

Abstract
Intestinal metaplasia has been investigated extensively as a possible premalignant condition for stomach cancer but its pathogenesis is still not fully understood. In the present study, we examined the relationship between endocrine and mucous cell marker expression periodically after Helicobacter pylori infection in the Mongolian gerbil model. The numbers of chromogranin A (CgA)‐positive, gastrin‐positive and gastric inhibitory polypeptide (GIP)‐positive cells in H. pylori‐infected groups was increased significantly compared with the non‐infected case. However, CgA‐positive and gastrin‐positive cells then decreased from 50 through 100 experimental weeks after H. pylori infection, whereas GIP‐positive cells increased. Coexistence of gastrin‐positive and GIP‐positive cells was detected in the same gastric and intestinal mixed phenotypic glandular‐type glands. In conclusion, the endocrine cell phenotype is in line with that of the mucous counterpart in the glands of H. pylori‐infected Mongolian gerbil stomach, supporting the concept that development of intestinal metaplasia is due to the abnormal differentiation of a stem cell. (Cancer Sci 2006; 97: 1015–1022)
The Mongolian gerbil (MG) is useful for examining the link between Helicobacter pylori infection and human gastric disorders, as the lesions induced by H. pylori in this experimental animal resemble those apparent in man.( 1 ) In our animal model, we have previously demonstrated that eradication at early stages of inflammation is effective in preventing H. pylori‐related stomach carcinogenesis.( 2 ) Wong et al.( 3 ) have demonstrated similar results in a human randomized‐controlled trial of H. pylori eradication in China, and pointed out the importance of analyses of the factors that determine irreversibility − in other words, the point of no return. Thus, for the prevention of stomach cancer in MG, it is very important to estimate the histological and genetic alternations in the glandular stomach periodically and continuously after H. pylori infection, which is impossible in humans because of imprecise information on the time of infection with bacteria.
Several studies have demonstrated that changes in endocrine and mucous cells are observed in intestinal metaplasia (IM) in the human pyloric mucosa associated with H. pylori infection.( 4 , 5 , 6 ) In the MG model, alterations in the endocrine cell population are also found during H. pylori infection.( 7 , 8 , 9 ) Regarding the cellular differentiation of endocrine cells in the gastrointestinal tract, gastrin is detectable predominantly in the pyloric glands of the stomach, whereas gastric inhibitory polypeptide (GIP) is characteristic of the duodenum and small intestine.( 10 , 11 , 12 , 13 , 14 ) Therefore, gastrin could be a gastric endocrine cell marker, in contrast to GIP as an intestinal example.( 14 ) We have recently documented clear evidence that the phenotypes of endocrine cells are associated strongly with those of mucous cells in human IM as well as in normal gastric glands,( 14 ) supporting the hypothesis that abnormal differentiation of stem cells underlies the development of IM in the human stomach.( 15 ) With investigations of the histogenesis of H. pylori‐related lesions, it is very interesting to focus on relationships between endocrine and mucous cells periodically in the MG model from the viewpoint of phenotypic expression.
In the present study, we therefore examined the expression of endocrine cell markers by immunohistochemistry and the quantitative real‐time reverse transcription–polymerase chain reaction (RT‐PCR) using a gland isolation technique, and evaluated the relationship between endocrine and mucous cell marker expression at 50, 75 and 100 weeks after H. pylori infection in the MG model.
Materials and Methods
Samples. Seventy specific pathogen‐free male MG (Seac Yoshitomi, Fukuoka, Japan), aged 7 weeks, and H. pylori (ATCC 43504; American Type Culture Collection, Rockville, MD, USA) were used for this study. The bacteria were grown from freezer stocks for 72 h and harvested in Brucella broth. Samples (0.8 mL) containing approximately 1.0 × 108 colony‐forming units per mL were used as the inoculum, as described earlier.( 16 , 17 , 18 , 19 ) Uninfected gerbils underwent sham inoculation using the same sterile Brucella broth.
The animals were divided into two major groups: H. pylori‐infected (Hp[+]), and non‐infected (Hp[–]) groups, and each group was subclassified with reference to time of death at 50, 75 and 100 weeks. Finally, the animals (n = 70) were divided into Hp(+)‐50‐week (n = 18), Hp(+)‐75‐week (n = 6), Hp(+)‐100‐week (n = 17), Hp(–)‐50‐week (n = 19), Hp(–)‐75‐week (n = 6) and Hp(–)‐100‐week (n = 4) groups (Fig. 1).
Figure 1.

Experimental design. Hp, Helicobacter pylori.
After 24 h fasting, all animals were subjected to deep ether anesthesia, laparotomized and exsanguinated from the inferior vena cava, with excision of their stomachs.( 16 ) The numbers of stomach samples used for immunohistochemical analysis were 10 for Hp(+)‐50‐week, six for Hp(+)‐75‐week, nine for Hp(+)‐100‐week, eight for Hp(–)‐50‐week, six for Hp(–)‐75‐week and four for Hp(–)‐100‐week groups. The fundic and pyloric regions, duodenum, and small and large intestines of five Hp(–) ‐50‐week gerbils were used as controls for immunohistochemical analyses. RNA extraction from the mucosa was also carried out for the fundic and pyloric regions, duodenum, and small and large intestines of three Hp(–)‐50‐week animals. For RNA extraction from isolated glands, pyloric regions were used from eight Hp(+)‐50‐week and eight Hp(+)‐100‐week gerbils. With the three Hp(–)‐50‐week cases, RNA extraction from isolated glands was carried out for the pyloric region and jejunum. After death, successful H. pylori infection was confirmed by remarkable elevation of serum IgG titers and/or histological inflammatory change in all H. pylori‐infected MG (data not shown).
