

Abstract
Dipeptidyl peptidase IV (DPPIV/CD26) is a multifunctional cell surface aminopeptidase that is widely expressed in different cell types. Recent studies have suggested that DPPIV plays an important role in tumor progression in several human malignancies. In the current study, we investigated the role of DPPIV in paclitaxel resistance in epithelial ovarian carcinoma (EOC) cells. We first examined the correlation between expression levels of DPPIV and sensitivity to paclitaxel in various EOC cell lines. Subsequently, to clarify the cellular functions of DPPIV, we investigated the role of this molecule in the sensitivity of EOC to paclitaxel in vitro and in vivo using stably DPPIV‐transfected EOC cells. We identified a positive correlation between DPPIV expression and paclitaxel sensitivity in various EOC cell lines. In addition, we observed a significant increase in the paclitaxel sensitivity of DPPIV‐overexpressing EOC cells. Furthermore, no apparent alteration in paclitaxel sensitivity was noted by the addition of a specific inhibitor of DPPIV activity in DPPIV‐transfected or natively DPPIV‐overexpressing EOC cells. In a subcutaneous murine model treated with paclitaxel, on Day 39, the tumor size of the DPPIV‐transfected cell‐inoculated group was as large as that of the vector‐transfected cell‐inoculated group. In contrast, on Day 61, the former was smaller than the latter. The present findings show that DPPIV may be involved in the increased sensitivity to paclitaxel of EOC cells regardless of the involvement of DPPIV activity. (Cancer Sci 2009)
Dipeptidyl peptidase IV (DPPIV) is a cell surface aminopeptidase which was originally characterized as a T‐cell differentiation antigen (CD26),( 1 ) and it has been reported to be present on epithelial cells of various tissues, including the lung, liver, kidney, intestine, prostate, and placenta.( 2 , 3 , 4 ) Since DPPIV cleaves amino‐terminal dipeptides from polypeptides with either l‐proline or l‐alanine in position 2, this enzyme is capable of degrading various bioactive peptides, cytokines, and several chemokines such as stromal cell‐derived factor‐1α (SDF‐1α) or RANTES (regulated on activation, normal T‐cell expressed, and secreted).( 3 , 5 , 6 ) Therefore, a variety of reports have shown that DPPIV is involved in various cellular processes such as the regulation of immune‐system‐mediated responses, signal transduction, and interaction with molecules of the extracellular matrix.( 7 , 8 ) In addition, a number of studies have provided evidence indicating that DPPIV may play a role in tumor suppression including an antiproliferative effect, and decreased invasive ability.( 9 , 10 , 11 , 12 ) Moreover, a recent report from Aytac et al. showed that leukemic T cells overexpressing DPPIV, through its associated enzyme activity, displayed enhanced sensitivity to doxorubicin, associated with the disruption of cell cycle‐related events.( 13 ) Furthermore, Sato et al. demonstrated that DPPIV enhanced sensitivity to apoptosis induced by doxorubicin and etoposide with increased susceptibility to the mitochondrial pathway of apoptosis and enhanced topoisomerase II alpha expression.( 14 )
Epithelial ovarian carcinoma (EOC) is the leading cause of mortality among cancers of the female reproductive system.( 15 ) The majority of patients with EOC have advanced intraperitoneal metastatic disease at diagnosis since this carcinoma frequently remains clinically silent. Today, paclitaxel‐platinum‐based chemotherapy has become the standard regimen for EOC worldwide. Nevertheless, despite the comparatively high‐level sensitivity of EOC to paclitaxel, the prognosis of advanced or recurrent cases remains poor since most deaths are the result of metastasis that is refractory to these chemotherapeutic agents. To overcome such paclitaxel resistance, a variety of additional molecular‐targeting therapies combined with paclitaxel have been investigated.( 16 , 17 ) However, the detailed mechanism underlying the resistance remains unclear, and the effect of combined treatment is not satisfactory.
Previously, we demonstrated that DPPIV expression in EOC cell lines was negatively correlated with the invasive potential. In addition, DPPIV overexpression in EOC cells induced a marked change in the cellular morphology from a fibroblastic to an epithelioid pattern and a significant decrease in the invasive potential in vitro and in vivo. ( 10 ) In the current study, we focused on the association of DPPIV with the susceptibility of EOC to paclitaxel. We first examined the correlation between expression levels of DPPIV and sensitivity to paclitaxel in various EOC cell lines. Subsequently, to clarify the cellular functions of DPPIV, we investigated the role of this molecule in the sensitivity of EOC to paclitaxel in vitro and in vivo using stably DPPIV‐transfected EOC cells. Herein, the possible application of this enzyme as a means of enhancing the chemosensitivity of EOC to paclitaxel is proposed.
Materials and Methods
Cell culture. We used 11 human EOC cell lines. SKOV‐3, NOS‐2, TAOV, NOS‐4, HRA, RMG‐I, and RMG‐II cells were cultured and maintained as described previously.( 10 , 18 , 19 ) NOS‐1 cells were established in our institute. ES‐2, HEY, and KOC‐7C cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). Furthermore, acquired paclitaxel‐resistant NOS‐2 cells (NOS‐PR cells) were established in our institute, as described previously.( 20 ) These cell lines were maintained in RPMI‐1640 (Sigma, St. Louis, MO, USA) supplemented with 10% fetal calf serum (FCS) and penicillin streptomycin at 37°C in a humidified atmosphere of 5% CO2.
Enzyme activity assay. DPPIV enzyme activity was measured spectrophotometrically using Gly‐Pro‐p‐nitroanilide‐tosylate (Peptide Institute, Osaka, Japan) as a DPPIV substrate, as described previously.( 10 ) We used diprotin A (DPA) (Peptide Institute) as a specific inhibitor of DPPIV enzyme activity.
