

Abstract
Array‐based comparative genomic hybridization (array‐CGH) has good potential for the high‐throughput identification of genetic aberrations in cell genomes. In the course of a program to screen a panel of 21 oral squamous‐cell carcinoma (OSCC) cell lines for genome‐wide copy‐number aberrations by array‐CGH using our in‐house bacterial artificial chromosome arrays, we identified a frequent homozygous deletion at 4q35 loci with approximately 1 Mb in extent. Among the seven genes located within this region, the expression of the melatonin receptor 1 A (MTNR1A) messenger RNA (mRNA) was not detected or decreased in 35 out of the 39 (89%) OSCC cell lines, but was detected in immortalized normal oral epithelial cell line, and was restored in gene‐silenced OSCC cells without its homozygous loss after treatment with 5‐aza‐2′‐deoxycytidine. The hypermethylation of the CpG (cytosine and guanine separated by phosphate) island in the promoter region of MTNR1A was inversely correlated with its expression in OSCC lines without a homozygous deletion. Methylation of this CpG island was also observed in primary OSCC tissues. In an immunohistochemical analysis of 50 primary OSCC tumors, the absence of immunoreactive MTNR1A was significantly associated with tumor size and a shorter overall survival in patients with OSCC tumors, and seems to be an independent prognosticator in a multivariate analysis. Exogenous restoration of MTNR1A expression inhibited the growth of OSCC cells lacking its expression. Together with the known tumor‐suppressive function of melatonin and MTNR1A in various tumors, our results indicate MTNR1A to be the most likely target for epigenetic silencing at 4q35 and to play a pivotal role during oral carcinogenesis. (Cancer Sci 2008; 99: 1390–1400)
Oral cancer, predominantly oral squamous‐cell carcinoma (OSCC), is the most common head and neck neoplasm, affecting 270 000 people worldwide each year.( 1 ) In Japan, OSCC is relatively common, responsible for more than 5500 deaths in 2003.( 2 ) Despite of the recent progress in the diagnosis of and therapeutic modalities for OSCC, the 5‐year survival rate has not improved in more than two decades.( 3 ) Therefore, a more comprehensive understanding of the molecular pathogenesis of OSCC is urgently needed to identify new targets for the effective therapy, and to recognize the early state of OSCC and/or premalignant lesions. Generally carcinogenesis, including OSCC,( 4 ) is considered to arise through the progressive accumulation of multiple genetic abnormalities to impair the functions of oncogenes or tumor‐suppressor genes (TSG). Besides genetic alterations, evidence has emerged that the DNA methylation of 5′‐CpG (cytosine and guanine separated by phosphate) islands has been shown to be a major cause of inactivation of TSG in human cancers,( 5 ) including OSCC.( 6 ) Although various TSG, such as CDKN2A, CDH1, MGMT, RARB, RASSF1A, DAPK, MLH1 and LRP1B, were reported to be inactivated and contribute to the oral carcinogenesis through DNA hypermethylation in OSCC,( 6 , 7 ) more genes need to be identified to understand the complete enigma of molecular pathogenesis of OSCC and to provide precise diagnostic methods and appropriate therapeutic approaches to this lethal disease.
Recently, genome‐wide screening for DNA copy‐number alterations using array‐based methods, including array‐based comparative genomic hybridization (array‐CGH), has been proved as an effective tool to explore OSCC‐associated oncogenes or TSG.( 8 , 9 , 10 , 11 , 12 , 13 ) Interestingly, some TSG, which have been identified from homozygously deleted regions in the cancer genome by array‐based approaches at first, are predominantly silenced by epigenetic mechanisms including DNA methylation, indicating that homozygous losses are good landmarks to explore novel epigenetically silenced TSG. Indeed, we have previously identified PRTFDC1 as the putative tumor suppressor for OSCC, which is frequently silenced through DNA methylation, from a homozygous loss at 10p12 detected in OSCC cell line by array‐CGH using in‐house bacterial artificial chromosome (BAC)‐based array.( 13 ) Therefore, it is expected that copy‐number analyses using another panel of materials including cell lines and/or using arrays with higher resolution will provide more landmarks to identify more candidates for TSG, which will be useful as diagnostic markers and/or therapeutic targets, inactivated through either genetic or epigenetic mechanism in OSCC.
In the present study, we identified melatonin receptor 1 A (MTNR1A), which encodes one of two high affinity forms of a receptor for pineal hormone melatonin, as a candidate target for the 4q35 homozygous deletion detected by array‐CGH analyses using our in‐house BAC‐arrays (MCG Cancer Array‐800 and MCG Whole Genome Array‐4500) against a panel of OSCC cell lines, which were different from lines we analyzed in a previous array‐CGH study.( 13 ) Interestingly, expression of this gene was frequently silenced in OSCC cell lines without its homozygous loss, although it was present in normal oral mucosa, suggesting that MTNR1A might be inactivated epigenetically in OSCC and contribute to oral carcinogenesis. In several cancers, indeed, it has been reported that melatonin treatment or ectopic expression of MTNR1A has a growth suppressive effect on cancer cells in vitro and in vivo,( 14 , 15 , 16 , 17 , 18 , 19 , 20 ) even though the intracellular mechanisms behind the antiproliferative actions of melatonin remain unclear. To assess this hypothesis, we examined the role of DNA methylation within the CpG island around exon 1 of MTNR1A in regard to its expression in OSCC, and also the clinicopathological significance of MTNR1A expression in primary OSCC.
Materials and Methods
Cell lines and primary tumors. Twenty‐five OSCC cell lines (OM‐1, OM‐2, TSU, ZA, NA, Ca9‐22, HOC‐313, HOC‐815, HSC‐2, HSC‐3, HSC‐4, HSC‐5, HSC‐6, HSC‐7, KON, SKN‐3, HO‐1‐N‐1, KOSC‐2, Sa3, SAS, KOSC‐3, HO‐1‐u‐1, T3M1Clone2, T3M1 CL‐10, and HSQ89) established from surgically resected tumors in Tokyo Medical and Dental University,( 21 ) or purchased from the Japanese Collection of Research Bioresources (Osaka, Japan) or RIKEN BioResource Center (Ibaragi, Japan) and 14 Tosca series of OSCC cell lines (TOSCa‐2S, 7, 18, 23, 24, 30, 32, 36, 45, 50, 52, 55, 58S1 and 65) established from surgically resected tumors in Showa University (Tokyo, Japan) were maintained in appropriate media supplemented with 10% fetal bovine serum, and 100 units/mL penicillin and 100 µg/mL streptomycin. The normal oral epithelial cell‐derived cell line, RT7, kindly provided by Professor Nobuyuki Kamata (Hiroshima University, Hiroshima, Japan) was maintained in keratinocyte serum‐free medium containing epidermal growth factor and bovine pituitary extract (Invitrogen, Carlsbad, CA, USA).( 22 ) As a control, we performed a primary culture of oral gingival epithelial cells under the approval for this study as described previously.( 13 )
Primary OSCC tumor samples to isolate genomic DNA were obtained during surgery from 16 patients who were treated at the National Cancer Institute or Chulalongkorn University (Bangkok, Thailand). None of the patients had been administered preoperative radiation, chemotherapy or immunotherapy. Tissues from the patients were immediately frozen in liquid nitrogen and stored at –80°C until required. Primary OSCC tumor samples for immunohistochemistry (IHC) were obtained during surgery from 50 patients who were treated at the National Defense Medical College Hospital (Saitama, Japan) and samples were embedded in paraffin for IHC. For double analyses, such as methylation analysis and IHC, in the paired cancerous‐ and non‐cancerous tissues of primary OSCC to compare DNA methylation and protein expression level, six cases treated at the Tokyo Medical and Dental University Hospital (Tokyo, Japan) were employed. Written consent was always obtained in the formal style and after approval by the local ethics committees.
Array‐CGH analysis. MCG Cancer Array‐800 and Whole Genome Array‐4500 were used for the array‐CGH. (13 ) Hybridizations were carried out as described elsewhere.( 12 ) Hybridized slides were scanned using GenePix 4000B (Axon Instruments, Foster City, CA, USA) and acquired images were analyzed with GenePix Pro 6.0 imaging software (Axon Instruments). Average ratios that deviated significantly (>2 SD) from 0 (log2 ratio, <–0.4 and >0.4) were considered abnormal.
Screening for homozygous deletions by genomic polymerase chain reaction (PCR) using cell lines. We screened DNA from 39 OSCC cell lines for homozygous losses by genomic PCR. All primer sequences used in this study are listed in supplementary Table S1.
Reverse transcription (RT)‐PCR. Single‐stranded complementary DNA (cDNA) was generated from total RNA and amplified with primers specific for each gene (Suppl. Table S1). The glyceraldehyde‐3‐phosphate dehydrogenase gene (GAPDH) was amplified to allow estimation of the efficiency of cDNA synthesis.
Mutation analysis. We looked for the mutations in MTNR1A by means of direct sequencing, using primers designed for genomic sequences around each exon (Suppl. Table S1). Any base changes detected in tumor samples were confirmed by sequencing each product in both directions.
Drug treatment. Cells were treated with 0, 5 or 10 µM of 5‐aza‐2′‐deoxycytidine (5‐aza‐dCyd) for 5 days without or with 100 ng/mL of trichostatin A (TSA) for the last 12 h.
Methylation analysis. Genomic DNA was treated with sodium bisulfite and subjected to PCR using primer sets designed to amplify regions of interest (Suppl. Table S1). For the combined bisulfite restriction analysis (COBRA),( 23 ) PCR products were digested with BstUI for Regions1 and 3, with TaqI for Region 2 and electrophoresed. For bisulfite sequencing, PCR products were subcloned and then sequenced.