Each tissue sample was fixed in 95% ethanol plus 1% acetic acid, sectioned at 4 µM, and stained with hematoxylin and eosin for histochemical examination.( 18 , 19 )
Immunohistochemistry. Immunohistochemical staining was carried out with the polyclonal antibodies listed in Table 1.( 14 ) The precise procedures for immunohistochemical techniques were as described previously.( 14 , 15 , 18 , 19 , 20 , 21 , 22 ) Briefly, 4 µm‐thick consecutive sections were deparaffinized and hydrated through a graded series of ethanol. After inhibition of endogenous peroxidase activity by immersion in 3% H2O2/methanol solution, sections were incubated with primary antibodies, washed thoroughly in phosphate‐buffered saline (PBS), then incubated with biotinylated secondary antibody followed by the avidin‐biotinylated horseradish peroxidase complex (Vectastain Elite ABC kit; Vector Laboratories, Burlingame, CA, USA). Finally, immune complexes were visualized by incubation with 0.01% H2O2 and 0.05% 3.3′‐diaminobenzidine tetrachloride (DAB). Nuclear counterstaining was accomplished with Mayer's hematoxylin. Two independent investigators (YT and TT) judged the histology and immunohistochemical staining of each marker.
Table 1.
Phenotypic markers for gastrointestinal endocrine and mucous cells
| Cell types | Tissue specificity | Marker | Source of antibody |
|---|---|---|---|
| Endocrine | Ubiquitous | Chromogranin A | Dako (Glostrup, Denmark) |
| Gastric | Gastrin | Yanaihara Institute (Fujinomiya, Japan) | |
| Intestinal | Gastric inhibitory polypeptide | Yanaihara | |
| Mucous | Gastric | Periodic acid–Schiff † staining (mucin stained red) | |
| Intestinal | Alcian blue ‡ staining (mucin stained blue) |
Periodic acid‐schiff (Nakalai Tesque, Kyoto, Japan),
‡ Alcian blue (Wako Pure Chemical Industries Ltd, Osaka, Japan)
Classification of glandular ducts in the stomachs of MG. The endocrine cells in each glandular duct were identified as cells with chromogranin A (CgA) cytoplasmic expression. Gastrin is a marker of gastric endocrine cells, whereas GIP is a typical intestinal endocrine cell marker (Table 1).( 14 )
Regarding the phenotypes of glandular ducts with reference to mucous cell differentiation, we used Alcian blue–periodic acid–Schiff staining (AB‐PAS) for identifying gastric surface mucous cells with mucin stained red and goblet cells stained blue (Table 1).( 21 , 23 ) Non‐neoplastic glandular ducts in the stomach were divided histologically and phenotypically into three types: gastric phenotypic glandular (G‐type gland), gastric‐and‐intestinal‐mixed phenotypic glandular (GI‐type gland), and intestinal phenotypic glandular (I‐type gland) ducts.( 15 , 20 )
Multiple staining with AB‐PAS, gastrin and GIP. Multiple staining with AB‐PAS, gastrin and GIP was achieved as follows. After staining with AB‐PAS, sections were incubated with anti‐gastrin antibody, followed by application of the biotin‐labeled goat antirabbit IgG and the peroxidase‐labeled avidin–biotin complex method. Binding was visualized with DAB. After thorough washing with Tris‐buffered saline, incubation was with the anti‐GIP antibody, followed by application of alkaline phosphatase‐labeled antirabbit IgG, and development with nitroblue tetrazolium chloride (NBT) and 5‐bromo‐4‐chloro‐3‐indolyl‐phosphate (BCIP) (BCIP/NBT substrate system for immunohistochemistry and in situ hybridization; Dako, Glostrup, Denmark) using the indirect immuno‐alkaline phosphatase method. With this multiple staining, gastrin‐positive and GIP‐positive cells were stained brown and dark purple, respectively. Glands were classified by AB‐PAS staining into G‐type, GI‐type and I‐type glands.
Gland isolation. Gland isolation was carried out as described previously.( 20 , 24 ) Fresh gastrointestinal tissues were obtained from MG, washed thoroughly in calcium‐ and magnesium‐free PBS to remove the luminal contents and cut into 2–4‐cm squares. These were then incubated in calcium‐ and magnesium‐free Hanks’ balanced salt solution containing 30 mM ethylenediamine‐tetraacetate (pH 7.0) and shaken for 15–20 min at 37°C. The gastrointestinal epithelium was then shaven off using the back of a knife, and harvested. The isolated glands were fixed in 70% ethanol and stored at −20°C until RNA extraction.
Sequencing of CgA, gastrin and GIP cDNA in MG. Total RNA from MG stomach and jejunum mucosae were extracted using ISOGEN (Nippon Gene, Toyama, Japan).( 20 ) First strand cDNA was synthesized basically as described previously( 25 ) using random primers with the Thermoscript RT‐PCR System (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions. To obtain partial cDNA sequences for CgA in MG, the most homologous regions were selected by comparison of the rat (DNA Bank of Japan [DDBJ]/European Molecular Biology Laboratory [EMBL]/GenBank accession number, NM_021655) and mouse (NM_007693) sequences (Table 2). To determine gastrin cDNA sequences, those of rat (NM_012849) and mouse (NM_010257) were compared for selection of one primer pair (Table 2). Regarding GIP, the most homologous regions were selected by comparison with the rat (X66724) and mouse (NM_008119) sequences (Table 2). After successful RT‐PCR amplification using cDNA from gerbil stomach and jejunum as a template, sequencing was carried out using the BigDye Terminator version 3.1 Cycle Sequencing Kit (Applied Biosystems, Foster, CA, USA) utilizing ABI Prism 3100 (Applied Biosystems) according to the manufacturer's instructions.
Table 2.
Primer sequences of chromogranin A (CgA), gastrin and gastric inhibitory polypeptide (GIP) in Mongolian gerbils
| Gene | Direction | Sequence | Product length (bp) |
|---|---|---|---|
| CgA | Sense | 5′‐CAAAAGGGGACACCAAGGTGATGAAGTG‐3′ | 153 |
| Antisense | 5′‐TCAGCAGATTCTGGTGTCGCAGGATAGA‐3′ | ||
| Gastrin | Sense | 5′‐GGAAGCCCCGCTCCCAGCTACAGGATG‐3′ | 171 |
| Antisense | 5′‐TCCGTGGCCTCTGCTTCTTGGACAGGTC‐3′ | ||
| GIP | Sense | 5′‐AGTGATTACAGCATCGCCATGGACAA‐3′ | 243 |
| Antisense | 5′‐CCAGGCCAGTAGCTCTTGAATCAGAAGG‐3′ | ||
| GAPDH | Sense | 5′‐AACGGCACAGTCAAGGCTGAGAACG‐3′ | 118 |
| Antisense | 5′‐CAACATACTCGGCACCGGCATCG‐3′ |
GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase.