Generation of stable SKOV‐3 clones expressing DPPIV. Vector construction and generation of stable SKOV‐3 clones expressing DPPIV were described previously.( 10 ) Briefly, transfections were carried out using Lipofectamine according to the manufacturer’s instructions (Invitrogen, San Diego, CA, USA). SKOV‐3 cells were transfected with pcDNA3.1 (–), and pcDNA3.1 (–) with DPPIV cDNA was inserted. Stable transfectants were selected by growth in medium supplemented with 400 μg/mL of G418 (Sigma‐Aldrich, St. Louis, MO, USA). Several hundred clones resistant to G418 were obtained. Since we hoped to eliminate any effects that could be attributed to clonal variation, polyclonal cells from these transfectants were used in the following experiments.( 10 )
Flow cytometric analysis. Fluorescence‐activated cell sorting (FACS) was performed to quantify the expression levels of DPPIV on the cell surface of EOC cells. Then, the cells were incubated with phycoerythrin‐conjugated monoclonal antibodies specific for DPPIV (Pharmingen, San Diego, CA, USA) for 30 min at 4°C, and washed three times with phosphate‐buffered saline (PBS). FACS data were acquired using a FACS Calibur (Becton Dickinson, San Jose, CA, USA), and analyzed using CELL Quest software (Becton Dickinson).
Cell proliferation assay. SKpc and SKDP cells were plated in triplicate at a density of 1500 cells in a 100 μL volume in 96‐well plates, and cultured for 1 to 4 or 5 days with/without 10% FCS, as described previously.( 10 ) Cell viability was assayed using a modified tetrazolium salt MTT assay employing a CellTiter 96 AQueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Absorbance was measured at 492 nm by a microplate reader (Multiskan Bichromatic; Labsystems, Helsinki, Finland).
Paclitaxel chemosensitivity assay. The paclitaxel chemosensitivity assay was performed as described previously.( 21 ) Briefly, cells were seeded in triplicate in 96‐well plates at a density of 5000 cells in a volume of 200 μL of culture media containing 10% FCS. After incubation for 24 h at 37°C, the medium was replaced with fresh medium with or without various concentrations of paclitaxel (Bristol Myers Squib, Tokyo, Japan). After an additional 72 h, cell viability was assayed using the Cell Titer 96 Aqueous One Solution Cell Proliferation Assay kit (Promega), as described above in ‘Cell proliferation assay’. IC50 values indicate the concentrations resulting in a 50% reduction in growth as compared with control cell growth.
Cell cycle analysis. Cells were incubated in culture media alone or culture media and paclitaxel (70 ng/mL) at 37°C for 8 h. The cells were then collected, washed twice with PBS, and resuspended in PBS containing 10 μg/mL propidium iodide (PI), 0.5% Tween 20, and 0.1% RNase at room temperature for 30 min. Samples were then analyzed for their DNA content using a FACS Calibur (Becton Dickinson). Cell debris and fixation artifacts were gated out and G0/G1, S, and G2‐M populations were quantified using CELL Quest software (Becton Dickinson).
Western blot analysis. Immunoblot analysis was performed as described previously.( 22 ) Cells were seeded on 10‐cm dishes in RPMI‐1640 containing 10% FCS. After the cells reached subconfluency, the medium was replaced by fresh RPM‐I1640 with or without paclitaxel (100 ng/mL). After 24 h of incubation, cell lysates were collected. Twenty μg of total cell lysate were electrophoresed on an SDS polyacryamide gel and transferred electrophoretically to Immobilon membranes (Millipore, Bedford, MA, USA). After blocking, the membrane was incubated for 1 h with the respective antihuman primary antibody at the recommended dilution (anti‐DPPIV: Bender MedSystems, Vienna, Austria; anti‐Apaf and Bcl‐2: BD Transduction Laboratories, Franklin Lakes, NJ, USA; anti‐Bcl‐XL, Bad, and Bax: Cell Signaling, Danvers, MA, USA; TWIST, and β‐actin: Santa Cruz Biotechnology, Santa Cruz, CA, USA). The primary antibodies were washed in Tween/PBS, and then incubated with horseradish peroxidase–conjugated secondary antibody. Proteins were visualized using enhanced chemiluminiscence (ECL) reagent (Amersham Biosciences, Tokyo, Japan), followed by exposure to X‐ray film.
Assay for apoptosis. To quantify apoptosis, Annexin V and PI staining was performed, followed by FACS. Preparation of the cells and the assay for apoptosis was performed using the MEBCYTO apoptosis kit (MBL International, Woburn, MA, USA), as described previously.( 21 )
In vivo experiment. A total of 2.0 × 106 SKpc and SKDP cells were subcutaneously inoculated in 0.2 mL of serum‐free RPMI.1640 medium through a 24‐gage needle into the lower flank of 5‐week‐old nude mice (Japan SLC, Nagoya, Japan). The subcutaneous tumor size was followed with or without the intraperitoneal (i.p.) administration of paclitaxel (20 mg/kg/body weight). To mice treated with paclitaxel, the i.p. injection of paclitaxel was initiated 20 days after tumor inoculation, and repeated twice a week for, at most, 6 weeks. Each group consisted of seven mice. The tumor was measured with calipers and the volume was calculated using the formula: π/6 × (largest diameter) × (smallest diameter)2. Data are presented as the mean ± SD. Furthermore, apoptotic events after treatment with paclitaxel were determined by the TdT‐mediated dUTP nick end‐labeling (TUNEL) assay in xenograft tumor sections. Briefly, 4‐ μm paraffin‐embedded tissue sections were prepared from the murine xenograft tumors. The slides were then subjected to TUNEL staining using an MEBSTAIN Apoptosis Kit according to the manufacturer’s protocol (MBL International). The stained slides were analyzed using an BZ8000 (Keyence, Tokyo, Japan) fluorescence microscope. Apoptotic cells were identified by positive TUNEL staining. All procedures were performed in accordance with the regulations for animal experiments from the Division of Experimental Animals of Nagoya University, Japan.
Statistical analysis. For data from in vitro experiments, statistical comparisons among groups were performed using the Student’s t‐test. Differences between groups were considered significant at P < 0.05. Data are expressed as the mean ± SD. In addition, Spearman’s correlation test was used to compare the IC50 values of paclitaxel and the mean fluorescence intensities of DPPIV among various EOC cell lines by FACS analysis. The time‐course of tumor growth was compared across the treatment groups with the use of two‐way anova, with group and time as the variables.