For the methylation‐specific PCR (MSP) analysis, sodium bisulfite‐treated DNA was subjected to PCR using primers specific to the methylated (MSP) and unmethylated (USP) forms of DNA sequences of interest (Suppl. Table S1). DNA from cell lines recognized as dominantly unmethylated (HSC‐5) and methylated (NA) by bisulfite sequencing were used as negative and positive controls for methylated alleles, respectively.
Promoter reporter assay. DNA fragments around the MTNR1A CpG island were obtained by PCR and ligated into the vector pGL3‐Basic (Promega, Madison, WI, USA). Reporter assay was performed as described elsewhere,( 13 ) using each construct or a control empty vector and an internal control pRL‐hTK vector (Promega).
IHC and scoring method. Indirect IHC was performed with formalin‐fixed, paraffin‐embedded tissue sections as described elsewhere.( 24 ) Briefly, antigens were retrieved by microwave pretreatment in high pH buffer (Dako, Carpinteria, CA, USA) for 10 min at 95°C. The slides were incubated with an anti‐MTNR1A antibody (1:50 dilution; LS‐A216, Medical & Biological Laboratories, Nagoya, Japan) overnight at 4°C, then reacted with a Histofine simple stain, MAX PO(G) (Nichirei, Tokyo, Japan). Antigen–antibody reactions were visualized with 0.2% diaminobenzidine tetrahydrochloride and hydrogen peroxide. The slides were counterstained with Mayer's hematoxylin.
Formalin‐fixed SKN3 cells expressing MTNR1A messenger RNA (mRNA), in which >50% of cells showed cytoplasmic or plasma membrane staining of MTNR1A protein, and SAS cells lacking MTNR1A mRNA expression, in which none of the cells showed staining of MTNR1A protein, were used as positive and negative controls, respectively, for IHC. The percentage of the total cell population that expressed MTNR1A was evaluated for each case at ×200 magnification. The results of the immunohistologic staining were classified as negative (1–10% of tumor cells stained) or positive (>10% of tumor cells stained). Two observers, who were blinded to the clinical outcomes of the patients, evaluated the slides independently; if a significant discrepancy emerged between their judgments, a consensus was reached after discussion.
Transient transfection, Western blotting and colony‐formation assays. Plasmid expressing N‐terminally FLAG and C‐terminally 3 × Myc‐tagged MTNR1A (pCMV‐3Tag4A‐FLAG‐MTNR1A) was obtained by cloning a N‐terminally FLAG‐tagged full coding sequence of MTNR1A into the pCMV‐3Tag4A vector (Stratagene, La Jolla, CA, USA) in‐frame along with the Myc‐epitope. pCMV‐3Tag4A‐FLAG‐MTNR1A or the control empty vector (pCMV‐3Tag4A‐mock) was transfected into cells for colony formation assays as described elsewhere.( 13 ) The expression of MTNR1A protein in transiently transfected cells was confirmed 24 h after transfection by Western blotting using cell extracts prepared by ProteoExtract Transmembrane Protein Extraction Kit (Novagen Madison, WI, USA) and anti‐Myc tag antibody (Cell Signaling Technology, Beverly, MA, USA).( 13 ) After 2 weeks of incubation with appropriate concentrations of G418 in six‐well plates, cells were fixed with 70% ethanol and stained with crystal violet.
Statistical analysis. The clinicopathological variables pertaining to the corresponding patients were analyzed for statistical significance by χ2 or Fisher's exact test. For the analysis of survival, Kaplan–Meier survival curves were constructed for groups based on univariate predictors and differences between the groups were tested with the log‐rank test. Univariate and multivariate survival analyses were performed using the likelihood ratio test of the stratified Cox proportional‐hazards model. Differences between subgroups were tested by the non‐parametric Mann–Whitney U‐test. Differences were assessed with a two‐sided test, and considered significant at the P < 0.05 level.
Results
Array‐CGH analysis of OSCC cell lines. We assessed copy‐number alterations among the 21 OSCC cell lines by array‐CGH using our in‐house BAC arrays, MCG Cancer Array‐800 and MCG Whole Genome Array‐4500. Copy‐number gains and losses were seen to some extent in all 21 lines (data not shown). As shown in Figure 1a, our array‐CGH predicted frequent copy‐number gains (>60%) for 3q, 7p, 8q, 20q and Xq and frequent losses (>60%) for 3p, 4q and 18q, which were mostly consistent with results of our previous array‐CGH analysis of a different panel of 18 OSCC cell lines,( 13 ) and were similar to published results of conventional‐CGH and array‐CGH analyses of primary OSCC.( 9 , 25 , 26 , 27 )
Figure 1.
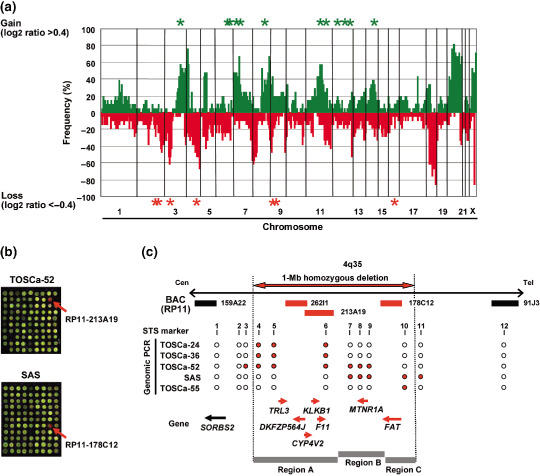
(a) Genome‐wide frequencies of copy‐number gains (above 0, green) and losses (below 0, red) detected by MCG Cancer Array‐800 in 21 oral squamous‐cell (OSCC) cell lines. Clones are ordered from chromosomes 1–22, X and Y and within each chromosome on the basis of the University of California Santa Cruz mapping position (http://genome.ucsc.edu/March, 2006 Assembly). Green asterisks (*), regions with high‐level amplification (log2 ratio >2); red asterisks (*), regions with homozygous deletion (log2 ratio <–2) detected with either MCG Cancer Array‐800 or MCG Whole Genome Array‐4500. (b) Representative image of array‐based comparative genomic hybridization (MCG Whole Genome Array‐4500) of the TOSCa‐52 and SAS cell line. Remarkable decreases in copy‐number ratio (log2 ratio <–2) of RP11–213A19 in TOSCa‐52 and RP11–178C12 in SAS cells at 4q35 were detected as clear red signals (red arrows). (c) Map of 4q35 around the region homozygously deleted in the TOSCa‐24, 36, 52, 55, and SAS cell lines. Bacterial artificial chromosomes (BAC) spotted on the array are shown by horizontal bars; red bars, BACs with log2 ratio <–2 indicating homozygous deletion; black bars, BACs with log2 ratio >–2 in these cell lines. The detailed mapping of homozygously deleted and retained regions in five cell lines was performed by genomic PCR using sequence‐tagged site markers (1, SHGC‐80058; 2, RH48688; 3, SHGC‐156018; 4, RH45826; 5, SGGC‐140659; 6, GDB:315917; 7, SHGC‐50729; 8, SHGC‐140376; 9, WI‐3160; 10, SHGC‐79933; 11, WI‐4200; and 12, RH42876). Homozygously deleted and retained regions are indicated as red closed and open circles, respectively. Three independent regions with homozygous deletion of more than one cell line (Regions a–c) were detected among five cell lines. Approximately 1‐Mb homozygously deleted region in the five cell lines as determined by STS markers is indicated as a horizontal red closed arrow. Genes located around this region, which are homozygously deleted (seven genes) or retained (one gene) in these five cell lines, are indicated as red or black arrows, respectively, that show positions and directions of transcription.
Since most of the common genetic aberrations had already been identified in OSCC, we paid our attention to the remarkable patterns of chromosomal abnormalities, such as high‐level amplifications (log2 ratio >2) and homozygous deletions (log2 ratio <–2), which are likely to be the landmarks of oncogenes and TSG, respectively.( 8 , 9 ) High‐level amplifications were detected in eight of the 21 cell lines (38.1%) and 13 loci were presented (Table 1). Among them, 7p11.2 around EGFR and 11q13 around CCND1, FGF3, and FGF4 were detected in more than two cell lines. Homozygous deletions were detected in 16 of 21 cell lines (76.2%), and seven loci were found in total (Table 2). Six of these loci were detected in more than two cell lines, and most of the loci were located around known TSG, such as LRP1B (2q22.1), FHIT (3p14.2), CDKN2A (9p21.3), PTPRD (9p24.1–23) and WWOX (16q23.1), which are inactivated in various cancers. Interestingly, homozygous loss around RP11–262I1, 213A19, 178C12 spotted on our BAC‐array at 4q35 was most frequently observed in 5 cell lines (TOSCa‐24, 36, 52, 55, and SAS cells; Fig. 1b,c and data not shown), prompting us to focus on this region for exploration of novel TSG. Although FAT has recently been reported as a candidate target for the region spanning this homozygous deletion,( 11 ) the reported homozygous deletions of the FAT gene were mostly intragenic in both cell lines and primary tumors of OSCC, whereas homozygous deletion, which we identified in the present study, located around FAT (Table 2), suggesting that other target genes for 4q35 homozygous loss in OSCC may exist besides of FAT.
Table 1.