Real‐time RT‐PCR. The oligo(dT)‐primed cDNA was synthesized as described previously, using the Thermoscript RT‐PCR System.( 20 , 25 ) Relative quantitative real‐time RT‐PCR of CgA, gastrin and GIP was carried out using the LightCycler system (Roche Diagnostics, Mannheim, Germany) with a SYBR Green PCR Kit (Qiagen, Hilden, Germany).( 20 ) The primer sequences of each marker are shown in Table 2. Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH; DDBJ/EMBL/GenBank accession number AB040445) was used as an internal control. Relative quantification was carried out as established earlier using an internal control, without the necessity for external standards.( 25 ) Values are expressed as a percentage of the CgA‐positive and gastrin‐positive cells in the pyloric region, and of GIP‐positive cells in the duodenum (both set as 100%). Specificity of the PCR reaction was confirmed using the melting program provided with the LightCycler software. To further confirm that there was no obvious primer dimer formation or amplification of any extra bands, the samples were electrophoresed in 2.5% agarose gels and visualized with ethidium bromide after the LightCycler reaction. Total RNA samples without RT provided a control for PCR amplification (data not shown).
Statistical analysis. The data were analyzed between groups using the Mann–Whitney U‐test. The Kruskal–Wallis test was applied to establish the significance of differences among G‐, GI‐ and I‐type glands with reference to gastrin and GIP expression among the Hp(+)‐50‐week, Hp(+)‐75‐week and Hp(+)‐100‐week groups. P‐values < 0.05 were considered statistically significant.
Results
Progression of intestinal metaplasia: average numbers of G‐, GI‐ and I‐type glands in H. pylori‐infected MG. Fig. 2 shows the average numbers of G‐, GI‐ and I‐type glands per 1 cm of the glandular stomach mucosa (number/cm) in the Hp(–)‐50‐week (n = 8), Hp(+)‐50‐week (n = 10), Hp(+)‐75‐week (n = 6) and Hp(+)‐100‐week (n = 9) cases. The average numbers of G‐type glands were 387.8 ± 62.8 (mean ± SD), 398.5 ± 91.3, 368.8 ± 128.9 and 401.2 ± 83.9, respectively. The average numbers of GI‐type glands were 0 ± 0, 0.018 ± 0.039, 0.092 ± 0.083 and 0.079 ± 0.125, respectively, whereas those of I‐type glands were 0 ± 0, 0.47 ± 0.32, 3.09 ± 1.40 and 2.83 ± 0.82, respectively. Thus, no GI‐ or I‐type glands were observed in Hp(–)‐50‐week animals as controls.
Figure 2.
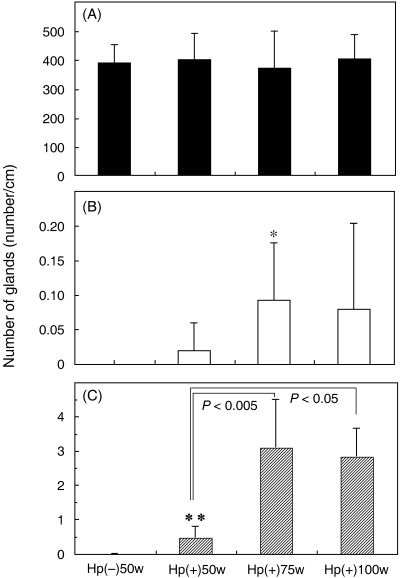
The average numbers (mean ± SD) of (A) glandular (G)‐type, (B) gastric and intestinal mixed (GI)‐type and (C) intestinal (I)‐type glandular ducts in Hp(–)‐50‐week (n = 8), Hp(+)‐50‐week (n = 10), Hp(+)‐75‐week (n = 6), and Hp(+)‐100‐week (n = 9) cases. *P < 0.05, **P < 0.005 vs the average Hp(–)‐50‐week number.
The number of I‐type glands in Hp(+)‐50‐week, Hp(+)‐75‐week and Hp(+)‐100‐week stomachs was increased significantly compared with the Hp(–)‐50‐week case (P < 0.005). In the Hp(+) groups, the average number of I‐type glands increased significantly from week 50 to week 75, and then did not change at week 100. Regarding the GI‐type glands, there was a significant difference between Hp(–)‐50‐week and Hp(+)‐75‐week (P < 0.05).
Immunolocalization of CgA, gastrin and GIP in the normal alimentary tract of MG. Expression of CgA, gastrin and GIP in the normal gastrointestinal tract was estimated by immunohistochemistry in Hp(–)‐50‐week animals (n = 5) (Fig. 3). The average numbers of CgA‐positive cells per 1 cm of the mucosa (number/cm) in fundic regions, pyloric regions, duodenum, small intestines and large intestines were 124.1 ± 19.3 (mean ± SD), 106.7 ± 38.6, 30.7 ± 14.9, 16.9 ± 5.2 and 44.3 ± 25.2, respectively. The expression of CgA decreased gradually from the fundic region to the small intestine and increased from the small to large intestine (Fig. 4A). The average numbers of gastrin‐positive cells in fundic regions, pyloric regions, duodenum, small intestines and large intestines were 1.1 ± 1.3, 99.8 ± 39.9, 2.6 ± 3.4, 0 ± 0 and 0 ± 0, respectively. No gastrin expression was detected in the small and large intestines. The average numbers of GIP‐positive cells in fundic regions, pyloric regions, duodenum, small intestines and large intestines were 0.6 ± 0.6, 1.1 ± 0.5, 25.2 ± 12.1, 22.3 ± 7.5 and 0.9 ± 0.2, respectively. GIP expression was observed frequently in the small but not the large intestine.
Figure 3.