Results
DPPIV/CD26 expression in EOC cell lines. We first examined DPPIV expression in EOC cell lines by FACS analysis. DPPIV was generally expressed in cell lines, although the extent of its expression varied from cell to cell. The mean DPPIV fluorescence intensities of SKOV‐3, ES‐2, HRA, KOC‐7C, and HEY cells were 12, 4, 4, 12, and 8, respectively. These EOC cell lines were negative or showed limited expressions of DPPIV. On the other hand, those of NOS‐2, TAOV, NOS4, RMG‐I, RMG‐II, and NOS‐1 cells were 684, 293, 292, 119, 167, and 746, respectively (Fig. 1A). These cell lines were positive for DPPIV expression, which is consistent with data obtained by enzyme activity analysis (Fig. 1B). We next examined the sensitivity to paclitaxel using these EOC cell lines. The IC50 values of SKOV‐3, ES‐2, HRA, KOC‐7C, and HEY cells were 43.0, 41.2, 50.2, 28.1, and 41.8, respectively. In contrast, those of NOS‐2, TAOV, NOS4, RMG‐I, RMG‐II, and NOS‐1 cells were 6.9, 9.1, 22.1, 10.5, 12.3, and 4.9, respectively. Representative data of SKOV‐3, and ES‐2, NOS2, and TAOV cells are shown in Figure 1(C). Figure 1(D) shows the correlation between the mean fluorescence intensity of DPPIV and IC50 values in 11 EOC cell lines. The results revealed that the expression of DPPIV and IC50 of paclitaxel were negatively correlated (P = 0.0047, r = −0.761).
Figure 1.
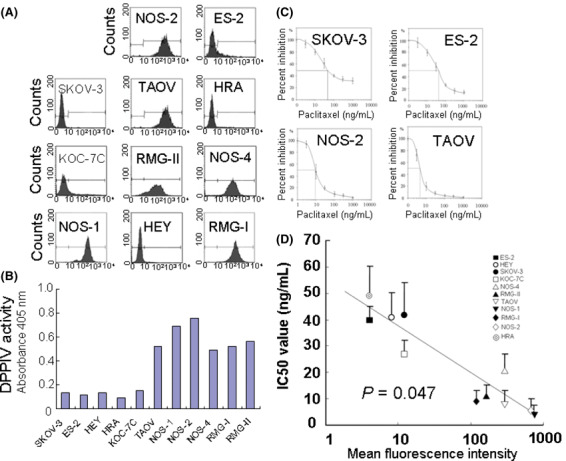
The correlation between expressions of dipeptidyl peptidase IV (DPPIV) and sensitivity to paclitaxel in 11 epithelial ovarian cancer (EOC) cell lines. (A) The DPPIV expressions of 11 EOC cell lines are indicated as FACS data. The mean fluorescence intensities (MFIs) of DPPIV in the EOC cell lines SKOV‐3, HEY, HRA, ES‐2, KOC‐7C, TAOV, NOS‐1, NOS‐2, NOS4, RMG‐I, and RMG‐II cells were 12, 8, 4, 4, 12, 293, 746, 684, 292, 119, and 167, respectively. (B) DPPIV activity of 11 EOC cell lines. DPPIV enzyme activity was measured at 405 nm after 45‐min incubation at 37°C. (C) Sensitivity to paclitaxel of several representative EOC cell lines (SKOV‐3, ES‐2, NOS‐2, and TAOV cells). IC50 values of these cell lines were 43.0, 41.2, 6.9, and 9.1, respectively. (D) The correlation between the MFI of DPPIV and IC50 value in 11 EOC cell lines. The IC50 values of these 11 cell lines were negatively correlated with DPPIV expression, as shown by the MFIs (P = 0.047, r = −0.761).
The effect of DPPIV overexpression on chemosensitivity to paclitaxel. To investigate the effect of DPPIV transfection on chemosensitivity to paclitaxel in EOC cells, we overexpressed DPPIV in SKOV‐3 cells (SKDP cells). While vector‐transfected SKpc cells expressed little DPPIV on the cell surface, SKDPIV cells expressed a markedly high level of DPPIV using Western blot analysis (Fig. 2A). Figure 2(B) shows the paclitaxel sensitivity curve between SKDP and SKpc cells. The IC50 values of SKDP and SKpc cells were 6.2 and 40.0, respectively. The IC50 value of vector‐transfected SKpc cells was almost the same as that of parental SKOV‐3 cells. On the contrary, the transfection of DPPIV enhanced the sensitivity to paclitaxel up to the level of natively paclitaxel‐sensitive EOC cells, such as NOS‐2 or TAOV cells. To assess the effect of DPPIV on the in vitro proliferation potential, using these two lines, we performed growth assays on plastic dishes. As a result, we confirmed that the growth rates between these two lines were almost the same as each other on the plastic dishes (Fig. 2C). Subsequently, to investigate the mechanism of DPPIV‐induced enhanced chemosensitivity, we assessed whether DPPIV transfection had an effect on paclitaxel‐induced cell‐cycle arrest in SKpc and SKDP cells. Cells were treated with paclitaxel (70 ng/mL) for 8 h, followed by PI staining and flow cytometric analysis. As shown in Figure 2(D), the enhancement of paclitaxel sensitivity observed in SKDP cells was attributable to an increase in the percentage of cells arresting at G2‐M. Furthermore, to investigate the involvement of DPPIV activity, we assessed whether the inhibition of DPPIV activity using DPA influenced the paclitaxel sensitivity of either DPPIV‐transfected SKDP cells or natively DPPIV‐overexpressing NOS‐2 cells. The DPPIV activity of SKDP cells was approximately 10‐times higher than that of SKpc cells, and it was inhibited by adding 100 μm of DPA (Fig. 3A). Although DPA treatment did not alter the surface expression levels of DPPIV (Fig. 3B), the addition of DPA (100 μm) did not induce any apparent alteration of the sensitivity to paclitaxel in either SKDP or NOS‐2 cells (Fig. 3C,D). According to this preliminary data, we did not identify a positive correlation between DPPIV activity and paclitaxel sensitivity in EOC cells.
Figure 2.