High‐level amplifications (log2 ratio >2.0) detected in oral squamous‐cell carcinoma (OSCC) cell lines by comparative genomic hybridization (CGH)‐array analysis using MCG Cancer Array‐800 and Whole Genome Array‐4500
| Locus no. | Bacterial artificial chromosome (BAC) | Locus † | Cell line (Total 21) | Possible target gene ‡ | Number of known gene ‡ | ||
|---|---|---|---|---|---|---|---|
| Chromosomal band | Position | n | Name | ||||
| 1 | RP11‐201D16 | 3q28‐29 | chr3: 193,773,254‐193,974,704 | 1 | TOSCa‐58S1 | FGF12 | 1 |
| 2 | RP11‐81B13 | 6q15 | chr6: 88,549,158‐88,724,237 | 1 | TOSCa‐2S | None | 16 |
| RP11‐52B15 | 6q15 | chr6: 89,876,894‐89,877,257 | 1 | TOSCa‐2S | |||
| 3 | RP11‐88H8 | 6q16.3 | chr6: 102,875,956‐103,063,942 | 1 | TOSCa‐2S | None | 0 |
| 4 | RP11‐59D10 | 6q22.31 | chr6: 120,850,056‐121,030,474 | 1 | TOSCa‐55 | None | 9 |
| RP11‐311B1 | 6q22.31 | chr6: 122,660,488‐122,751,648 | 1 | TOSCa‐55 | |||
| RP11‐374A22 | 6q22.31 | chr6: 124,066,898‐124,228,515 | 1 | TOSCa‐55 | |||
| 5 | RP11‐11D14 | 7p12.3 | chr7: 47,568,937‐47,569,205 | 1 | TOSCa‐32 | None | 6 |
| 6 | RP11‐90N11 | 7p11.2 | chr7: 53,898,011‐54,057,902 | 1 | TOSCa‐2S | EGFR | 9 |
| RP11‐14K11 | 7p11.2 | chr7: 54,954,314‐54,954,933 | 3 | TOSCa‐2S, 32, 50 | |||
| RP11‐339F13 | 7p11.2 | chr7: 55,222,879‐55,348,131 | 3 | TOSCa‐2S, 32, 50 | |||
| RP11‐97P11 | 7p11.2 | chr7: 55,431,590‐55,575,284 | 3 | TOSCa‐2S, 32, 50 | |||
| RP11‐34J24 | 7p11.2 | chr7: 55,596,472‐55,596,912 | 3 | TOSCa‐2S, 32, 50 | |||
| RP11‐80L24 | 7p11.2 | chr7: 55,843,666‐55,977,803 | 3 | TOSCa‐2S, 32, 50 | |||
| 7 | RP11‐89K10 | 8q24.21 | chr8: 127,636,702‐127,799,435 | 1 | TOSCa‐45 | MYC | 2 |
| RP11‐80K22 | 8q24.21 | chr8: 128,735,610‐128884782 | 1 | TOSCa‐45 | |||
| RP11‐237F24 | 8q24.21 | chr8: 128,817,498‐128,822,855 | 1 | TOSCa‐45 | |||
| 8 | RP11‐35F11 | 11q12.3 | chr11: 62,933,912‐63,103,603 | 1 | TOSCa‐65 | CCND1, FGF3, FGF4 | 185 |
| RP11‐80B24 | 11q13.1 | chr11: 63,230,447‐63,411,884 | 1 | TOSCa‐65 | |||
| RP11‐804L21 | 11q13.3 | chr11: 69,333,918‐69,343,129 | 4 | TOSCa‐2S, 45, 55, 65 | |||
| 9 | RP11‐258C1 | 11q22.1 | chr11: 99,810,209‐99,988,933 | 1 | TOSCa‐55 | BIRC2, BIRC3, YAP1 | 29 |
| RP11‐40B14 | 11q22.1 | chr11: 99,932,373‐100,132,253 | 1 | TOSCa‐55 | |||
| RP11‐681O17 | 11q22.1 | chr11: 100,365,386‐100,536,934 | 1 | TOSCa‐55 | |||
| RP11‐750P5 | 11q22.2 | chr11: 101,623,735‐102,191,046 | 1 | TOSCa‐55 | |||
| RP11‐28I24 | 11q22.2 | chr11: 101,722,105‐101,886,737 | 1 | TOSCa‐55 | |||
| RP11‐315O6 | 11q22.2 | chr11: 101,724,723‐101,939,121 | 1 | TOSCa‐55 | |||
| RP11‐817J15 | 11q22.2 | chr11: 101,922,842‐102,095,829 | 1 | TOSCa‐55 | |||
| RP11‐750P5 | 11q22.2 | chr11: 102,010,717‐102,191,009 | 1 | TOSCa‐55 | |||
| RP11‐88H18 | 11q22.2‐22.3 | chr11: 102,253,679‐102,424,014 | 1 | TOSCa‐55 | |||
| RP11‐33F6 | 11q22.3 | chr11: 102,496,919‐102,497,333 | 1 | TOSCa‐55 | |||
| RP11‐2I22 | 11q22.3 | chr11: 102,613,696‐102,771,594 | 1 | TOSCa‐55 | |||
| RP11‐51M23 | 11q22.3 | chr11: 104,480,486‐104,480,901 | 1 | TOSCa‐55 | |||
| 10 | RP11‐319E16 | 12p13.32‐13.31 | chr12: 5,163,936‐5344,301 | 1 | SAS | KRAS2 | 230 |
| RP11‐451H11 | 12p13.31 | chr12: 5,771,103‐5927,033 | 1 | SAS | |||
| RP11‐166G2 | 12p13.31 | chr12: 5,782,525‐5783,146 | 1 | SAS | |||
| RP11‐96K24 | 12p13.1 | chr12: 13,644,252‐13,809,265 | 1 | SAS | |||
| RP11‐295I5 | 12p12.1 | chr12: 25,121,736‐25,317,791 | 1 | SAS | |||
| RP11‐64J22 | 12p12.1 | chr12: 25,882,999‐25,883,554 | 1 | SAS | |||
| RP11‐877E17 | 12p12.1 | chr12: 25,986,021‐26,163,998 | 1 | SAS | |||
| RP11‐283G6 | 12p12.1‐11.23 | chr12: 26,122,383‐26,326,610 | 1 | SAS | |||
| RP11‐53C3 | 12p12.1 | chr12: 26,255,248‐26,255,675 | 1 | SAS | |||
| RP11‐666F17 | 12p11.23 | chr12: 26,671,081‐26,857,010 | 1 | SAS | |||
| RP11‐89L4 | 12p11.23 | chr12: 26,832,206‐26,832,866 | 1 | SAS | |||
| RP11‐73I12 | 12p11.21 | chr12: 31,300,995‐31,457,338 | 1 | SAS | |||
| RP11‐517B23 | 12p11.21 | chr12: 31,362,925‐31,533,973 | 1 | SAS | |||
| 11 | RP11‐837P6 | 12q21.33‐22 | chr12: 91,147,160‐91,214,336 | 1 | TOSCa‐2S | None | 3 |
| 12 | RP11‐205M16 | 12q24.32 | chr12: 125,875,266‐126,030,621 | 1 | TOSCa‐2S | None | 0 |
| 13 | RP11‐2L22 | 14q23.1 | chr14: 58,681,521‐58,851,787 | 1 | TOSCa‐58S1 | HIF1A | 60 |
| RP11‐79M1 | 14q23.1 | chr14: 59,822,513‐59,822,977 | 1 | TOSCa‐58S1 | |||
| RP11‐193F5 | 14q23.1 | chr14: 60,271,223‐60,505,149 | 1 | TOSCa‐58S1 | |||
| RP11‐471N20 | 14q23.1 | chr14: 60,286,133‐60,457,690 | 1 | TOSCa‐58S1 | |||
| RP11‐64I19 | 14q23.1 | chr14: 60,680,813‐60,681,315 | 1 | TOSCa‐58S1 | |||
| RP11‐88L10 | 14q23.1 | chr14: 60,704,698‐60,870,460 | 1 | TOSCa‐58S1 | |||
| RP11‐436G5 | 14q23.1 | chr14: 61,022,970‐61,237,453 | 1 | TOSCa‐58S1 | |||
| RP11‐79I3 | 14q23.1 | chr14: 61,309,602‐61,449,452 | 1 | TOSCa‐58S1 | |||
| RP11‐145O12 | 14q23.2 | chr14: 62,681,383‐62,848,934 | 1 | TOSCa‐58S1 | |||
| RP11‐445J13 | 14q23.2 | chr14: 62,884,544‐63,060,428 | 1 | TOSCa‐58S1 | |||
| RP11‐14C21 | 14q23.2 | chr14: 63,712,417‐63,865,860 | 1 | TOSCa‐58S1 | |||
| RP11‐712C19 | 14q23.2 | chr14: 63,767,389‐63,819,456 | 1 | TOSCa‐58S1 | |||
| RP11‐44K16 | 14q23.2‐23.3 | chr14: 63,974,866‐64,135,774 | 1 | TOSCa‐58S1 | |||
| RP11‐63G22 | 14q23.3 | chr14: 64,463,311‐64,463,893 | 1 | TOSCa‐58S1 | |||
| RP11‐840I19 | 14q23.3 | chr14: 64,611,598‐64,638,980 | 1 | TOSCa‐58S1 | |||
| RP11‐305I24 | 14q23.3 | chr14: 66,806,287‐67,002,089 | 1 | TOSCa‐58S1 | |||
| RP11‐156E22 | 14q23.3 | chr14: 66,847,838‐67,014,199 | 1 | TOSCa‐58S1 | |||
| RP11‐79B13 | 14q24.1 | chr14: 67,744,258‐67,904,880 | 1 | TOSCa‐58S1 | |||
| RP11‐108B17 | 14q24.1 | chr14: 67,980,187‐68,130,021 | 1 | TOSCa‐58S1 | |||
Based on University of California Santa Cruz Genome Browser, March 2006 Assembly.
Genes located around BAC, whose homozygous deletion was validated by genomic polymerase chain reaction (PCR) and/or fluorescence in situ hybridization (FISH).
Table 2.
