

Abstract
Although dendritic cells (DC) have been well demonstrated as a strong cellular adjuvant for a tumor vaccine, there are several limitations for clinical application. A protein‐based vaccine using a potent adjuvant is an appealing approach for tumor antigen‐specific immunotherapy because of their simplicity, safety, efficacy and capacity for repeated administration. CpG‐oligodeoxynucleotides (ODN) have been used as adjuvants to stimulate innate and adaptive immune responses for cancer treatment. The authors evaluated the adjuvant effects of CpG‐ODN in a vaccine incorporating recombinant fusion protein of the HIV TAT PTD domain and carcinoembryonic antigen (TAT‐CEA). Mice vaccinated with TAT‐CEA and CpG‐ODN (TAT‐CEA + CpG) showed enhanced CEA‐specific immunity, including cytotoxic T‐lymphocytes (CTL) activity and interferon (IFN)‐γ secreting T cells compared with CEA and CpG‐ODN (CEA + CpG) or TAT‐CEA vaccination alone. Vaccination with TAT‐CEA + CpG elicited Th1‐based responses, as indicated by the higher ratio of immunoglobulin (Ig)G2a antibody/IgG1 antibodies specific for CEA. The survival rate was significantly increased after vaccination with TAT‐CEA + CpG in a tumor model using MC38/CEA2. Furthermore, the TAT‐CEA ± CpG vaccine groups showed similar antitumor immunity to the CEA peptide‐pulsed DC (CEA peptide/DC) vaccine groups. These data suggest that coadministration of TAT fusion protein with CpG‐ODN may serve as a potential formulation for enhancing antitumor activity. (Cancer Sci 2008; 99: 1034–1039)
Oligodeoxynucleotides (ODN) containing unmethylated CpG motifs can activate B cells, dendritic cells (DC), and natural killer (NK) cells, as well as induce a Th1 cell‐like pattern of cytokine production.( 1 , 2 , 3 , 4 ) Moreover, CpG‐ODN enhances humoral responses, driving them toward immunoglobulin (Ig)G2a, IgG2b, and IgG3 isotypes (Th1 type indicator),( 5 ) and enhances cytotoxic T‐lymphocytes (CTL) activity.( 6 ) Co‐administering CpG‐ODN with protein antigens, subunit vaccines, or DNA vaccines is both safe and effective in enhancing immunity in an animal challenge model.( 7 , 8 , 9 ) CpG‐ODN are internalized to endosomes, leading to acidification and recognition by the TLR9 trigger and nuclear factor (NF)‐κB activation.( 10 , 11 )
The HIV‐1 TAT peptide can transport macromolecules into cells and allows proteins to be used as immunogens for major histocompatibility complex (MHC) class I‐restricted CTL responses.( 12 ) TAT peptides from derived PTD protein are transported rapidly from the extracellular milieu into the cytosol.( 10 , 13 ) These cationic peptides can efficiently deliver 20‐ to 200‐kDa proteins, peptides, liposomes, or antisense oligonucleotides into cells.( 10 ) Therefore, the adjuvant effect of ODN could enhance immune responses by binding them to TAT peptides fused with antigen proteins.
Carcinoembryonic antigen (CEA) is a 180‐kDa oncofetal glycoprotein and soluble tumor marker. It is extensively expressed in the vast majority of colorectal, gastric, and pancreatic carcinomas, in approximately 50% of breast cancers, and in 70% of non‐small cell lung cancers.( 14 ) Several vaccine approaches, including peptides, viral vectors and dendritic cells, have been performed for human and experimental studies for the treatment of tumors expressing CEA.( 15 , 16 , 17 ) The authors have previously reported that DC transduced with recombinant adenoviruses expressing CEA, or DC pulsed with peptide of CEA, have been demonstrated to stimulate strong tumor‐antigen‐specific CTL responses in vitro,( 18 ) and to induce antitumor immunity in vivo in animal models,( 19 ) respectively. Thus, the DC‐based vaccine approach is well established for human and experimental studies. However, DC are sparse in peripheral blood mononuclear cells (PBMC) and do not proliferate readily in vitro, despite the report of Romani et al.( 20 ) Therefore heavy leukapheresis is sometimes required for DC preparation in cancer patients, who quite often have damaged bone marrow (the source of the cells) because of treatments with radiation and/or antitumor drugs.
Protein‐based vaccines such as TAT‐fusion protein have become an appealing approach to inducing antigen‐specific immunotherapy because of their simplicity, safety, efficacy and capacity for repeated administration. In the present study, the authors demonstrated that the combination of TAT‐CEA fusion protein and CpG‐ODN showed a significantly enhanced induction of tumor‐specific CTL and prolonged survival in an MC38/CEA2 tumor model.