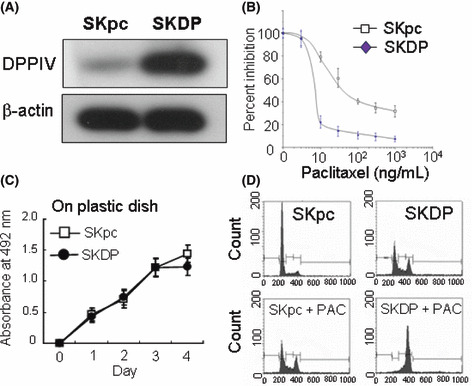
The effect of dipeptidyl peptidase IV (DPPIV) overexpression on susceptibility to paclitaxel. (A) Enhanced expression of DPPIV in DPPIV‐transfected cells. Western blot analysis using total cell lysates. (B) Transfection of DPPIV expression in SKOV‐3 cells induced a marked increase in chemosensitivity to paclitaxel. The IC50 values of SKDP and SKpc cells were 6.2 and 40.0, respectively. (C) Effect of DPPIV transfection on cell proliferation on plastic dishes using SKDP and SKpc cells. Four‐day proliferation curves of SKpc and SKDP cells in the presence of 10% FCS. Analysis of the proliferation rate was performed as described in the Materials and Methods. No significant differences between SKpc and SKDP cells were observed. Bars, SD. (D) Effect of DPPIV transfection on paclitaxel‐mediated cell‐cycle arrest at G2‐M. SKpc and SKDP cells were incubated at 37°C in media containing paclitaxel (70 ng/mL) for 8 h. Data are representative of three separate experiments.
Figure 3.
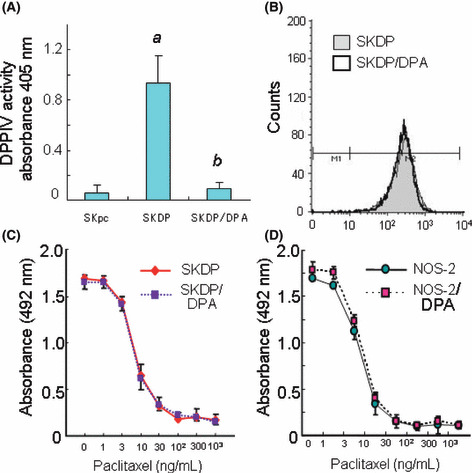
The effect of a dipeptidyl peptidase IV (DPPIV) inhibitor (diprotin A, DPA) on chemosensitivity to paclitaxel in either DPPIV‐transfected SKDP or natively DPPIV‐overexpressing NOS‐2 cells. (A) The DPPIV activity of SKDP cells with or without DPA (100 μm). (a) P < 0.001, SKpc versus SKDP cells; (b) P < 0.001, SKDP versus SKDP/DPA cells. (B) The surface expressions of DPPIV in the presence or absence of DPA (100 μm, 72 h). DPA‐treatment did not alter the surface expression levels of DPPIV in SKDP cells. White peak, SKDP/DPA cells; gray peak, SKDP cells. (C) Inhibition of DPPIV activity by the addition of DPA (100 μm) in SKpc and SKDP cells. (D) Inhibition of DPPIV activity by the addition of DPA (100 μm) in NOS‐2 cells.
Effect of DPPIV transfection on paclitaxel‐induced apoptosis of EOC cells. We subsequently assessed whether the transfection of DPPIV had an effect on the paclitaxel‐induced apoptosis of SKpc and SKDP cells. Cells were treated with paclitaxel (100 ng/mL) for 48 h, followed by Annexin V/PI staining and flow cytometric analysis. As shown in Figure 4(A), SKpc cells exhibited mild apoptosis on paclitaxel administration, while SKDP cells showed moderate to marked apoptosis. The quantitative data reveal that, compared to SKpc cells showing 6.2% apoptotic cells on paclitaxel treatment, SKDP cells showed a marked increase in apoptosis, accounting for 19.1%. Subsequently, we examined whether the expressions of apoptosis markers varied in the presence or absence of treatment with paclitaxel using Western blot analysis. In the absence of paclitaxel, the expression levels of Bcl‐2, Bcl‐XL, Bax, and Bad were very similar between SKpc and SKDP cells. On the other hand, in the presence of paclitaxel, the expression levels of these proteins were markedly changed. Thus, the expression levels of Bcl‐2 and Bcl‐XL were decreased and those of Bax and Bad were increased in SKDP compared to SKpc cells (Fig. 4B). Furthermore, we examined whether the expression of TWIST, a master regulator of epithelial–mesenchymal transition (EMT), and possibly linked to paclitaxel resistance, was altered among these transfectants. Western blot analysis demonstrated that the down‐regulated TWIST expression was observed in SKDP compared to that in SKpc cells. In addition, these tendencies were not altered on exposure to paclitaxel (Fig. 4C).
Figure 4.
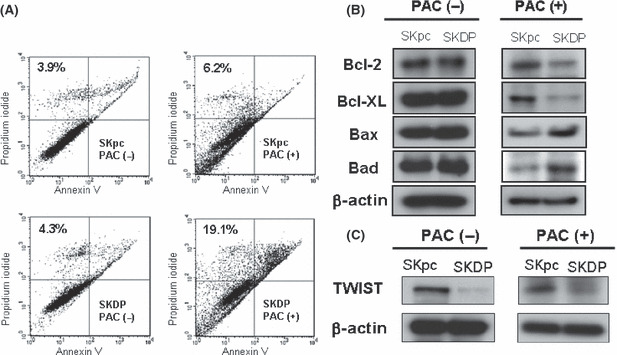
Effect of dipeptidyl peptidase IV (DPPIV) transfection on the paclitaxel‐induced apoptotic death of epithelial ovarian carcinoma (EOC) cells. (A) Detection of paclitaxel‐induced apoptosis using Annexin V and propidium iodide (PI) assays of SKpc and SKDP cells treated with 100 ng/mL of paclitaxel for 48 h. The percentage of apoptotic cells calculated by flow cytometric analysis is presented. Each value (%) indicates Annexin V (+)/ PI (+) cells. (B) Change in expressions of apoptosis‐related proteins with or without paclitaxel treatment using Western blot analysis. (C) Western blot analysis demonstrated down‐regulated TWIST expression in SKDP compared to SKpc cells in the presence or absence of paclitaxel.