Homozygous deletions (log2 ratio < –2.0) detected in oral squamous‐cell carcinoma (OSCC) cell lines by array‐ comparative genomic hybridization (CGH) using MCG Cancer Array‐800 and Whole Genome Array‐4500
| Locus no. | Bacterial artificial chromosome (BAC) | Locus † | Cell line (Total 21) | Possible target gene ‡ | Number of known gene ‡ | ||
|---|---|---|---|---|---|---|---|
| Chromomal band | Position | n | Name | ||||
| 1 | RP11‐91A11 | 2q21.2 | chr2: 133,466,940‐133,622,645 | 1 | TOSCa‐7 | None | 1 |
| 2 | RP11‐29N17 | 2q22.1 | chr2: 141,492,118‐141,492,588 | 3 | TOSCa‐45, T3M1 Clone2, T3M1CL‐10 | LRP1B | 1 |
| 3 | RP11‐94D19 | 3p14.2 | chr3: 60,756,984‐60,927,060 | 2 | TOSCa‐32, 50 | FHIT | 1 |
| 4 | RP11‐262I1 | 4q35.1‐35.2 | chr4: 187,244,507‐187,380,943 | 3 | TOSCa‐24, 36, 52 | FAT | 7 |
| RP11‐213A19 | 4q35.2 | chr4: 187,358,363‐187,519,645 | 3 | TOSCa‐24, 36, 52 | |||
| RP11‐178C12 | 4q35.2 | chr4: 187,742,000‐187,910,017 | 2 | TOSCa‐55, SAS | |||
| RP11‐182A9 | 4q35.2 | chr4: 187,810,108‐187,977,806 | 1 | TOSCa‐55 | |||
| 5 | RP11‐176P17 | 9p23 | chr9: 9,504,447‐9653,533 | 4 | TOSCa‐24, 30, 36, HO‐1‐u‐1, KOSC‐3 | PTPRD | 1 |
| RP11‐91E3 | 9p23 | chr9: 9,554,544‐9689,968 | 2 | TOSCa‐24, HO‐1‐u‐1, KOSC‐3 | |||
| RP11‐6H18 | 9p23 | chr9: 9,722,988‐9862,247 | 2 | TOSCa‐24, HO‐1‐u‐1, KOSC‐3 | |||
| RP11‐19G1 | 9p23 | chr9: 9,932,058‐10,130,641 | 2 | HO‐1‐u‐1, KOSC‐3 | |||
| 6 | RP11‐113D19 | 9p21.3 | chr9: 20,996,404‐21,158,458 | 1 | HSQ89 | CDKN2A, CDKN2B, MTAP | 4 |
| RP11‐344A7 | 9p21.3 | chr9: 21,506,374‐21,676,219 | 1 | HSQ89 | |||
| RP11‐145E5 | 9p21.3 | chr9: 21,998,414‐22,155,946 | 2 | TOSCa‐58S1, HSQ89 | |||
| RP11‐408N14 | 9p21.3 | chr9: 22,155,847‐22,309,629 | 1 | TOSCa‐58S1, HSQ89 | |||
| RP11‐441I5 | 9p21.3 | chr9: 22,309,530‐22,479,595 | 1 | TOSCa‐58S1, HSQ89 | |||
| RP11‐11J1 | 9p21.3 | chr9: 22,479,496‐22,579,721 | 1 | TOSCa‐58S1, HSQ89 | |||
| RP11‐782K2 | 9p21.3 | chr9: 22,584,981‐22,585,358 | 1 | TOSCa‐58S1, HSQ89 | |||
| RP11‐33O15 | 9p21.3 | chr9: 22,823,087‐22,823,490 | 1 | TOSCa‐58S1, HSQ89 | |||
| 7 | RP11‐61L1 | 16q23 | chr16: 77344719‐77345302 | 2 | TOSCa‐32, 50 | WWOX | 1 |
Based on University of California Santa Cruz Genome Browser, March 2006 Assembly.
Genes located around BAC, whose homozygous deletion was validated by genomic polymerase chain reaction (PCR) and/or fluorescence in situ hybridization (FISH).
Identification of target genes involved in homozygous deletion at 4q35. To define the extent of the homozygous deletion in TOSCa‐24, 36, 52, 55, and SAS cells, which showed homozygous loss at 4q35 in array‐CGH, we first performed genomic PCR with a series of sequence‐tagged site markers (Fig. 1c). Three independent regions with homozygous deletion in more than one cell line were detected among five cell lines (Regions A–C in Fig. 1c) with an approximately 1 Mb in extent, and seven genes, TRL3, DKFZP564J, KLKB1, CYP4V2, F11, MTNR1A and FAT, located within the whole region were observed in five cell lines according to the information archived by genome databases (http://www.ncbi.nlm.nih.gov/ and http://genome.ucsc.edu/, March 2006 Assembly). To confirm homozygous deletion of those genes and to identify other cell lines harboring cryptic homozygous loss, we next performed genomic PCR with primer sets for these seven genes in the 39 OSCC cell lines, 21 lines used for array‐CGH in the present study and 18 lines used in the previous study (Fig. 2a).( 13 ) We detected a complete loss of TRL3 in two cell lines, DKFZP564J in three lines, CYP4V2 in three lines, KLKB1 in three lines, F11 in three lines, MTNR1A in two lines and FAT in two lines.
Figure 2.
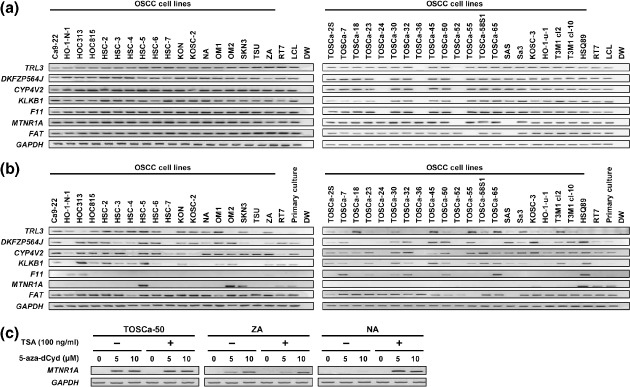
(a) Genomic polymerase chain reaction (PCR) analyses of genes located around the 4q35 homozygously deleted region in 39 oral squamous‐cell carcinoma (OSCC) cell lines. Homozygous deletions of TRL3, DKFZP564J, CYP4V2, KLKB1, F11, melatonin receptor 1 A (MTNR1A) and FAT were detected in two or three OSCC lines. RT7, an immortalized cell line derived from normal oral epithelial cells; LCL, normal lymphoblastoid cell line. (b) Messenger RNA (mRNA) expression of TRL3, DKFZP564J, CYP4V2, KLKB1, F11, MTNR1A and FAT in OSCC cell lines, the RT7 cell line, and primary cultured normal oral mucosa detected by reverse transcription–polymerase chain reaction (RT‐PCR). Notably, 33 of the 37 OSCC cell lines (89%) without a homozygous deletion of MTNR1A showed decreased expression of this gene, whereas RT7, an immortalized normal oral epithelial cell line, and primary cultured normal oral mucosa (primary culture) expressed the MTNR1A mRNA. (c) Representative results of RT‐PCR to reveal restored MTNR1A expression after demethylation in OSCC cell lines lacking its expression. mRNA was isolated after 5 days treatment with various concentrations of 5‐aza‐dCyd for 5 days and/or 100 ng/mL TSA for the last 12 h. GAPDH was used as an internal control.
Frequent silencing MTNR1A expression and its restoration after DNA demethylation in OSCC cell lines. Next we determined the mRNA expression levels of those seven genes in 39 OSCC cell lines along with an immortalized cell line derived from normal oral epithelial cells, RT7, and primary cultured normal oral mucosa. Among seven genes, FAT was expressed in most of the OSCC cell lines (Fig. 2b), and TRL3, KLKB1 and F11 were silenced more or less in OSCC cell lines and not expressed in the RT7 cell line and primary cultured normal oral mucosa, suggesting that these genes are unlikely to be targets for inactivation in OSCC cells. The discrepancy between our results and previously reported deletion and expression data of FAT can be explained by the fact that this gene shows intragenic homozygous loss in OSCC cell lines.( 11 ) CYP4V2 and DKFZP564J were expressed in primary cultured normal oral mucosa but infrequently silenced in OSCC lines. On the other hand, MTNR1A was frequently silenced in OSCC lines without homozygous deletion, but clearly expressed in RT7 and primary cultured normal oral mucosa, suggesting that MTNR1A is one of the targets for 4q35 homozygous deletion and the silencing of this gene may result from mechanisms other than the genomic deletion. Since no mutation was detected in any of the coding exons (exons 1 and 2) of MTNR1A in all of the cell lines tested except those with homozygous loss of this gene (data not shown), we speculated that epigenetic mechanisms including DNA methylation may contribute to the silencing of this gene in OSCC. We searched for the CpG island around the transcription start site, in which aberrant methylation can to silence TSG in the cancer genome,( 5 , 6 ) using the CpGPLOT program (http://www.ebi.ac.uk/emboss/cpgplot/), and identified one CpG island in the MTNR1A gene, prompting us to focus on MTNR1A for further DNA methylation analyses. Although CYP4V2 and DKFZP564J also have CpG islands around their exon 1, no restoration of their expression was observed after the treatment with 5‐aza‐dCyd alone and no hypermethylation within the CpG islands was detected by COBRA in cell lines without homozygous deletion but with no or decreased expression of those genes in our preliminary study (Suppl. Fig. S1), suggesting that DNA methylation may not contribute to the silencing of those genes in OSCC.
