 1999; 18: 1007–14. [DOI] [PubMed] [Google Scholar]
- 22. Iwakawa R, Kohno T, Anami Y et al. Association of p16 homozygous deletions with clinicopathological characteristics and EGFR/KRAS/p53 mutations in lung adenocarcinoma. Clin Cancer Res 2008; 14: 3746–53. 18559592 [Google Scholar]
- 23. Matsumoto S, Iwakawa R, Takahashi K et al. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene 2007; 26: 5911–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Forbes SA, Tang G, Bindal N et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res 2010; 38(Database issue): D652–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schwarz E, Freese UK, Gissmann L et al. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 1985; 314: 111–4. [DOI] [PubMed] [Google Scholar]
- 26. Baker CC, Phelps WC, Lindgren V, Braun MJ, Gonda MA, Howley PM. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J Virol 1987; 61: 962–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yee C, Krishnan‐Hewlett I, Baker CC, Schlegel R, Howley PM. Presence and expression of human papillomavirus sequences in human cervical carcinoma cell lines. Am J Pathol 1985; 119: 361–6. [PMC free article] [PubMed] [Google Scholar]
- 28. Yokota J, Tsukada Y, Nakajima T et al. Loss of heterozygosity on the short arm of chromosome 3 in carcinoma of the uterine cervix. Cancer Res 1989; 49: 3598–601. [PubMed] [Google Scholar]
- 29. Kohno T, Takayama H, Hamaguchi M et al. Deletion mapping of chromosome 3p in human uterine cervical cancer. Oncogene 1993; 8: 1825–32. [PubMed] [Google Scholar]
- 30. Pett M, Coleman N. Integration of high‐risk human papillomavirus: a key event in cervical carcinogenesis? J Pathol 2007; 212: 356–67. [DOI] [PubMed] [Google Scholar]
- 31. Berglind H, Pawitan Y, Kato S, Ishioka C, Soussi T. Analysis of p53 mutation status in human cancer cell lines: a paradigm for cell line cross‐contamination. Cancer Biol Ther 2008; 7: 699–708. [DOI] [PubMed] [Google Scholar]
- 32. Petitjean A, Mathe E, Kato S et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 2007; 28: 622–9. [DOI] [PubMed] [Google Scholar]
- 33. Yousem SA, Ohori NP, Sonmez‐Alpan E. Occurrence of human papillomavirus DNA in primary lung neoplasms. Cancer 1992; 69: 693–7. [DOI] [PubMed] [Google Scholar]
- 34. Wistuba II, Behrens C, Milchgrub S et al. Comparison of molecular changes in lung cancers in HIV‐positive and HIV‐indeterminate subjects. JAMA 1998; 279: 1554–9. [DOI] [PubMed] [Google Scholar]
- 35. Shimizu E, Coxon A, Otterson GA et al. RB protein status and clinical correlation from 171 cell lines representing lung cancer, extrapulmonary small cell carcinoma, and mesothelioma. Oncogene 1994; 9: 2441–8. [PubMed] [Google Scholar]
- 36. Cheng YW, Wu MF, Wang J et al. Human papillomavirus 16/18 E6 oncoprotein is expressed in lung cancer and related with p53 inactivation. Cancer Res 2007; 67: 10686–93. [DOI] [PubMed] [Google Scholar]
- 37. Wang Y, Wang A, Jiang R et al. Human papillomavirus type 16 and 18 infection is associated with lung cancer patients from the central part of China. Oncol Rep 2008; 2: 333–9. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Prevalence of human papillomavirus (HPV) 16, 18, and 33 in lung adenocarcinomas in East Asia.
Table S2. Primer sequences for detection of human papillomavirus (HPV) DNA in cancer cell DNA.
Table S3‐1. p53 status defined in our studies but not registered in the COSMIC database.
Table S3‐2. Concordance of p53 status defined in our studies and registered in the COSMIC database.
Table S3‐3. Discordance of p53 status defined in our studies and registered in the COSMIC database.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item