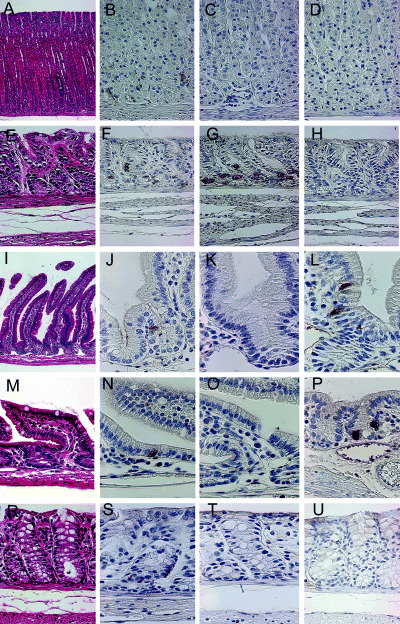
Immunohistochemical staining of chromogranin A (CgA), gastrin and gastric inhibitory polypeptide (GIP) in (A–D) normal fundic and (E–H) pyloric regions, (I–L) duodenum, (M–P) small intestines and (R–U) large intestines of Mongolian gerbils. Expression of CgA was observed in the bottom of (B) normal fundic, (F) pyloric, (J) duodenal, (N) small intestinal and (S) colonic glandular ducts. (C,D) No gastrin or GIP was observed in the fundic glands. In the pyloric glands, (G) expression of gastrin was detected clearly, but (H) no GIP expression was observed. In the duodenum and small intestine, (L,P) GIP expression was detected, but (K,O) no gastrin expression was observed. Neither (T) gastrin nor (U) GIP were detected in the large intestine. Original magnification: (A) ×100; (B–U) ×400.
Figure 4.
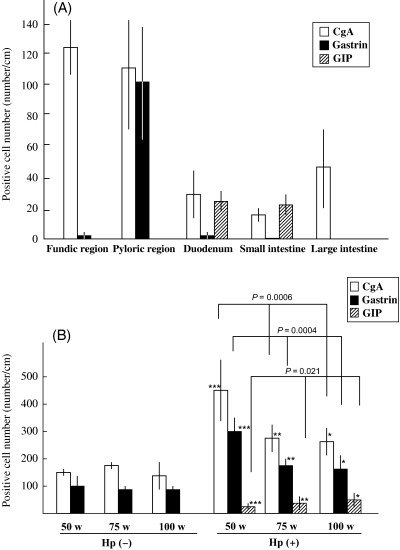
Immunohistochemical analysis of chromogranin A (CgA)‐, gastrin‐ and gastric inhibitory polypeptide (GIP)‐positive cells. (A) Normal gastrointestinal tract. (B) Helicobacter pylori‐infected Mongolian gerbils. *P < 0.01, **P < 0.005, ***P < 0.0005 compared with each control group.
Immunohistochemical analysis of CgA, gastrin and GIP in the pyloric regions of H. pylori‐infected MG stomachs. For immunohistochemical analyses of CgA, gastrin and GIP, stomach samples of eight Hp(–)‐50‐week, six Hp(–)‐75‐week, four Hp(–)‐100‐week, ten Hp(+)‐50‐week, six Hp(+)‐75‐week and nine Hp(+)‐100‐week animals were used (Fig. 4B). The average numbers of CgA‐positive cells per 1 cm of the pyloric region (number/cm) were 156.7 ± 6.4, 175.7 ± 2.5, 136.3 ± 60.4, 446.1 ± 104.2, 271.9 ± 175.4 and 250.0 ± 60.0, respectively. There was no significant variation in the number of CgA‐positive cells in the controls at the three time points. Values were elevated in each H. pylori‐infected group, but decreased gradually from Hp(+)‐50‐week through Hp(+)‐75‐week to Hp(+)‐100‐week (P = 0.0006). The average number of gastrin‐positive cells in Hp(–)‐50‐week, Hp(–)‐75‐week, Hp(–)‐100‐week, Hp(+)‐50‐week, Hp(+)‐75‐week and Hp(+)‐100‐week were 96.1 ± 46.7, 83.7 ± 8.6, 92.7 ± 11.5, 295.1 ± 54.1, 175.4 ± 24.5 and 162.7 ± 44.7, respectively. The average number of GIP‐positive cells was 1.9 ± 1.4, 2.1 ± 0.2, 2.8 ± 1.8, 20.0 ± 5.0, 35.5 ± 15.0 and 40.4 ± 24.3, respectively.
Determination of partial nucleotide sequences of CgA, gastrin and GIP. Partial nucleotide sequences of neuroendocrine markers in the MG were determined and deposited at the DNA Data Bank of Japan (DDBJ) (http://www.ddbj.nig.ac.jp/Welcome‐e.html). DDBJ/EMBL/GenBank The accession numbers for CgA, gastrin and GIP are AB253527, AB253528 and AB253529, respectively.
Expression of CgA, gastrin and GIP mRNA in the normal alimentary tract. The expression of CgA, gastrin and GIP mRNA in the normal gastrointestinal tract was also estimated by real‐time RT‐PCR in Hp(–)‐50‐week animals (n = 3) (Fig. 5A), values being expressed as a percentage of those in the pyloric mucosa of stomach for CgA and gastrin, and relative to that in the duodenum for GIP. The average relative expression levels of CgA in fundic regions, pyloric regions, duodenum, small intestines and large intestines were 74.9% (35.05%[mean − SD]−160.11%[mean + SD]), 100% (59.87–167.02%), 10.25% (6.29–16.69%), 9.39% (7.62–11.56%) and 57.04% (51.50–63.17%), respectively. The corresponding figures for gastrin were 1.69% (0.45–6.34%), 100% (39.36–254.08%), 1.12% (0.15–8.34%), 0.0036% (0.00032–0.041%) and 0.015% (0.000024–8.76%), and for GIP were 0.00019% (0.000059–0.00064%), 1.30% (0.46–3.67%), 100% (61.69–162.11%), 22.85% (7.00–74.60%) and 0.0088% (0.0014–0.057%). In line with the immunohistochemical and real‐time RT‐PCR findings, gastrin and GIP were used as gastric and intestinal endocrine cell markers, respectively, in MG.
Figure 5.