We first treated OSCC cell lines (TOSCa‐50, ZA, and NA) lacking MTNR1A expression with 5‐aza‐dCyd, to investigate whether DNA demethylation could restore the expression of MTNR1A mRNA. Induction of MTNR1A mRNA expression occurred after treatment with 5‐aza‐dCyd in TOSCa‐50 and ZA cells (Fig. 2c). In addition, we observed an induction of MTNR1A mRNA expression by 5‐aza‐dCyd along with TSA in NA cell, although treatment with TSA alone had no effect on the expression, suggesting that histone deacetylation may play some synergistic role in the transcriptional silencing of MTNR1A among methylated OSCC cells.
Methylation of the MTNR1A CpG island in OSCC cell lines. To demonstrate the potential role of the methylation within a CpG island in silencing of MTNR1A, we next assessed the methylation status of each CpG site around the MTNR1A CpG island (Regions 1–3 in Fig. 3a) in OSCC cell lines with or without MTNR1A expression and the RT7 cell line, by means of bisulfite sequencing. All regions tended to be extensively methylated in MTNR1A‐nonexpressing OSCC cell lines (NA, HSC‐6, ZA, HSC‐4 and TOSCa‐65), whereas all regions were hypomethylated in MTNR1A‐expressing OSCC cell lines (HSC‐5) and in RT7 (Fig. 3a). We compared the methylation and expression status of MTNR1A in a large number of OSCC lines by COBRA. Consistent with the results of bisulfite sequencing, the unmethylated allele was predominantly detected in all regions among the OSCC cell lines expressing MTNR1A (HSQ89, HSC‐5, OM2 and SKN3) and the RT7 cell line, whereas most of the OSCC cell lines lacking MTNR1A expression without the homozygous deletion showed dominant hypermethylation in almost all regions (Fig. 3b).
Figure 3.
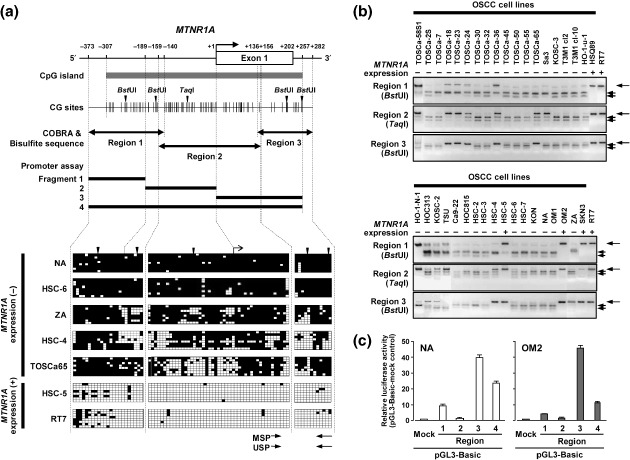
(a) Schematic map of the CpG (cytosine and guanine separated by phosphate) island (horizontal gray thick bar) around exon 1 (open box) of melatonin receptor 1 A (MTNR1A) and representative results of bisulfite sequencing. CpG sites are indicated by vertical ticks on the expanded axis. The transcription start site is marked at +1. The fragments examined in a promoter assay are indicated by black thick lines. The regions examined in the combined bisulfite restriction analysis (COBRA) and bisulfite sequencing are indicated by black closed arrows. For the COBRA, restriction sites for BstUI and TaqI are indicated by black downward arrowheads. Representative results of bisulfite sequencing of the MTNR1A CpG island were examined in MTNR1A‐expressing oral squamous‐cell carcinoma (OSCC) cell line (+) and MTNR1A‐nonexpressing OSCC lines (–). Each square indicates a CpG site: open squares, unmethylated; solid squares, methylated. Polymerase chain reaction (PCR) primers for methylation‐specific PCR (MSP) and unmethylation‐specific PCR (USP) are indicated by arrows. (b) Representative results of the COBRA of the MTNR1A CpG island in OSCC cell lines and RT7 after restriction with BstUI for Regions 1 and 3 and TaqI for Region 2. Arrows show fragments specifically restricted at sites recognized as methylated CpG, whereas arrowheads show undigested fragments indicating unmethylated CpG. (c) Promoter activity of the MTNR1A CpG island. pGL3 basic empty vectors (mock) and constructs containing one of three different sequences around the highly methylated region of MTNR1A (Fragments 1–4; 118 bp, 190 bp, 257 bp and 565 bp in size, respectively, in Fig. 3a) were transfected into NA and OM2 cells. Luciferase activity was normalized versus an internal control. The data presented are the means ± SD for three separate experiments, each performed in triplicate.
Promoter activity of the sequence around the MTNR1A CpG island. Since the sequence around the MTNR1A CpG island seems to be a target for methylation‐mediated gene silencing, we tested four fragments covering a putative critical region for promoter activity (Fig. 3a) by promoter reporter assay using two OSCC cell lines: OM2 expressing MTNR1A and NA lacking MTNR1A expression (Fig. 2b). Fragment 3 containing exon 1 and intron 1 within Region 2 and Region 3 showed a remarkable transcriptional activity (Fig. 3c), whereas Fragments 1 and 2 containing sequences upstream (5′) to exon 1, a putative regulatory region for transcription, showed little or almost no transcriptional activity, suggesting that Fragment 3 may contain critical sequences for gene silencing through methylation.
Methylation of the MTNR1A promoter region in primary OSCC tumors. To determine whether the aberrant methylation of MTNR1A also takes place in primary tumors, we designed primer sets for MSP based on methylation pattern in OSCC cell lines and critical region for promoter activity within CpG island, and performed MSP in a panel of 16 primary OSCC tumors (Fig. 4a). Since (a) restriction sites in COBRA are poor in critical region (Fig. 3a) and (b) thereby we need highly sensitive method to detect methylated sequences around the critical region in heterogeneous primary tumor tissues containing contaminated normal cells, we used methylation‐specific MSP analysis instead of COBRA to screen primary samples. Consistent with the results of the bisulfite sequencing and COBRA (Fig. 3a,b), a representative cell line lacking MTNR1A expression (NA) showed a predominantly methylated pattern, whereas the MTNR1A‐expressing cell line (HSC‐5) had a predominantly unmethylated pattern. We detected MTNR1A hypermethylation in 10 of the 16 primary OSCC tissues (62%, Fig. 4a). To confirm the results of the MSP analysis quantitatively, we performed bisulfite sequencing in some of representative cases (Fig. 4b). Aberrant hypermethylation was observed in OSCC tissues that showed a methylation pattern in the MSP analysis (Fig. 4a). In a panel of six paired cancerous and non‐cancerous OSCC tumors, tumors tended to show a hypermethylation pattern compared with corresponding non‐cancerous mucosa in MSP analysis (Suppl. Fig. S2A) and bisulfite sequencing (Suppl. Fig. S2B and data not shown).
Figure 4.
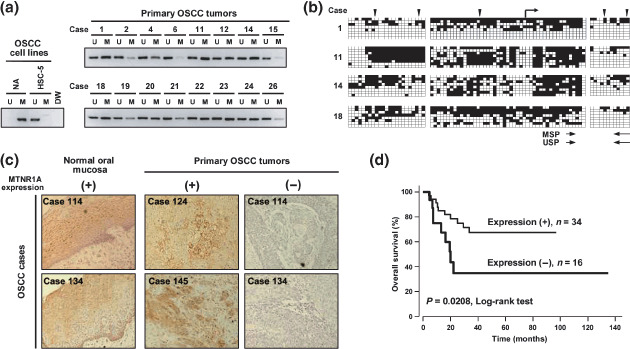
Methylation status of the CpG (cytosine and guanine separated by phosphate) island and expression levels of melatonin receptor 1 A (MTNR1A) in primary tumors of oral squamous‐cell carcinoma (OSCC). (a) Representative results of a methylation‐specific polymerase chain reaction (MSP) analysis of the MTNR1A promoter region in primary OSCC tissues. Parallel amplification reactions were done using primers specific for methylated (M) and unmethylated (U) sequences by MSP. NA and HSC‐5 cell lines were used for predominantly methylated and unmethylated controls, respectively. (b) Results of bisulfite sequencing of the MTNR1A CpG island representative cases analyzed by MSP in primary OSCC tumors. See legend for Fig. 3 A for interpretation. Polymerase chain reaction (PCR) primers for methylation specific PCR (MSP and unmethylation‐specific PCR [USP]) are indicated by arrows. (c) Representative results of immunohistochemical staining of MTNR1A protein. Tumors in two cases showed positive MTNR1A staining in ≥10% of cancer cells (positive, +), whereas two cases showed positive MTNR1A staining in <10% of cancer cells (negative, –). In neighboring non‐neoplastic mucosa, positive immunoreactivity of MTNR1A was detected. MTNR1A immunoreactivity was present mainly in the cytoplasm and/or plasma membrane of normal epithelial cells and also of immunopositive malignant cells. Magnifications are ×200. (d) Kaplan–Meier curve for overall survival rates of 50 patients with primary OSCC tumors. Patients with negative MTNR1A expression in tumors showed significantly worse prognosis compared with those with positive expression (P = 0.0208).
Association between expression level of MTNR1A protein and clinicopathological characteristics in primary cases of OSCC. To clarify the clinical significance of MTNR1A expression status in primary cases of OSCC, the expression level of MTNR1A protein was evaluated by immunohistochemistry. Positive MTNR1A immunoreactivity was detected in normal oral epithelia (Fig. 4c and Suppl. Fig. S2C). The expression of MTNR1A protein tended to be silenced in tumors through hypermethylation in critical regions for promoter activity within the CpG island compared with corresponding normal mucosa (Suppl. Fig. S2A,B). In 50 cases with OSCC, 34 OSCC specimens showed positive staining of MTNR1A, whereas 16 cases showed negative immunoreactivity of MTNR1A (Table 3). The relationship between the expression of MTNR1A protein and the clinicopathological characteristics is summarized in Table 3. The protein expression of MTNR1A was significantly associated with T category in tumor–lymph node–metasteses (TNM) classification: OSCC tended to lack the MTNR1A expression in T3 and T4 tumors (P = 0.007, χ2 test). However, the MTNR1A protein expression in each tumor was not associated with the age and sex of patients, differentiation status, stage and N category of TNM classification of tumor. In overall survival (Fig. 4d), negative MTNR1A immunoreactivity in tumor cells was significantly associated with a worse overall survival (P = 0.0208, log‐rank test) in all stages.