The effect of serum starvation on the proliferation of SKpc and SKDP cells. We subsequently hypothesized that if DPPIV overexpression led to an enhanced susceptibility to paclitaxel, it might be related to sensitivity to other cell stresses such as serum starvation. Figure 5(A) shows 5‐day proliferation curves of SKpc and SKDP cells under serum‐starved conditions. The proliferation rates were markedly lower in SKDP than in SKpc cells in serum‐free media. SKpc cells were capable of growing even in serum‐free media until 96 h after the beginning of starvation. In contrast, SKDP cells failed to proliferate in these media. Moreover, we examined whether the expression of apoptosis markers was affected under the serum‐starved condition using Western blot analysis. The expression levels of Apaf, Bax, and Bad were upregulated in SKDP cells compared to those in SKpc cells after 96‐h exposure to the serum‐free condition. Furthermore, the expression level of BCl‐2 was decreased (Fig. 5B).
Figure 5.
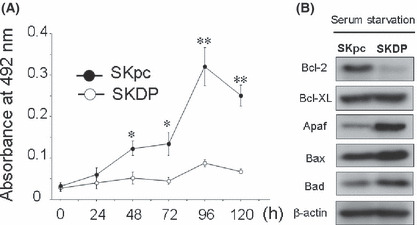
The effect of serum starvation on the proliferation and expression levels of several apoptosis‐related proteins in SKpc and SKDP cells. (A) Five‐day proliferation curves of SKpc and SKDP cells under serum‐starved conditions. Although SKpc cells were capable of growing even in serum‐free media until 96 h after the beginning of starvation, SKDP cells failed to proliferate in these media. *P < 0.001; **P < 0.0001. (B) Change in the expression of apoptosis‐related proteins under serum‐starved conditions for 96 h using Western blot analysis.
Downregulation of DPPIV expression and enzyme activity in EOC cells with acquired paclitaxel resistance. We established and confirmed the resistance to paclitaxel of NOS‐PR cells.( 32 ) Figure 6(A) shows the paclitaxel‐sensitivity assay of parental NOS‐2 and resistant NOS‐PR cells. More pronounced chemoresistance to paclitaxel was noted in NOS‐PR compared to NOS‐2 cells. The IC50 values of NOS‐2 and NOS‐PR cells were 9.3 and 367.3 ng/mL, respectively. The IC50 value of NOS‐PR cells was considered to correspond to clinical paclitaxel resistance. We subsequently compared the surface expression levels of DPPIV between the NOS‐PR and parental NOS‐2 lines. With regard to the morphology, NOS‐2 cells displayed an epithelioid and cobblestone appearance (Fig. 6B, left panel). In contrast, NOS‐PR cells showed a loss of cell polarity, causing a spindle‐shaped morphology, and the increased formation of pseudopodia (Fig. 6B, right panel). We already demonstrated that NOS‐2 cells displayed a markedly high level of DPPIV, as shown in Figure 1(A). On the other hand, the DPPIV expression level of NOS‐PR was lower than that of NOS‐2 cells (Fig. 6C). Moreover, to confirm the enzyme activity of DPPIV protein, we also analyzed aminopeptidase activity. The DPPIV activity of NOS‐PR cells was approximately one‐sixth lower than that of parental NOS‐2 cells in terms of absorbance (Fig. 6D). The DPPIV activities of both lines were almost completely inhibited by adding 100 μm of DPA. Furthermore, the inhibition of DPPIV activity using DPA did not influence either the cell morphology or paclitaxel sensitivity of both lines (data not shown).
Figure 6.
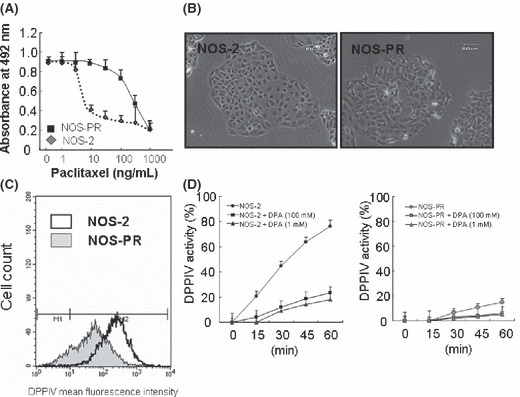
Downregulation of dipeptidyl peptidase IV (DPPIV) expression and enzyme activity in epithelial ovarian carcinoma (EOC) cells with acquired paclitaxel resistance. (A) Paclitaxel sensitivity assay of parental NOS‐2 and resistant NOS‐PR cells. More pronounced chemoresistance to paclitaxel was noted in NOS‐PR compared to NOS‐2 cells. The IC50 values of NOS‐2 and NOS‐PR cells were 9.3 and 367.3 ng/mL, respectively. (B) The surface expression levels of DPPIV between the NOS‐PR and parental NOS‐2 lines. The expression level of DPPIV of NOS‐PR cells was lower than that of NOS‐2 cells. (C) Cell morphology of NOS‐2 and NOS‐PR cells. NOS‐2 cells displayed an epithelioid and cobblestone appearance (left panel). NOS‐PR cells showed the loss of cell polarity, leading to a spindle‐shaped morphology (right panel). (D) The DPPIV activity of NOS‐2 and NOS‐PR cells. DPPIV activities of both lines were almost completely inhibited by the addition of 100 μm of DPA.
The effect of DPPIV overexpression on subcutaneous tumor formation in nude mice treated with paclitaxel. Finally, we investigated whether DPPIV‐sensitized tumors formed in nude mice receiving following subcutaneous xenografting. Subcutaneous tumor formation was observed at least approximately 1 week after the inoculation of mice with SKpc or SKDP cells. Subsequently, periodic i.p. treatment with PBS or paclitaxel was performed, as described in the Materials and Methods. Figure 7(A,B) shows the general appearance of mice treated with paclitaxel 39 or 61 days after inoculation with SKpc (left mouse) or SKDP cells (right mouse). Figure 7(C) shows the time‐course of tumor‐growth curves of both groups on paclitaxel treatment. Among the paclitaxel‐treated mice, the subcutaneous tumor size following inoculation with SKDP cells was macroscopically almost the same as that in those inoculated with SKpc cells until 39 days after cell inoculation. In contrast, the difference between the sizes of both tumors increased from that time (P < 0.0001; Fig. 7C). This suggests that the enhancement of paclitaxel sensitivity in vivo might be in part due to the overexpression of DPPIV. Moreover, even in the absence of paclitaxel treatment, reduced in vivo tumor formation was observed in the SKDP cell‐inoculated compared to the SKpc cell‐inoculated group. Furthermore, TUNEL staining was performed for the resected murine subcutaneous tumor on Day 61. Positive TUNEL staining indicating apoptotic change was more clearly identified in the tumor formed by SKDP cells than that by SKpc cells (Fig. 7D).