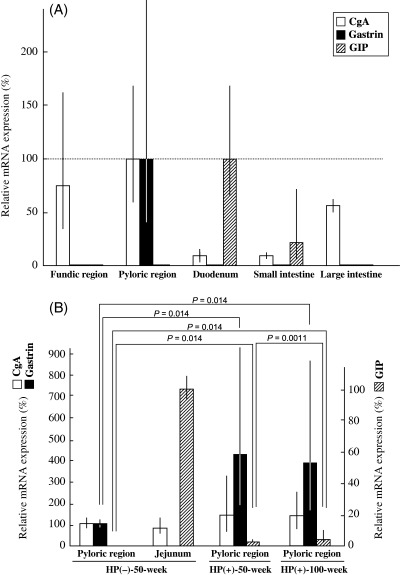
Real‐time reverse transcription–polymerase chain reaction analysis of chromogranin A (CgA), gastrin and gastric inhibitory polypeptide (GIP) in the pyloric region of Mongolian gerbils. (A) Normal alimentary tract. (B) Isolated pyloric glands from Helicobacter pylori‐infected stomachs.
Alteration of CgA, gastrin and GIP mRNA expression in isolated pyloric glands from H. pylori‐infected MG. We used gland isolation to avoid contamination of epithelial cell elements with stromal or inflammatory cells. Isolated glands were obtained from the pyloric regions of Hp(–)‐50‐week (n = 3), Hp(+)‐50‐week (n = 8) and Hp(+)‐100‐week (n = 8) animals. Examples were also collected as controls of GIP mRNA expression from Hp(–)‐50‐week jejunums (n = 3). Values are expressed as the percentage of those in isolated glands obtained from the pyloric region for CgA and gastrin and in the jejunum for GIP (Fig. 5B). Average relative expression levels for CgA mRNA were 100% (81.50–122.70%), 80.11% (54.32–118.14%), 136.60% (55.78–334.55%) and 147.55% (82.65–263.43%) in the normal Hp(–)‐50‐week pyloric region and the jejunum, and in the H. pylori‐infected pyloric regions of Hp(+)‐50‐week and Hp(+)‐100‐week animals, respectively. Those for gastrin mRNA were 100% (89.95–111.17%), 0.21% (0.10–0.44%), 436.46% (205.35–927.65%) and 399.88% (182.48–876.31%), respectively, and for GIP mRNA were 0.00057% (0.000037–0.0086%), 100% (92.68–107.90%), 0.29% (0.14–0.57%) and 3.38% (1.40–8.17%), respectively.
The expression of gastrin mRNA in H. pylori‐infected groups (Hp[+]‐50‐week and Hp[+]‐100‐week) was increased significantly compared with the Hp(–)‐50‐week case. However, there was no significant difference in gastrin mRNA expression between Hp(+)‐50‐week and Hp(+)‐100‐week groups. The expression of GIP mRNA in H. pylori‐infected groups (Hp[+]‐50‐week and Hp[+]‐100‐week) was also increased significantly compared with Hp(–)‐50‐week. In addition, there was a significant difference in GIP between Hp(+)‐50‐week and Hp(+)‐100‐week (Fig. 5B). Regarding CgA mRNA expression, there was no statistically significant variation among the groups.
Colocalization of gastrin‐ and GIP‐positive endocrine cells of GI‐type glands. The presence of gastrin‐ and GIP‐positive cells was double‐immunohistochemically evaluated with AB‐PAS staining (Fig. 6) to directly compare the localization of endocrine and mucous G‐type and I‐type markers. The numbers of gastrin‐positive and GIP‐positive cells per gland (number/gland) were analyzed in 60 G‐type, 15 GI‐type and 60 I‐type glands of H. pylori‐infected groups. The average numbers of gastrin‐positive cells were 1.48 ± 0.57, 0.53 ± 0.51 and 0.03 ± 0.18 in G‐, GI‐ and I‐type glands, respectively. Those for GIP‐positive cells were 0.15 ± 0.36, 1.00 ± 0.53 and 1.68 ± 0.65, respectively (Fig. 7). Gastrin‐positive cells decreased gradually from G‐ through GI‐, to I‐type glands (P < 0.0001), whereas GIP‐positive cells were correlated inversely (P < 0.0001) (Fig. 7). Coexistence of gastrin‐ and GIP‐positive cells was detected in the same gland in GI‐type glands (Fig. 6).
Figure 6.
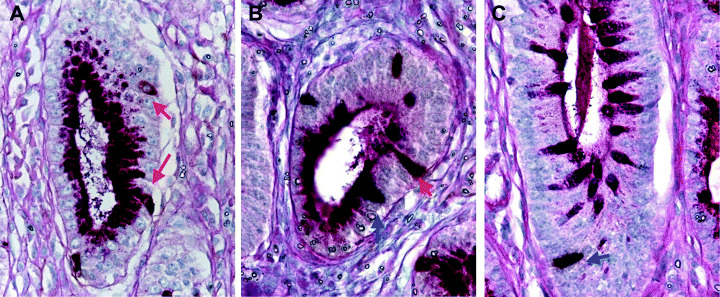
Expression of gastrin and gastric inhibitory polypeptide (GIP) in (A) glandular (G)‐type, (B) gastric and intestinal mixed (GI)‐type and (C) intestinal (I)‐type glands. (A) In the G‐type glands, gastrin‐positive cells were identified by brown cytoplasmic staining (red arrow), but no GIP expression was observed. (B) In the GI‐type glands, gastrin‐ and GIP‐positive cells were identified by brown (red arrow) and dark purple (blue arrow) cytoplasmic staining. (C) In the I‐type glands, GIP expression was detected by dark purple staining (blue arrow), but no gastrin expression was apparent. Original magnification: (A–C) ×500.
Figure 7.