Table 3.
Correlation between clinical background and expression of melatonin receptor 1 A (MTNR1A) protein
| n | MTNR1A expression † | P‐value* | ||
|---|---|---|---|---|
| Positive (%) | Negative (%) | |||
| Total | 50 | 34 (68) | 16 (32) | |
| Age (years) | ||||
| < 60 | 25 | 18 (72) | 7 (28) | 0.762 |
| ≥ 60 | 25 | 16 (64) | 9 (36) | |
| Gender | ||||
| Male | 36 | 22 (61) | 14 (39) | 0.175 |
| Female | 14 | 12 (86) | 2 (14) | |
| Differentiation | ||||
| Well | 23 | 18 (78) | 5 (22) | 0.258 |
| Poor‐Moderate | 27 | 16 (59) | 11 (41) | |
| Stage | ||||
| I + II | 24 | 18 (75) | 6 (25) | 0.474 |
| III + IV | 26 | 16 (62) | 10 (38) | |
| TNM classification | ||||
| T category | ||||
| T1 + T2 | 36 | 29 (81) | 7 (19) | 0.007 |
| T3 + T4 | 14 | 5 (36) | 9 (64) | |
| N category | ||||
| N0 | 32 | 20 (63) | 12 (37) | 0.351 |
| N1‐3 | 18 | 14 (78) | 4 (22) | |
NOTE: Statistically significant values are in boldface type.
P‐values are from χ2 or Fisher's exact test and were statistically significant when <0.05 (two sided).
MTNR1A expression was evaluated by immunohistochemical analysis as described in Materials and Methods.
In Cox proportional hazard regression analysis for overall survival (Table 4), univariate analysis demonstrated that the MTNR1A expression status, T category of TNM classification, tumor stage and the differentiation status were significantly associated with the overall survival (Table 4). Multivariate analysis using a stepwise Cox regression procedure revealed that the MTNR1A expression status and tumor stage were identified as independently selected predictive factors for overall survival in both forward and backward procedures (P = 0.0498 and 0.0099, respectively).
Table 4.
Cox proportional hazard regression analysis for overall survival
| Factor | Univariate | Multivariate † P‐value* | |
|---|---|---|---|
| Hazard ratio (95% confidence interval) | P‐value* | ||
| Age (years) | |||
| ≥60 versus <60 | 1.060 (0.430–2.612) | 0.899 | – |
| Gender | |||
| Male versus Female | 1.080 (0.410–2.843) | 0.877 | – |
| Differentiation | |||
| Poor‐Moderate versus Well | 2.899 (1.041–8.065) | 0.042 | – |
| Stage | |||
| III + IV versus I + II | 4.184 (1.493–11.765) | 0.007 | 0.0099 |
| TNM classification | |||
| T category | |||
| T3 + T4 versus T1 + T2 | 3.509 (1.416–8.696) | 0.007 | – |
| N category | |||
| N1‐3 versus N0 | 2.193 (0.890–5.405) | 0.088 | – |
| MTNR1A expression ‡ | |||
| Negative versus Positive | 2.816 (1.128–7.029) | 0.027 | 0.0498 |
NOTE: Statistically significant values are in boldface type.
P‐values are from two‐sided tests and were statistically significant when <0.05.
Forward‐ and backward‐stepwise analyses were used for multivariate analysis.
MTNR1A expression was evaluated by immunohistochemical analysis as described in Materials and Methods.
Suppressive effect of MTNR1A expression on growth of OSCC cells. To gain further insight into the potential role of MTNR1A in oral carcinogenesis, we investigated whether restoration of MTNR1A expression would suppress growth of OSCC cells in which the gene had been silenced. We performed colony‐formation assays using the full coding sequence of MTNR1A with FLAG‐ and Myc‐tags cloned into a mammalian expression vector (pCMV‐3Tag4A‐FLAG‐MTNR1A) and cells lacking expression of MTNR1A (HSC‐7 and NA cell lines). As shown in Figure 5 and 2 weeks after transfection and subsequent selection of drug‐resistant colonies, the numbers of large colonies produced by MTNR1A‐transfected HSC‐7 and NA cells significantly decreased compared with those produced by empty vector‐transfected counterparts.
Figure 5.
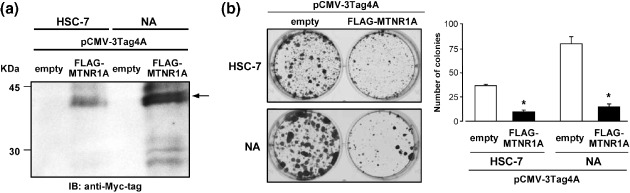
Effects of restoration of melatonin receptor 1 A (MTNR1A) expression on growth of oral squamous‐cell carcinoma (OSCC) cells. (a) A FLAG‐ and Myc‐tagged construct containing MTNR1A (pCMV‐3Tag4A‐FLAG‐MTNR1A) or empty vector (pCMV‐3Tag4A‐mock) as a control was transfected into HSC‐7 or NA cells, which lack expression of the MTNR1A gene. Western‐blotting using 10 µg of membrane protein extracts prepared from cells 24 h after transfection and anti‐Myc tag antibody demonstrated that transiently pCMV‐3Tag4A‐FLAG‐MTNR1A‐transfected cells expressed epitope‐tagged MTNR1A protein. (b) Two weeks after transfection and subsequent selection of drug‐resistant colonies in 6‐well plates, the colonies formed by MTNR1A‐transfected cells were less numerous than those formed by mock‐transfected cells (left). In quantitative analysis of colony formation (right), colonies larger than 2 mm were counted, and results are presented as the means ± SD (bars) of three separate experiments, each performed in triplicate. Statistical analysis used the Mann–Whitney U‐test: *P < 0.05 versus empty‐vector transfected cells.
Discussion
In this study, we identified MTNR1A as one of the targets for the inactivation of OSCC from a homozygously deleted region at 4q35 detected in OSCC cell lines during genome‐wide screening by array‐CGH analysis using our in‐house BAC arrays, although possible involvement of other target genes for this homozygous loss remain unclear. Expression of MTNR1A was detected in normal oral epithelial‐cell‐derived immortalized cell lines but frequently silenced through methylation of CpG sites around the MTNR1A CpG island exhibiting promoter activity in our panel of oral cancer cell lines, suggesting that MTNR1A may be one of targets for inactivation in OSCC. Hypermethylation of the MTNR1A promoter region was also frequently detected in primary oral cancers. The negative immunoreactivity of MTNR1A protein in primary OSCC tumors appeared to be associated with the tumor size and shorter overall survival time even after stratification with other clinicopathological factors by multivariate analysis. In addition, forced expression of MTNR1A in OSCC cells lacking its endogenous expression suppressed cell growth. These results suggest that the genetic and/or epigenetic inactivation of MTNR1A plays a pivotal role in the pathogenesis of OSCC.
The MTNR1A gene encodes one of two high affinity forms of a receptor for melatonin, the primary hormone secreted by the pineal gland. This receptor is a G‐protein coupled, 7‐transmembrane receptor that is known to be responsible for melatonin effects on mammalian circadian rhythms and reproductive alterations affected by day length.( 28 , 29 ) The pineal hormone, melatonin, most widely recognized for its role in sleep and circadian rhythm regulation, has been demonstrated to exert oncostatic effects both in vivo and in vitro in various types of malignancies, such as cancers of the breast, prostate, skin and liver, and gliomas,( 19 , 20 , 30 ) although the most studied model of melatonin's oncostatic effect is breast cancer, especially the estrogen‐dependent human breast cancer cell and the carcinogen‐induced mammary carcinoma in the rat.( 14 , 15 , 16 , 17 , 18 , 19 , 20 ) These studies show varying results ranging from the oncostatic effects of melatonin to no effect at all, albeit under different experimental conditions, such as cell/tissue types, doses, time of dosage and length of treatment. In addition, the extra‐ or intracellular mechanism by which melatonin suppresses cancer growth remains largely undetermined, although several mechanisms have been proposed: (a) repression of the hypothalamic–pituitary–reproductive axis; (b) enhancement of immune function; and (c) direct antiproliferative effects on cancer cells through various receptors including MTNR1A or its antioxidant effect.( 18 , 19 , 20 , 31 ) The evidence that melatonin's growth‐inhibitory effects on human breast cancer cells and NIH 3T3 cells are MTNR1A receptor dependent,( 32 , 33 ) led to the finding that MTNR1A mediates melatonin‐induced growth‐inhibitory and gene‐modulatory effects at least in part. Indeed, our colony‐formation assays using MTNR1A‐transfected OSCC cells revealed growth‐suppressive and/or antiproliferative activity of the MTNR1A protein in OSCC. However, altered expression of MTNR1A and/or its underlying mechanisms in cancer cells, including OSCC cells, has been largely unknown. This is the first report to show that MTNR1A is frequently silenced in OSCC cells, which correlates with malignant phenotype and poor prognosis in primary cases, through genetic and/or epigenetic mechanisms. It is known that melatonin is generated and released by not only the pineal gland but also by entero‐endocrine cells in the gastrointestinal tract including the oral cavity and by the salivary gland,( 31 , 34 ) suggesting that melatonin seems to reach to oral epithelial cells and exert its tumor suppressive activity through MTNR1A or by other pathways.