Figure 7.
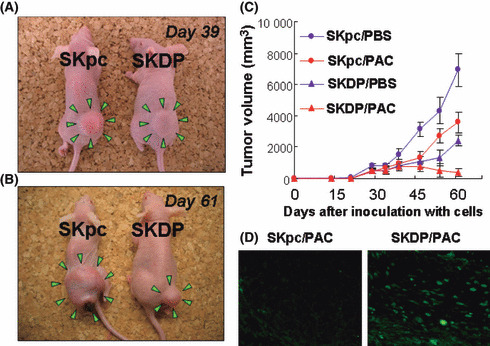
The effect of the overexpression of dipeptidyl peptidase IV (DPPIV) on subcutaneous tumor formation in nude mice with or without treatment with paclitaxel. A total of 2.0 × 106 SKpc and SKDP cells were subcutaneously implanted in the lower flank of nude mice. Subsequent intraperitoneal (i.p.) administration of paclitaxel (20 mg/kg) or PBS was initiated 20 days after tumor inoculation, and repeated twice a week for a maximum of 6 weeks. (A,B) Among paclitaxel‐treated mice, the general appearance of mice 39 (A) and 61 (B) days after inoculation with SKpc (left mouse) and SKDP (right mouse) cells. On Day 39, the tumor size of the SKDP cell‐inoculated group was as large as that of the SKpc cell‐inoculated group. In contrast, on Day 61, the former was smaller than the latter. (C) Tumor growth curves of the four groups: Red closed red circle (SKpc/PAC‐group), SKpc‐inoculated group treated with paclitaxel (n = 7); closed red triangle (SKDP/PAC‐group), SKDP‐inoculated group treated with paclitaxel (n = 7); closed blue circle (SKpc/PBS‐group), SKpc‐inoculated group treated with PBS (n = 7); closed blue triangle (SKDP/PBS‐group), SKDP‐inoculated group treated with PBS (n = 7). On Day 61: P < 0.0001, SKpc/PAC‐group versus SKDP/PAC‐group; P < 0.0001, SKpc/PBS‐group versus SKDP/PBS‐group. Data are presented as the mean ± SD. (D) TUNEL staining was performed for the resected murine subcutaneous tumor on Day 61. Apoptotic cells were identified by positive TUNEL staining (left panel, SKpc/PAC; right panel, SKDP/PAC).
Discussion
Paclitaxel exerts its effect through the stabilization of microtubules, induction of cell‐cycle arrest in G2‐M, and activation of proapoptotic signaling.( 23 , 24 ) In the present study, we first showed that the surface expressions of DPPIV were positively correlated with sensitivities to paclitaxel in various EOC cell lines. This suggested that reduced DPPIV expression may be involved in the intrinsic resistance of EOC cells. Based on this observation, we subsequently demonstrated that the overexpression of DPPIV results in paclitaxel‐induced apoptosis, and, consequently, promotes the susceptibility of EOC cells to paclitaxel. Thus far, Yamochi, et al. reported that in a B‐cell lymphoma line, DPPIV expression was associated with the increased phosphorylation of p38 and its upstream regulators mitogen‐activated protein kinase kinase 3/6 and apoptosis signal‐regulating kinase 1, and that the p38 signaling pathway plays a role in the regulation of topoisomerase IIA expression, resulting in greater in vitro and in vivo tumor sensitivity to doxorubicin.( 11 ) Regarding the link between DPPIV and chemosensitivity, their results were consistent with ours. However, to our knowledge, this is the first report showing a link between the expression of DPPIV and chemosensitivity to paclitaxel in solid tumors, especially EOC cells in vitro and in vivo, although the detailed mechanism remains under investigation.
Regarding the involvement of DPPIV enzyme activity in sensitivity to paclitaxel, the present data using DPPIV‐transfected or natively DPPIV‐overexpressing cells showed that sensitivity to paclitaxel did not change even on the simultaneous addition of a DPPIV inhibitor. DPPIV is a multifunctional molecule that has been reported to play an important role in cellular differentiation, signal transduction by tyrosine phosphorylation of a subset of small molecular mass proteins, and pathways, independent of its enzymatic activity.( 25 , 26 , 27 , 28 ) Thus, regarding the link between DPPIV and sensitivity to paclitaxel, there may be a mechanism independent of DPPIV activity. We could not conclude that these data directly indicate the reduced involvement of DPPIV activity in the paclitaxel sensitivity of EOC, since a major limitation of this study was that we unfortunately did not perform an experiment using CD26‐positive/DPPIV‐negative mutants.
Considering the capability of DPPIV to cleave various selected biological factors, including neuropeptides, cytokines, and chemokines, which promote cancer metastasis, it is possible that DPPIV activity may contribute to antichemoresistance through proteolytically modifying peptides involved in anti‐apoptotic signaling, and/or their precursors. Interestingly, our data demonstrated that under serum starvation DPPIV‐transfected EOC cells did not proliferate with enhanced expressions of Apaf, Bax, and Bad; in contrast, vector‐transfected cells did. This implies that DPPIV expression is involved in sensitivity to other cell stresses such as deficient growth factors, as well as exposure to paclitaxel. Wesley et al. showed that, in non‐small‐cell lung carcinoma cells, DPPIV induces cell cycle arrest and promotes the susceptibility of tumor cells to apoptosis upon serum withdrawal, and that both wild‐type and mutant DPPIV in the enzyme activity region showing reduced levels of protease activity had similar effects on the induction of apoptosis.( 29 ) Their data were consistent with ours, regardless of the involvement of DPPIV activity. However, if this result was due to increased DPPIV enzyme activity, the underlying mechanism may be based on serum withdrawal, possibly through the inactivation of some unidentified, essential growth factors involving DPPIV. For example, SDF‐1α, a chemokine of the CXC subfamily, is a good substrate for DPPIV because it contains the preferred essential N‐terminal X‐Pro motif. Our earlier data demonstrated the involvement of the SDF‐1α/CXCR4 axis in the enhanced peritoneal metastasis of EOC.( 30 ) In glioma cells, SDF‐1αprevented the apoptosis of tumor cells with the phosphorylation of Akt, a kinase associated with survival when serum is withdrawn from the culture medium, besides enhanced chemotaxis, in accordance with this better known role of chemokines.( 31 ) Therefore, although the detailed mechanisms are still under investigation, at present, we think that DPPIV may be involved in signaling cascades regulating cell survival, protection from apoptosis, and paclitaxel resistance via both aminopeptidase‐dependent and ‐independent mechanisms.