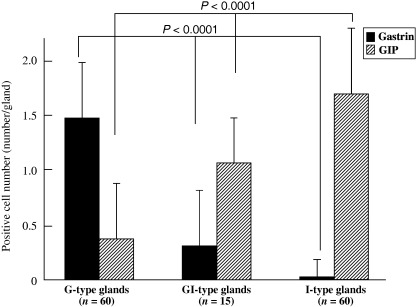
Expression of gastrin and gastric inhibitory polypeptide (GIP) in glandular (G)‐type, gastric and intestinal mixed (GI)‐type and intestinal (I)‐type glands. The number of gastrin‐positive cells decreased gradually from G‐type through GI‐type to I‐type glands (P < 0.0001), correlating inversely with the number of GIP‐positive cells (P < 0.0001).
Discussion
To our knowledge, this is the first report of expression of endocrine and mucous cell markers observed periodically in the glandular stomach of H. pylori‐infected MG, although several studies have shown that long‐term H. pylori colonization produces hyperplasia of gastrin‐producing antral G‐cells and carcinoid tumors in MG.( 7 , 9 , 26 ) In the present study, the immunohistochemical data demonstrated that the numbers of CgA‐ and gastrin‐positive cells in H. pylori‐infected groups was increased significantly compared with the non‐infected condition, but both demonstrated a gradual decrease over time, despite the lack of any significant variation in CgA or gastrin mRNA expression between Hp(+)‐50‐week and Hp(+)‐100‐week. In humans, there have been several reports of no significant differences in the number of G‐cells and G‐cell density in the stomach mucosa between H. pylori‐infected and ‐uninfected healthy volunteers,( 27 , 28 , 29 ) although the number of G‐cells was significantly less in patients with both H. pylori infection and duodenal ulcer than in either infected or uninfected controls.( 27 , 28 , 29 , 30 ) Kamada et al. reported that G‐cell number in H. pylori‐associated gastritis mucosa was decreased in comparison with an uninfected case.( 30 ) Diamaline et al. suggested that CgA production in enterochromaffin‐like cells of the rat stomach was part of the functional response of these cells to circulating gastrin.( 31 ) We consider that the expression of some factors, including CgA and gastrin, is influenced by the time after H. pylori infection, and further analyses in the MG model may explain the discrepancy with human reports. Regarding the relationship between IM and the change in endocrine cells, several reports demonstrated that G‐cells disappeared and I‐type endocrine cells conversely appeared in human IM mucosa,( 4 , 5 , 6 ) and that many Glicentin‐positive, intestinal phenotype cells were found at the IM gland level,( 6 , 32 ) in line with our gland isolation findings. However, few GIP‐positive cells were observed in non‐infected groups. H. pylori infection may trigger intestinalization of the stomach mucosa.( 21 ) We have showed previously that the phenotypes of endocrine cells are associated strongly with those of mucous cells in human IM,( 14 ) and the GI‐type glands in the present study had both gastrin‐ and GIP‐positive cells in the H. pylori‐infected MG glandular stomach. This evidence supports the concept that all of the different types of mucous and endocrine cells may be generated from a single stem cell.( 15 )
Regarding intestinalization of the stomach mucosa, changes in the expression of various genes, especially homeobox examples determining cell structures and functions, may be involved. The fetal stomach, which develops from the foregut, displays areas of I‐type mucosa with goblet cells and epithelial cells with striated borders in the antrum and cardia.( 33 ) It is important to consider the correlation between expression of phenotypes and organ‐specific genes. We earlier showed that Sox2 and Cdx1/2 are gastric‐ and intestinal‐specific transcription factors, respectively.( 15 , 20 ) In isolated pyloric and intestinal metaplastic glands, the phenotypes of mucous cells were found to be associated strongly with these specific transcription factors. In isolated GI‐type glands, Sox2 and Cdx1/2 were both observed, as well as gastric and intestinal mucous cell markers such as MUC5AC, MUC6, MUC2 and villin. Recently, La Rosa et al. demonstrated that Cdx2 may be a sensitive and specific marker of midgut endocrine cells and endocrine tumors.( 34 ) We think that Cdx2 might be important in the regulation of intestinal phenotype endocrine cell markers such as GIP, glicentin and glucagon‐like polypeptide‐1 because of its localization. Jenny et al. reported previously that neurogenin3, a basic helix‐loop‐helix transcription factor, is required for endocrine cell fate specification in multipotent intestinal progenitor cells, whereas gastric endocrine development is both neurogenin3 dependent and independent.( 35 ) Thus, specific transcription factors, including Cdx2, might play an important role in the intestinalization of endocrine cells as well as mucous cells, because a phenotypic link was here observed sequentially between mucous and endocrine cells. Moreover, recently, several reports have demonstrated that impaired expression of the stomach morphogenic factor Sonic hedgehog (Shh) by parietal cells and increased expression of the transcriptional activators of intestinal and pancreatic differentiation, namely CDX2 and PDX1, are crucial for the development of stomach atrophy and for intestinal, endocrine and pancreatic transdifferentiation processes.( 36 , 37 ) Suzuki et al. described that prolonged colonization by H. pylori led to extension of inflammation from the antrum to the corpus of the stomach with downregulation of Shh in gastric epithelial differentiation in the MG model.( 38 ) Again, evaluation of the expression of the specific transcription factors detailed above should be carried out periodically in the H. pylori‐infected MG model.
Our present data demonstrate that most glands in the stomach of H. pylori‐infected MG were G‐type. The numbers of GI‐ and I‐type glands were extremely low in comparison. In the non‐infected MG, no GI‐ or I‐type glands were detected. We have demonstrated previously that most stomach cancers present with gastric phenotypic expression in the glandular stomach of H. pylori‐infected MG treated with carcinogens.( 21 ) In the rat, Tatematsu et al. reported that pepsinogen 1‐altered pyloric glands, which are low in pepsinogen 1, are putative preneoplastic lesions in the glandular stomach.( 39 , 40 , 41 ) In humans, most early‐stage gastric cancers consist mainly of G‐type cancer cells, irrespective of histological type.( 15 , 42 , 43 , 44 ) Thus, it is important to note the possibility that precancerous lesions exist in G‐type as well as IM glands.( 21 )
In conclusion, the phenotype of endocrine cells is in line with that of their mucous cell counterparts in the glands of the H. pylori‐infected MG stomach, supporting the concept that the development of IM is due to abnormal differentiation of stem cells. For elucidation of stomach carcinogenesis, it is very important to evaluate factors related to H. pylori infection periodically in the MG model.