In our promoter assay, Fragment 3, which locates in exon 1 and part of intron 1 of MTNR1A, showed the highest and most remarkable promoter activity compared with fragments (Fragments 1 and 2) within upstream (5′) to exon 1 in either MTNR1A‐expressing or nonexpressing OSCC cell lines. This result consists with the methylation pattern observed in cell lines as well as primary tumors of OSCC (3, 4). Therefore, at least sequences within 5′ part of exon 1 seem to be the target for the methylation‐induced silencing of MTNR1A, although few studies, including ours, have shown that promoter activity can be observed in fragments, especially CpG islands, not containing a 5′ sequences upstream to transcriptional start sites.( 13 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 )
In immunohistochemical analysis of MTNR1A among our panel of 50 primary OSCC tumors, the expression status of the MTNR1A protein in each sample was associated with T category of TNM classification, but not with other clinicopathological variables, including age, sex, differentiation status, stage and N category of TNM classification. The most striking finding in our study was that the negative immunoreactivity of the MTNR1A protein was significantly associated with a worse clinical outcome even after stratification with other clinicopathological characteristics in multivariate analysis, suggesting that the inactivation of MTNR1A may involve in the malignant progression of OSCC at least partly in a stage‐independent manner, and might be useful as an independent prognosticator in patients with OSCC. In our preliminary immunohistochemical study, similar results were obtained in other primary head and neck squamous cell carcinoma (HNSCC), such as pharyngeal squamous cell carcinoma (n = 58; Nakamura et al. unpublished data), suggesting that inactivation of MTNR1A may generally contribute to the pathogenesis of HNSCC. Since no clinical specimens were available to compare the expression level of MTNR1A mRNA or protein with the methylation status of this gene, it remains unclear which mechanisms, such as methylation, deletion or mutation, are predominant to silence MTNR1A in primary OSCC tumors. However, only 2 of 39 OSCC cell lines showed homozygous deletion and no mutation was detected in any cell lines, suggesting that epigenetic mechanisms including DNA methylation may predominatly contribute to inactivate MTNR1A. In addition, the expression status of MTNR1A protein was not associated with tumor stage, but associated with the T category and not the N category in TNM classification, suggesting that the loss of MTNR1A expression and inactivation may occur in a specific subgroup of OSCC, even at an earlier stage, and contribute to higher proliferative and/or survival activity of tumor cells in this subgroup. Further study will be needed to test this hypothesis and to determine which is the most sensitive and specific method for detecting alteration of MTNR1A as useful clinical marker in this disease.
Recently, FAT, a member of the cadherin superfamily has been reported to be a putative TSG, which frequently shows intragenic (exonic) homozygous deletion in cell lines and primary tumors of OSCC, at 4q35.( 11 ) Our screening using in‐house BAC array‐based array‐CGH in a panel of cell lines and genomic PCR detected homozygous deletion of FAT in two of the 39 OSCC cell lines, although we did not evaluated homozygous loss in all exons of this gene. However, our reverse transcription–polymerase chain reaction (RT‐PCR) detected the expression of FAT in almost all OSCC cell lines except lines with homozygous deletion as well as in RT7 and primary cultured normal oral mucosa at the same level, suggesting that silencing of FAT through mutations, deletion, or other mechanisms including DNA methylation may rarely occur in OSCC. Indeed, Nakaya et al.( 11 ) showed that only OSCC cell lines with homozygous deletion of FAT lack its expression in RT‐PCR. Since one cell line (SAS) tested in the present study showed homozygous deletion of both FAT and MTNR1A, it is possible that inactivation of these two genes is synergistically involved in the pathogenesis of OSCC. It will be interesting to determine the expression pattern of the FAT and MTNR1A proteins in the same cohort of primry OSCC tumors, and evaluate their clinicopathlogical significance together or independently.
Supporting information
Fig. S1. (a) Representative results of reverse transcription–polymerase chain reaction (RT‐PCR) analysis to examine the DKFZP564J and CYP4V2 expression levels after demethylation in oral squamous‐cell carcinoma (OSCC) cell lines with no or decreased expression of those genes. Messenger RNA (mRNA) was isolated after treatment with various concentrations of 5‐aza‐dCyd for 5 days and/or 100 ng/mL trichostatin A (TSA) for 12 h. GAPDH was used as an internal control. (b) Methylation status of the DKFZP564J and CYP4V2 CpG (cytosine and guanine separated by phosphate) islands in a panel of OSCC cell lines and the RT7 cell line. (Top) Schematic map of the CpG islands (horizontal gray thick bars) around exon 1 (open boxes) of DKFZP564J and CYP4V2. CpG sites are indicated by vertical ticks on the expanded axis and the regions examined in the combined bisulfite restriction analysis (COBRA) are indicated by black closed arrows with their sizes and restriction sites for BstUI (gray downward arrowheads). Sequences of primers for COBRA are shown in supplementary Table 1. (Bottom) Representative results of COBRA for the DKFZP564J and CYP4V2 CpG islands in OSCC cell lines and RT7 cell line. CpGenome Universal Methylated DNA (Chemicon International, Temecula, CA, USA) was used as positive control (M). Arrowheads show fragments specifically restricted at sites recognized as methylated CpG, whereas arrows show undigested fragments indicating unmethylated CpG.
Fig. S2. Methylation status of the CpG (cytosine and guanine separated by phosphate) island and expression level of melatonin receptor 1 A (MTNR1A) in primary tumors of oral squamous‐cell carcinoma (OSCC). (a) Representative results of methylation‐specific polymerase chain reaction (MSP) analysis of the MTNR1A promoter region in surgically resected primary OSCC tumors (T) and corresponding non‐cancerous oral mucosa (N). Parallel amplification reactions were done using primers specific for methylated (M) and unmethylated (U) sequences by MSP. See legend for Fig. 4a for interpretation. (b) Results of quantitative bisulfite sequencing of the MTNR1A CpG island Region 2 and 3 in two representative cases analyzed by MSP in primary OSCC tumors. See legend for Fig. 3a and 4b for interpretation. Consistent with the pattern observed in MSP analysis, tumors (T) showed hypermethylated pattern especially in 3′ of Region 2 compared with corresponding non‐cancerous oral mucosa (N). Polymerase chain reaction (PCR) primers for methylation specific PCR (MSP and unmethylated‐specific PCR [USP]) are indicated by arrows. (c) Representative results of immunohistochemical staining of the MTNR1A protein. Tumors in two cases showed positive MTNR1A staining in <10% of cancer cells in tumor tissues, whereas positive immunoreactivity of MTNR1A was detected in neighboring non‐neoplastic mucosa. Magnifications are ×200.
Table S1. Primer sequences used in this study.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
We are grateful to Professor Masaki Noda (Hard Tissue Genome Research Center, Tokyo Medical and Dental University) for continuous encouragement throughout this work. We also thank Ayako Takahashi and Rumi Mori for technical assistance.
This study was supported in part by a grants‐in‐aid for Scientific Research and Scientific Research on Priority Areas, and 21st Century Center of Excellence (COE) Program for Molecular Destruction and Reconstitution of Tooth and Bone from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; a grant from Core Research for Evolutional Science and Technology (CREST) of Japan Science and Technology Corporation (JST); and a grant from the New Energy and Industrial Technology Development Organization (NEDO).