Generally, there are two types of resistance to antineoplastic agents: intrinsic and acquired chemoresistance. Clinically, recurrent EOC is frequently associated with acquired resistance to chemotherapeutic agents. In our earlier analysis, we established acquired paclitaxel‐resistant EOC cell lines by their continuous exposure to increasing concentrations of paclitaxel.( 32 ) Interestingly, in our current analysis, we further noted the decreased expression of DPPIV and activity in the acquired paclitaxel resistance of EOC cells compared to wild‐type parental cells. Namely, the downregulation of DPPIV was observed accompanied by the gaining of paclitaxel resistance. Although, at present, we are unable to clearly explain its underlying mechanism and significance, we thought that there might be a possible relation between the decreased expression of DPPIV and not only intrinsic but also acquired chemoresistance. We plan to elucidate the role of DPPIV expression and its enzyme activity in refractory, recurrent EOC in the future.
Regarding the link between the morphologies of the above‐mentioned EOC cell lines and paclitaxel sensitivity, weakly DPPIV‐expressing cells were spindle‐shaped, fibroblastic, and less paclitaxel‐sensitive, while those of highly DPPIV‐expressing EOC cells were epithelioid and had a cobblestone‐like appearance, and were comparatively sensitive to paclitaxel. Recent reports have shown that there are close links between the expression of TWIST, a key regulator of EMT through the modulation of E‐cadherin expression,( 33 ) and lower chemosensitivity to paclitaxel.( 34 ) In particular, Wang et al. demonstrated, through the analysis of eight nasopharyngeal carcinoma cell lines, that the upregulation of TWIST was associated with resistance to paclitaxel, and TWIST inactivation through small RNA interference led to an increased sensitivity to paclitaxel‐induced cell death.( 35 ) In our present results, DPPIV overexpression in EOC cells led to the downregulation of TWIST expression. Although they were very preliminary data, it is possible that the mechanism underlying DPPIV chemosensitivity is due to reduced TWIST expression, at least in part, regardless of aminopeptidase dependence. We plan to investigate the detailed correlation between DPPIV and TWIST in the next study.
In conclusion, the data obtained here suggest that DPPIV overexpression may lead to the reduced tumorigenicity and restoration of sensitivity to paclitaxel in EOC cells in vitro and in vivo. In addition, our finding that DPPIV expression may be an in vivo marker of tumor sensitivity to paclitaxel treatment may lead to future treatment strategies targeting DPPIV for selected human cancers in clinical practice.
References
- 1. Fleischer B. CD26: a surface protease involved in T‐cell activation. Immunol Today 1994; 15: 180–4. [DOI] [PubMed] [Google Scholar]
- 2. Heike M, Mobius U, Knuth A, Meuer S, Meyer zum Buschenfelde KH. Tissue distribution of the T cell activation antigen Ta1. Serological, immunohistochemical and biochemical investigations. Clin Exp Immunol 1988; 74: 431–4. [PMC free article] [PubMed] [Google Scholar]
- 3. Nemoto E, Sugawara S, Takada H, Shoji S, Horiuch H. Increase of CD26/dipeptidyl peptidase IV expression on human gingival fibroblasts upon stimulation with cytokines and bacterial components. Infect Immun 1999; 67: 6225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mizutani S, Sumi S, Narita O, Tomoda Y. Purification and properties of human placental dipeptidyl peptidase IV. Nippon Sanka Fujinka Gakkai Zasshi 1985; 37: 769–75. [PubMed] [Google Scholar]
- 5. Oravecz T, Pall M, Roderiquez G et al. Regulation of the receptor specificity and function of the chemokine RANTES (regulated on activation, normal T cell expressed and secreted) by dipeptidyl peptidase IV (CD26)‐mediated cleavage. J Exp Med 1997; 186: 1865–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mentlein R. Dipeptidyl‐peptidase IV (CD26) – role in the inactivation of regulatory peptides. Regul Pept 1999; 85 (1): 9–24. [DOI] [PubMed] [Google Scholar]
- 7. Hanski C, Huhle T, Reutter W. Involvement of plasma membrane dipeptidyl peptidase IV in fibronectin‐mediated adhesion of cells on collagen. Biol Chem Hoppe Seyler 1985; 366: 1169–76. [DOI] [PubMed] [Google Scholar]
- 8. Loster K, Zeilinger K, Schuppan D, Reutter W. The cysteine‐rich region of dipeptidyl peptidase IV (CD 26) is the collagen‐binding site. Biochem Biophys Res Commun 1995; 217: 341–8. [DOI] [PubMed] [Google Scholar]
- 9. Pethiyagoda CL, Welch DR, Fleming TP. Dipeptidyl peptidase IV (DPPIV) inhibits cellular invasion of melanoma cells. Clin Exp Metastasis 2000; 18: 391–400. [DOI] [PubMed] [Google Scholar]
- 10. Kajiyama H, Kikkawa F, Suzuki T, Shibata K, Ino K, Mizutani S. Prolonged survival and decreased invasive activity attributable to dipeptidyl peptidase IV overexpression in ovarian carcinoma. Cancer Res 2002; 62: 2753–7. [PubMed] [Google Scholar]
- 11. Yamochi T, Aytac U, Sato T et al. Regulation of p38 phosphorylation and topoisomerase IIalpha expression in the B‐cell lymphoma line Jiyoye by CD26/dipeptidyl peptidase IV is associated with enhanced in vitro and in vivo sensitivity to doxorubicin. Cancer Res 2005; 65: 1973–83. [DOI] [PubMed] [Google Scholar]