Acknowledgments
The authors thank Mr Harunari Tanaka for expert technical assistance and Dr Malcolm A. Moore for revision of the scientific English language. This study was supported in part by a Grant‐in‐Aid for the Third‐term Comprehensive 10‐year Strategy for Cancer Control and a Grant‐in‐Aid for Cancer Research from the Ministry of Health, Labour and Welfare, Japan, and a Grant‐in‐Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1. Hirayama F, Takagi S, Yokoyama Y, Iwao E, Ikeda Y. Establishment of gastric Helicobacter pylori infection in Mongolian gerbils. J Gastroenterol 1996; 31 (Suppl. 9): 24–8. [PubMed] [Google Scholar]
- 2. Nozaki K, Shimizu N, Ikehara Y et al. Effect of early eradication on Helicobacter pylori‐related gastric carcinogenesis in Mongolian gerbils. Cancer Sci 2003; 94: 235–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wong BC, Lam SK, Wong WM et al. Helicobacter pylori eradication to prevent gastric cancer in a high‐risk region of China: a randomized controlled trial. JAMA 2004; 291: 187–94. [DOI] [PubMed] [Google Scholar]
- 4. Bordi C, Ravazzola M. Endocrine cells in the intestinal metaplasia of gastric mucosa. Am J Pathol 1979; 96: 391–8. [PMC free article] [PubMed] [Google Scholar]
- 5. Mingazzini P, Carlei F, Malchiodi‐Albedi F et al. Endocrine cells in intestinal metaplasia of the stomach. J Pathol 1984; 144: 171–8. [DOI] [PubMed] [Google Scholar]
- 6. Tsutsumi Y, Nagura H, Watanabe K, Yanaihara N. A novel subtyping of intestinal metaplasia of the stomach, with special reference to the histochemical characterizations of endocrine cells. Virchows Arch A Pathol Anat Histopathol 1983; 401: 73–88. [DOI] [PubMed] [Google Scholar]
- 7. Kagawa J, Honda S, Kodama M, Sato R, Murakami K, Fujioka T. Enterocromaffin‐like cell tumor induced by Helicobacter pylori infection in Mongolian gerbils. Helicobacter 2002; 7: 390–7. [DOI] [PubMed] [Google Scholar]
- 8. Hirayama F, Takagi S, Yokoyama Y, Yamamoto K, Iwao E, Haga K. Long‐term effects of Helicobacter pylori eradication in Mongolian gerbils. J Gastroenterol 2002; 37: 779–84. [DOI] [PubMed] [Google Scholar]
- 9. Hirayama F, Takagi S, Iwao E, Yokoyama Y, Haga K, Hanada S. Development of poorly differentiated adenocarcinoma and carcinoid due to long‐term Helicobacter pylori colonization in Mongolian gerbils. J Gastroenterol 1999; 34: 450–4. [DOI] [PubMed] [Google Scholar]
- 10. Oberg K. Gastric neuroendocrine cells and secretory products. Yale J Biol Med 1998; 71: 149–54. [PMC free article] [PubMed] [Google Scholar]
- 11. Owen DA. Normal histology of the stomach. Am J Surg Pathol 1986; 10: 48–61. [DOI] [PubMed] [Google Scholar]
- 12. Sjolund K, Sanden G, Hakanson R, Sundler F. Endocrine cells in human intestine: an immunocytochemical study. Gastroenterology 1983; 85: 1120–30. [PubMed] [Google Scholar]
- 13. Hocker M, Wiedenmann B. Molecular mechanisms of enteroendocrine differentiation. Ann NY Acad Sci 1998; 859: 160–74. [DOI] [PubMed] [Google Scholar]
- 14. Otsuka T, Tsukamoto T, Mizoshita T et al. Coexistence of gastric‐ and intestinal‐type endocrine cells in gastric and intestinal mixed intestinal metaplasia of the human stomach. Pathol Int 2005; 55: 170–9. [DOI] [PubMed] [Google Scholar]
- 15. Tatematsu M, Tsukamoto T, Inada K. Stem cells and gastric cancer: role of gastric and intestinal mixed intestinal metaplasia. Cancer Sci 2003; 94: 135–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shimizu N, Inada K, Nakanishi H et al. Helicobacter pylori infection enhances glandular stomach carcinogenesis in Mongolian gerbils treated with chemical carcinogens. Carcinogenesis 1999; 20: 669–76. [DOI] [PubMed] [Google Scholar]
- 17. Shimizu N, Ikehara Y, Inada K et al. Eradication diminishes enhancing effects of Helicobacter pylori infection on glandular stomach carcinogenesis in Mongolian gerbils. Cancer Res 2000; 60: 1512–14. [PubMed] [Google Scholar]
- 18. Cao X, Tsukamoto T, Nozaki K et al. Earlier Helicobacter pylori infection increases the risk for the N‐methyl‐N‐nitrosourea‐induced stomach carcinogenesis in Mongolian gerbils. Jpn J Cancer Res 2002; 93: 1293–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cao X, Tsukamoto T, Nozaki K et al. Eradication of Helicobacter pylori induces apoptosis and inhibits proliferation of heterotopic proliferative glands in infected Mongolian gerbils. Cancer Sci 2004; 95: 872–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tsukamoto T, Inada K, Tanaka H et al. Down‐regulation of a gastric transcription factor, Sox2, and ectopic expression of intestinal homeobox genes, Cdx1 and Cdx2: inverse correlation during progression from gastric/intestinal‐mixed to complete intestinal metaplasia. J Cancer Res Clin Oncol 2004; 130: 135–45. [DOI] [PubMed] [Google Scholar]
- 21. Mizoshita T, Tsukamoto T, Takenaka Y et al. Gastric and intestinal phenotypes and histogenesis of advanced glandular stomach cancers in carcinogen‐treated, Helicobacter pylori‐infected Mongolian gerbils. Cancer Sci 2006; 97: 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tatematsu M, Tsukamoto T, Mizoshita T. Role of Helicobacter pylori in gastric carcinogenesis: the origin of gastric cancers and heterotopic proliferative glands in Mongolian gerbils. Helicobacter 2005; 10: 97–106. [DOI] [PubMed] [Google Scholar]