References
- 1. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55: 74–108. [DOI] [PubMed] [Google Scholar]
- 2. Nomura K, Sobue T, Nakatani H et al . Number of deaths and proportional mortality rates from malignant neoplasms by site in Japan (2003). In: Editorial Board of Cancer Statistics in Japan , eds. Cancer Statistics in Japan 2005. Tokyo: National Cancer Center, 2005; 36–9. [Google Scholar]
- 3. Lippman SM, Hong WK. Molecular markers of the risk of oral cancer. N Engl J Med 2001; 344: 1323–6. [DOI] [PubMed] [Google Scholar]
- 4. Scully C, Field JK, Tanzawa H. Genetic aberrations in oral or head and neck squamous cell carcinoma (SCCHN): 1. Carcinogen metabolism, DNA repair and cell cycle control. Oral Oncol 2000; 36: 256–63. [DOI] [PubMed] [Google Scholar]
- 5. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3: 415–28. [DOI] [PubMed] [Google Scholar]
- 6. Ha PK, Califano JA. Promoter methylation and inactivation of tumour‐suppressor genes in oral squamous‐cell carcinoma. Lancet Oncol 2006; 7: 77–82. [DOI] [PubMed] [Google Scholar]
- 7. Nakagawa T, Pimkhaokham A, Suzuki E, Omura K, Inazawa J, Imoto I. Genetic or epigenetic silencing of low density lipoprotein receptor‐related protein 1B expression in oral squamous cell carcinoma. Cancer Sci 2006; 97: 1070–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Snijders AM, Schmidt BL, Fridlyand J et al . Rare amplicons implicate frequent deregulation of cell fate specification pathways in oral squamous cell carcinoma. Oncogene 2005; 24: 4232–42. [DOI] [PubMed] [Google Scholar]
- 9. Baldwin C, Garnis C, Zhang L, Rosin MP, Lam WL. Multiple microalterations detected at high frequency in oral cancer. Cancer Res 2005; 65: 7561–7. [DOI] [PubMed] [Google Scholar]
- 10. Liu CJ, Lin SC, Chen YJ, Chang KM, Chang KW. Array‐comparative genomic hybridization to detect genomewide changes in microdissected primary and metastatic oral squamous cell carcinomas. Mol Carcinog 2006; 45: 721–31. [DOI] [PubMed] [Google Scholar]
- 11. Nakaya K, Yamagata HD, Arita N et al . Identification of homozygous deletions of tumor suppressor gene FAT in oral cancer using CGH‐array. Oncogene 2007; 26: 5300–8. [DOI] [PubMed] [Google Scholar]
- 12. Inazawa J, Inoue J, Imoto I. Comparative genomic hybridization (CGH)‐arrays pave the way for identification of novel cancer‐related genes. Cancer Sci 2004; 95: 559–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suzuki E, Imoto I, Pimkhaokham A et al . PRTFDC1, a possible tumor‐suppressor gene, is frequently silenced in oral squamous‐cell carcinomas by aberrant promoter hypermethylation. Oncogene 2007; 26: 7921–32. [DOI] [PubMed] [Google Scholar]
- 14. Tamarkin L, Cohen M, Roselle D, Reichert C, Lippman M, Chabner B. Melatonin inhibition and pinealectomy enhancement of 7,12 dimethylbenz (a) anthracene‐induced mammary tumors in the rat. Cancer Res 1981; 41: 4432–6. [PubMed] [Google Scholar]
- 15. Blask DE, Sauer LA, Dauchy LA, Holowachuk EW, Ruhoff MS. New insights into melatonin regulation of cancer growth. Adv Exp Med Biol 1999; 460: 337–43. [PubMed] [Google Scholar]
- 16. Yuan L, Collins AR, Dai J, Dubocovich ML, Hill SM. Overexpression of the mt1 melatonin receptor enhances melatonin's growth‐suppression of human breast cancer cells. Mol Cell Endocrinol 2002; 192: 147–56. [DOI] [PubMed] [Google Scholar]
- 17. Collins A, Yuan L, Kiefer TL, Cheng Q, Lai L, Hill SM. Overexpression of the MT1 melatonin receptor in MCF‐7 human breast cancer cells inhibits mammary tumor formation in nude mice. Cancer Lett 2003; 189: 49–57. [DOI] [PubMed] [Google Scholar]
- 18. Bartsch C, Bartsch H. The anti‐tumor activity of pineal melatonin and cancer enhancing life styles in industrialized societies. Cancer Causes Control 2006; 17: 559–71. [DOI] [PubMed] [Google Scholar]
- 19. Vijayalaxmi Thomas CR Jr, Reiter RJ, Herman TS. Melatonin: from basic research to cancer treatment clinics. J Clin Oncol 2002; 20: 2575–601. [DOI] [PubMed] [Google Scholar]
- 20. Jung B, Ahmad N. Melatonin in cancer management: progress and promise. Cancer Res 2006; 66: 9789–93. [DOI] [PubMed] [Google Scholar]
- 21. Akanuma D, Uzawa N, Yoshida MA, Negishi A, Amagasa T, Ikeuchi T. Inactivation patterns of the p16 (INK4a) gene in oral squamous cell carcinoma cell lines. Oral Oncol 1999; 35: 476–83. [DOI] [PubMed] [Google Scholar]
- 22. Fujimoto R, Kamata N, Yokoyama K et al . Establishment of immortalized human oral keratinocytes by gene transfer of a telomerase component. J Jpn Oral Muco Membr (Jpn) 2002; 8: 1–8. [Google Scholar]
- 23. Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res 1997; 25: 2532–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Imoto I, Tsuda H, Hirasawa A et al . Expression of cIAP1, a target for 11q22 amplification, correlates with resistance of cervical cancers to radiotherapy. Cancer Res 2002; 62: 4860–6. [PubMed] [Google Scholar]
- 25. Hermsen MA, Joenje H, Arwert F et al . Assessment of chromosomal gains and losses in oral squamous cell carcinoma by comparative genomic hybridisation. Oral Oncol 1997; 33: 414–18. [DOI] [PubMed] [Google Scholar]
- 26. Wolff E, Girod S, Liehr T et al . Oral squamous cell carcinomas are characterized by a rather uniform pattern of genomic imbalances detected by comparative genomic hybridisation. Oral Oncol 1998; 34: 186–90. [DOI] [PubMed] [Google Scholar]
- 27. Chen YJ, Lin SC, Kao T et al . Genome profiling of oral squamous cell carcinoma. J Pathol 2004; 204: 326–32. [DOI] [PubMed] [Google Scholar]
- 28. Brzezinski A. Melatonin in humans. New Eng J Med 1997; 336: 186–95. [DOI] [PubMed] [Google Scholar]
- 29. Dubocovich ML, Rivera‐Bermudez MA, Gerdin MJ, Masana MI. Molecular pharmacology, regulation and function of mammalian melatonin receptors. Front Biosci 2003; 8: d1093–108. [DOI] [PubMed] [Google Scholar]
- 30. Martin V, Herrera F, Carrera‐Gonzalez P et al . Intracellular signaling pathways involved in the cell growth inhibition of glioma cells by melatonin. Cancer Res 2006; 66: 1081–8. [DOI] [PubMed] [Google Scholar]
- 31. Cutando A, Gómez‐Moreno G, Arana C, Acuña‐Castroviejo D, Reiter RJ. Melatonin: potential functions in the oral cavity. J Periodontol 2007; 78: 1094–102. [DOI] [PubMed] [Google Scholar]
- 32. Ram PT, Dai J, Yuan L et al . Involvement of the mt1 melatonin receptor in human breast cancer. Cancer Lett 2002; 179: 141–50. [DOI] [PubMed] [Google Scholar]
- 33. Jones MP, Melan MA, Witt‐Enderby PA. Melatonin decreases cell proliferation and transformation in a melatonin receptor‐dependent manner. Cancer Lett 2000; 151: 133–43. [DOI] [PubMed] [Google Scholar]
- 34. Czesnikiewicz‐Guzik M, Konturek SJ, Loster B, Wisniewska G, Majewski S. Melatonin and its role in oxidative stress related diseases of oral cavity. J Physiol Pharmacol 2007; 58 (Suppl 3): 5–19. [PubMed] [Google Scholar]
- 35. Kolb A. The first intron of the murine â‐casein gene contains a functional promoter. Biochem Biophys Res Commun 2003; 306: 1099–105. [DOI] [PubMed] [Google Scholar]
- 36. Nakagawachi T, Soejima H, Urano T et al . Silencing effect of CpG island hypermethylation and histone modifications on O6‐methylguanine‐DNA methyltransferase (MGMT) gene expression in human cancer. Oncogene 2003; 22: 8835–44. [DOI] [PubMed] [Google Scholar]
- 37. Sonoda I, Imoto I, Inoue J et al . Frequent silencing of low density lipoprotein receptor‐related protein 1B (LRP1B) expression by genetic and epigenetic mechanisms in esophageal squamous cell carcinoma. Cancer Res 2004; 64: 3741–7. [DOI] [PubMed] [Google Scholar]
- 38. Misawa A, Inoue J, Sugino Y et al . Methylation‐associated silencing of the nuclear receptor 1I2 gene in advanced‐type neuroblastomas, identified by bacterial artificial chromosome array‐based methylated CpG island amplicon. Cancer Res 2005; 65: 10233–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. (a) Representative results of reverse transcription–polymerase chain reaction (RT‐PCR) analysis to examine the DKFZP564J and CYP4V2 expression levels after demethylation in oral squamous‐cell carcinoma (OSCC) cell lines with no or decreased expression of those genes. Messenger RNA (mRNA) was isolated after treatment with various concentrations of 5‐aza‐dCyd for 5 days and/or 100 ng/mL trichostatin A (TSA) for 12 h. GAPDH was used as an internal control. (b) Methylation status of the DKFZP564J and CYP4V2 CpG (cytosine and guanine separated by phosphate) islands in a panel of OSCC cell lines and the RT7 cell line. (Top) Schematic map of the CpG islands (horizontal gray thick bars) around exon 1 (open boxes) of DKFZP564J and CYP4V2. CpG sites are indicated by vertical ticks on the expanded axis and the regions examined in the combined bisulfite restriction analysis (COBRA) are indicated by black closed arrows with their sizes and restriction sites for BstUI (gray downward arrowheads). Sequences of primers for COBRA are shown in supplementary Table 1. (Bottom) Representative results of COBRA for the DKFZP564J and CYP4V2 CpG islands in OSCC cell lines and RT7 cell line. CpGenome Universal Methylated DNA (Chemicon International, Temecula, CA, USA) was used as positive control (M). Arrowheads show fragments specifically restricted at sites recognized as methylated CpG, whereas arrows show undigested fragments indicating unmethylated CpG.
Fig. S2. Methylation status of the CpG (cytosine and guanine separated by phosphate) island and expression level of melatonin receptor 1 A (MTNR1A) in primary tumors of oral squamous‐cell carcinoma (OSCC). (a) Representative results of methylation‐specific polymerase chain reaction (MSP) analysis of the MTNR1A promoter region in surgically resected primary OSCC tumors (T) and corresponding non‐cancerous oral mucosa (N). Parallel amplification reactions were done using primers specific for methylated (M) and unmethylated (U) sequences by MSP. See legend for Fig. 4a for interpretation. (b) Results of quantitative bisulfite sequencing of the MTNR1A CpG island Region 2 and 3 in two representative cases analyzed by MSP in primary OSCC tumors. See legend for Fig. 3a and 4b for interpretation. Consistent with the pattern observed in MSP analysis, tumors (T) showed hypermethylated pattern especially in 3′ of Region 2 compared with corresponding non‐cancerous oral mucosa (N). Polymerase chain reaction (PCR) primers for methylation specific PCR (MSP and unmethylated‐specific PCR [USP]) are indicated by arrows. (c) Representative results of immunohistochemical staining of the MTNR1A protein. Tumors in two cases showed positive MTNR1A staining in <10% of cancer cells in tumor tissues, whereas positive immunoreactivity of MTNR1A was detected in neighboring non‐neoplastic mucosa. Magnifications are ×200.
Table S1. Primer sequences used in this study.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item