Materials and Methods
Animals and cell lines. Female C57BL/6(H‐2Kb) mice aged 6–8 weeks were purchased from Orient (Kapyung, Kyounggi, Korea). The MC38/CEA2 adenocarcinoma cell line was kindly provided by Dr J. Schlom (Division of Tumor Immunology and Biology, NIH, USA). Murine colon adenocarcinoma cells expressing human CEA (MC38/CEA2) were generated by retroviral transduction of MC38 cells with CEA cDNA. The MC38/CEA2 cells, MC38 cells, and GL26 cells were cultured in complete Dulbecco's Modified Eagle Medium (DMEM; BioWhittaker, Walkersville, MD, USA) medium supplement with 10% heat‐inactivated fetal bovine serum (FBS), 100 U/mL penicillin, 100 µg/mL streptomycin, and 2 mmol L‐glutamine. To enhance the expression of major histocompatibility complex (MHC) class I, MC38/CEA2 cells were treated with 50 IU/mL of murine interferon (IFN)‐γ (Endogen Inc., Cambridge, MA, USA) for 48 h at 37°C before re‐stimulation and testing with a CTL cytotoxicity assay.
Production of TAT‐CEA fusion protein. The CEA cDNA was amplified from the LoVo cell line using a reverse transcriptase‐polymerase chain reaction (RT‐PCR) with a CEA domain 1 (A1‐B1) sense primer 5′‐ACG CCA TAT GCT GCT GAT CCA GAA CA‐3′ containing the XhoI site, and a CEA domain 1 antisense primer 5′‐ATA TGG ATC CCG TCG TGA CTG TGG TCC T‐3′ containing the BamHI site. The amplified DNA was digested with XhoI and BamHI, and the resulting DNA fragment was gel purified. The CEA DNA fragments were then cloned into the XhoI and BamHI sites of the pET‐15b vector (Novagen, Madison, WI, USA). The plasmid construct was transformed into Escherichia coli DH5α (Real Biotech Corp., Taipei, Taiwan) and selection performed against ampicillin. Cellular adhesion domains of the HIV‐1 TAT were generated via PCR using the plasmid pNL4‐3 as a template, with NdeI and XhoI restriction sites on the 5′‐ and 3′ terminals, respectively, producing a short TAT peptide, ‘YGRKKRRQRRR’. The resulting fragment was digested with NdeI and XhoI and subcloned into the corresponding restriction sites of pET15b‐CEA. The plasmid pET15b‐TAT‐CEA vector was purified and again transformed into BL21 (Real Biotech Corp., Taipei, Taiwan) cells, and incubated in Luria‐Bertani (LB) broth supplemented with ampicillin at a final concentration of 100 µg/mL.
CpG‐ODN. The immunostimulatory CpG‐ODN, designated 1826 (5′‐TCC ATG ACG TTC CTG ACG TT‐3′), has adjuvant activity with protein antigens. CpG‐ODN was purchased from Bioneer (Daejeon, Korea), and synthesized with a nuclease‐resistant phosphorothioate backbone. CpG‐ODN was dissolved in water and had undetectable endotoxin levels.
Protein expression and purification. The BL21 strain of E. coli is used for expression of genes in expression vectors containing the bacteriophage T7 promoter. BL21 cells were transformed with the pET‐15b plasmid encoding the TAT‐CEA insertion element. The cells were grown in LB broth containing 100 µg/mL ampicillin. Protein expression was induced by the addition of 1.5 mmol isopropyl β‐D thiogalactoside (IPTG) and incubation for 3 h. The sonicated supernatants were applied at room temperature to a pre‐equilibrated Ni‐NTA Superflow (Qiagen, Valencia, CA, USA) in Buffer Z plus 10 mmol imidazole, and separated by gravity. Next, refolding buffer was added that contained gradually lower concentrations of urea (6.0–0.1 mol) and gradually higher concentrations of imidazole. The final product was obtained at 0.1 mol urea and 1 mol imidazole, followed by overnight dialysis to remove residual salts.
For western blot analysis, purified proteins were loaded using 12.5% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and then transferred onto a nitrocellulose membrane. The membrane was incubated with 5% non‐fat milk in phosphate‐buffered saline (PBS) and then with an anti‐His monoclonal antibody (mAb) overnight at 4°C. After washing, the membranes were incubated with an alkaline phosphatase‐conjugated goat antimouse IgG antibody (Amersham Biosciences, Buckinghamshire, UK) for 1 h at room temperature. Immunoreactive bands were detected using the ECL western blotting analysis system (Amersham Biosciences).