- 23. Nozaki K, Shimizu N, Tsukamoto T et al. Reversibility of heterotopic proliferative glands in glandular stomach of Helicobacter pylori‐infected Mongolian gerbils on eradication. Jpn J Cancer Res 2002; 93: 374–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mizoshita T, Joh T, Oshima T et al. Differential expression of decay‐accelerating factor isoforms in the digestive tract of guinea pig. Life Sci 2002; 70: 867–76. [DOI] [PubMed] [Google Scholar]
- 25. Tsukamoto T, Fukami H, Yamanaka S et al. Hexosaminidase‐altered aberrant crypts, carrying decreased hexosaminidase alpha and beta subunit mRNAs, in colon of 1,2‐dimethylhydrazine‐treated rats. Jpn J Cancer Res 2001; 92: 109–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sugiyama A, Ikeno T, Maruta F et al. Long‐term Helicobacter pylori colonization produces G cell hyperplasia and carcinoid tumor in Mongolian gerbils. J Cell Mol Med 2000; 4: 308–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sumii M, Sumii K, Tari A et al. Expression of antral gastrin and somatostatin mRNA in Helicobacter pylori‐infected subjects. Am J Gastroenterol 1994; 89: 1515–19. [PubMed] [Google Scholar]
- 28. Sokic‐Milutinovic A, Todorovic V, Milosavljevic T, Micev M, Drndarevic N, Mitrovic O. Gastrin and antral G cells in course of Helicobacter pylori eradication: Six months follow up study. World J Gastroenterol 2005; 11: 4140–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Graham DY, Lew GM, Lechago J. Antral G‐cell and D‐cell numbers in Helicobacter pylori infection: effect of H. pylori eradication. Gastroenterology 1993; 104: 1655–60. [DOI] [PubMed] [Google Scholar]
- 30. Kamada T, Haruma K, Kawaguchi H, Yoshihara M, Sumii K, Kajiyama G. The association between antral G and D cells and mucosal inflammation, atrophy, and Helicobacter pylori infection in subjects with normal mucosa, chronic gastritis, and duodenal ulcer. Am J Gastroenterol 1998; 93: 748–52. [DOI] [PubMed] [Google Scholar]
- 31. Dimaline R, Evans D, Forster ER, Sandvik AK, Dockray GJ. Control of gastric corpus chromogranin A messenger RNA abundance in the rat. Am J Physiol 1993; 264: G583–8. [DOI] [PubMed] [Google Scholar]
- 32. Tsutsumi Y, Nagura H, Watanabe K. Immune aspects of intestinal metaplasia of the stomach: an immunohistochemical study. Virchows Arch A Pathol Anat Histopathol 1984; 403: 345–59. [DOI] [PubMed] [Google Scholar]
- 33. Salenius P. On the ontogenesis of the human gastric epithelial cells. A histologic and histochemical study. Acta Anat (Basel) 1962; 50 (Suppl. 46): 1–76. [PubMed] [Google Scholar]
- 34. La Rosa S, Rigoli E, Uccella S, Chiaravalli AM, Capella C. CDX2 as a marker of intestinal EC‐cells and related well‐differentiated endocrine tumors. Virchows Arch 2004; 445: 248–54. [DOI] [PubMed] [Google Scholar]
- 35. Jenny M, Uhl C, Roche C et al. Neurogenin3 is differentially required for endocrine cell fate specification in the intestinal and gastric epithelium. EMBO J 2002; 21: 6338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sakai H, Eishi Y, Li XL et al. PDX1 homeobox protein expression in pseudopyloric glands and gastric carcinomas. Gut 2004; 53: 323–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Faller G, Kirchner T. Immunological and morphogenic basis of gastric mucosa atrophy and metaplasia. Virchows Arch 2005; 446: 1–9. [DOI] [PubMed] [Google Scholar]
- 38. Suzuki H, Minegishi Y, Nomoto Y et al. Down‐regulation of a morphogen (sonic hedgehog) gradient in the gastric epithelium of Helicobacter pylori‐infected Mongolian gerbils. J Pathol 2005; 206: 186–97. [DOI] [PubMed] [Google Scholar]
- 39. Tatematsu M, Aoki T, Inoue T, Mutai M, Furihata C, Ito N. Coefficient induction of pepsinogen 1‐decreased pyloric glands and gastric cancers in five different strains of rats treated with N‐methyl‐N"‐nitro‐N‐nitrosoguanidine. Carcinogenesis 1988; 9: 495–8. [DOI] [PubMed] [Google Scholar]
- 40. Tatematsu M, Furihata C, Katsuyama T et al. Immunohistochemical demonstration of pyloric gland‐type cells with low‐pepsinogen isozyme 1 in preneoplastic and neoplastic tissues of rat stomachs treated with N‐methyl‐N’‐nitro‐N‐nitrosoguanidine. J Natl Cancer Inst 1987; 78: 771–7. [PubMed] [Google Scholar]
- 41. Tatematsu M, Mutai M, Aoki T, De Camargo JL, Furihata C, Ito N. Proliferation kinetics of pepsinogen altered pyloric gland cells in rats treated with N‐methyl‐N"‐nitro‐N‐nitrosoguanidine. Carcinogenesis 1989; 10: 907–11. [DOI] [PubMed] [Google Scholar]
- 42. Saito A, Shimoda T, Nakanishi Y, Ochiai A, Toda G. Histologic heterogeneity and mucin phenotypic expression in early gastric cancer. Pathol Int 2001; 51: 165–71. [DOI] [PubMed] [Google Scholar]
- 43. Koseki K, Takizawa T, Koike M, Ito M, Nihei Z, Sugihara K. Distinction of differentiated type early gastric carcinoma with gastric type mucin expression. Cancer 2000; 89: 724–32. [PubMed] [Google Scholar]
- 44. Egashira Y, Shimoda T, Ikegami M. Mucin histochemical analysis of minute gastric differentiated adenocarcinoma. Pathol Int 1999; 49: 55–61. [DOI] [PubMed] [Google Scholar]