- 12. Wesley UV, McGroarty M, Homoyouni A. Dipeptidyl peptidase inhibits malignant phenotype of prostate cancer cells by blocking basic fibroblast growth factor signaling pathway. Cancer Res 2005; 65: 1325–34. [DOI] [PubMed] [Google Scholar]
- 13. Aytac U, Claret FX, Ho L et al. Expression of CD26 and its associated dipeptidyl peptidase IV enzyme activity enhances sensitivity to doxorubicin‐induced cell cycle arrest at the G(2)/M checkpoint. Cancer Res 2001; 61: 7204–10. [PubMed] [Google Scholar]
- 14. Sato K, Aytac U, Yamochi T et al. CD26/dipeptidyl peptidase IV enhances expression of topoisomerase II alpha and sensitivity to apoptosis induced by topoisomerase II inhibitors. Br J Cancer 2003; 89: 1366–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brun JL, Feyler A, Chene G, Saurel J, Brun G, Hocke C. Long‐term results and prognostic factors in patients with epithelial ovarian cancer. Gynecol Oncol 2000; 78 (1): 21–7. [DOI] [PubMed] [Google Scholar]
- 16. Taxman DJ, MacKeigan JP, Clements C, Bergstralh DT, Ting JP. Transcriptional profiling of targets for combination therapy of lung carcinoma with paclitaxel and mitogen‐activated protein/extracellular signal‐regulated kinase kinase inhibitor. Cancer Res 2003; 63: 5095–104. [PubMed] [Google Scholar]
- 17. Lee JT Jr, Steelman LS, McCubrey JA. Phosphatidylinositol 3’‐kinase activation leads to multidrug resistance protein‐1 expression and subsequent chemoresistance in advanced prostate cancer cells. Cancer Res 2004; 64: 8397–404. [DOI] [PubMed] [Google Scholar]
- 18. Kajiyama H, Shibata K, Terauchi M et al. Neutral endopeptidase 24.11/CD10 suppresses progressive potential in ovarian carcinoma in vitro and in vivo. Clin Cancer Res 2005; 11: 1798–808. [DOI] [PubMed] [Google Scholar]
- 19. Kajiyama H, Kikkawa F, Khin E, Shibata K, Ino K, Mizutani S. Dipeptidyl peptidase IV overexpression induces up‐regulation of E‐cadherin and tissue inhibitors of matrix metalloproteinases, resulting in decreased invasive potential in ovarian carcinoma cells. Cancer Res 2003; 63: 2278–83. [PubMed] [Google Scholar]
- 20. Umezu T, Kajiyama H, Terauchi M et al. Establishment of a new cell line of endometrioid carcinoma of the ovary and its chemosensitivity. Hum Cell 2007; 20: 71–6. [DOI] [PubMed] [Google Scholar]
- 21. Yamashita M, Kajiyama H, Terauchi M et al. Involvement of aminopeptidase N in enhanced chemosensitivity to paclitaxel in ovarian carcinoma in vitro and in vivo. Int J Cancer 2007; 120: 2243–50. [DOI] [PubMed] [Google Scholar]
- 22. Shibata K, Kikkawa F, Nawa A, Suganuma N, Hamaguchi M. Fibronectin secretion from human peritoneal tissue induces Mr 92,000 type IV collagenase expression and invasion in ovarian cancer cell lines. Cancer Res 1997; 57: 5416–20. [PubMed] [Google Scholar]
- 23. Horwitz SB. Taxol (paclitaxel): mechanisms of action. Ann Oncol 1994; 5 (Suppl 6): S3–6. [PubMed] [Google Scholar]
- 24. Wang TH, Wang HS, Soong YK. Paclitaxel‐induced cell death: where the cell cycle and apoptosis come together. Cancer 2000; 88: 2619–28. [DOI] [PubMed] [Google Scholar]
- 25. Wesley UV, Albino AP, Tiwari S, Houghton AN. A role for dipeptidyl peptidase IV in suppressing the malignant phenotype of melanocytic cells. J Exp Med 1999; 190: 311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sedo A, Malik R, Krepela E. Dipeptidyl peptidase IV in C6 rat glioma cell line differentiation. Biol Chem 1998; 379 (1): 39–44. [DOI] [PubMed] [Google Scholar]
- 27. Sedo A, Kraml J. Dipeptidyl peptidase IV in cell proliferation and differentiation. Sb Lek 1994; 95: 285–8. [PubMed] [Google Scholar]
- 28. Iwata S, Morimoto C. CD26/dipeptidyl peptidase IV in context. The different roles of a multifunctional ectoenzyme in malignant transformation. J Exp Med 1999; 190: 301–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wesley UV, Tiwari S, Houghton AN. Role for dipeptidyl peptidase IV in tumor suppression of human non small cell lung carcinoma cells. Int J Cancer 2004; 109: 855–66. [DOI] [PubMed] [Google Scholar]
- 30. Kajiyama H, Shibata K, Terauchi M, Ino K, Nawa A, Kikkawa F. Involvement of SDF‐1alpha/CXCR4 axis in the enhanced peritoneal metastasis of epithelial ovarian carcinoma. Int J Cancer 2008; 122 (1): 91–9. [DOI] [PubMed] [Google Scholar]
- 31. Zhou Y, Larsen PH, Hao C, Yong VW. CXCR4 is a major chemokine receptor on glioma cells and mediates their survival. J Biol Chem 2002; 277: 49481–7. [DOI] [PubMed] [Google Scholar]
- 32. Kajiyama H, Shibata K, Terauchi M et al. Chemoresistance to paclitaxel induces epithelial‐mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol 2007; 31: 277–83. [PubMed] [Google Scholar]
- 33. Comijn J, Berx G, Vermassen P et al. The two‐handed E box binding zinc finger protein SIP1 downregulates E‐cadherin and induces invasion. Mol Cell 2001; 7: 1267–78. [DOI] [PubMed] [Google Scholar]
- 34. Zhang X, Wang Q, Ling MT, Wong YC, Leung SC, Wang X. Anti‐apoptotic role of TWIST and its association with Akt pathway in mediating taxol resistance in nasopharyngeal carcinoma cells. Int J Cancer 2007; 120: 1891–8. [DOI] [PubMed] [Google Scholar]
- 35. Wang X, Ling MT, Guan XY et al. Identification of a novel function of TWIST, a bHLH protein, in the development of acquired taxol resistance in human cancer cells. Oncogene 2004; 23: 474–82. [DOI] [PubMed] [Google Scholar]