Mouse vaccination and CTL induction. For induction of primary CTL in vivo, 50 µg of TAT‐CEA protein and 2 µmol of CpG‐ODN [TAT‐CEA + CpG] were vaccinated s.c. into syngeneic mice at day 0. Groups of mice were vaccinated with either 50 µg of CEA protein [CEA], 50 µg of CEA and 2 µmol of CpG ODN [CEA + CpG], 50 µg of TAT‐CEA protein without CpG‐ODN [TAT‐CEA], or PBS. Vaccination with DC pulsed with CEA peptide [CEA peptide/DC] was done as described previously.( 15 ) On day 7, mice were given a booster vaccination using the same protocol as above. Seven days after the booster vaccination, mice were killed and splenocytes were harvested, homogenized, and red blood cells (RBC) were lysed with hypotonic buffer. Non‐adherent splenocytes were separated from DC, macrophages, and monocytes by adherence to plastic for 90 min and used as effector cells. Splenocytes (2 × 107) were re‐stimulated with 4% paraformaldehyde and prefixed MC38/CEA2 cells for 5 days. The cells were then cultured in the presence of 10 IU/mL interleukin (IL)‐2 for 5 days at 37°C.
Generation of CEA RNA in vitro transcription. The pcDNA3‐CEA vector was linearized with SmaI and in vitro transcription was performed with the mMessage in mMachine™ Ultra T7 kit (Ambion, Austin, TX, USA) according to the manufacturer's instructions. mRNA concentration and quality were assessed using spectrophotometer and agarose gel electrophoresis. RNA samples were routinely evaluated using formaldehyde/agarose gel electrophoresis for size and integrity and stored at –70°C.
Preparation of DC transferred with CEA RNA. DC were generated from mouse bone marrow with recombinant murine granulocyte–macrophage colony‐stimulating factor (GM‐CSF; 20 ng/mL) and recombinant murine IL‐4 (20 ng/mL; R&D Systems, Minneapolis, MN, USA) as described previously.( 21 ) Prior to electroporation on day 7, immature DC were washed twice with serum‐free medium and resuspended to a final concentration of 1 × 107 cells/mL. The cell suspension (200 µL) was preincubated in a 0.2‐cm gap electroporation cuvette for 5 min on ice. Next, 20 µg of RNA was added and cells were pulsed with an ECM 830 electroporator (BTX, San Diego, CA, USA). The physical parameters were voltage of 300 V and pulsed time of 50 µs. After electroporation, the cells were immediately transferred into mature medium and incubated for 24 h at 37°C. Cell viability was more than 80% after electroporation. The transfection efficacy of electroporation on DC was determined with enhanced green fluorescent protein (EGFP) RNA using fluorescence‐activated cell sorting (FACS) analysis.
Cytotoxicity assay. The assay for cell‐mediated killing of target cells was performed in vitro using a standard 4‐h chromium assay at various effector/target ratios. Briefly, splenocytes were in vitro with 4% paraformaldehyde prefixed MC38/CEA2 cells in the presence of recombinant murine IL‐2 (10 IU/mL) for 5 days and used as effector cells. MC38/CEA2, MC38, and GL26 tumor cells were labeled with 100 3/7 mBq of [51Cr] sodium chromate/106 cell for 1 h, washed four times, and then added to quadruple wells of 96‐v‐bottom microtiter plates with various numbers of effector cells. The effector : target (E : T) ratios were 10:1, 20:1, and 40:1. After incubation for 4 h at 37°C, 100 µL of supernatant in each well was collected and radioactivity was counted using a gamma counter. The percentage of specific lysis was calculated as described previously.( 21 )
Enzyme‐linked immunospot (ELISPOT) assay. An ELISPOT kit was purchased from AID (Strasberg, Germany) and used according to the manufacturer's instructions. Briefly, the re‐stimulated splenocytes were seeded into a 96‐well microplate coated with antimouse IFN‐γ antibody at a concentration of 1 × 105 cells/well in cell culture medium. CEA RNA‐electroporated DC (5 × 104 cells/well) were added as a stimulus. The plates were incubated for 24 h at 37°C. After developing the spots, the reaction was quenched with distilled water, and the plates were inverted and allowed to dry overnight in the dark. The number of spots corresponding to the IFN‐γ‐secreting cells was determined using an automatic AID‐ELISPOT‐Reader.
Mouse Ig isotyping. Sera were tested for specific Ig subclass antibodies using the Mouse Immunoglobulin Screening/Isotyping Kit (Zymed Laboratories, Invitrogen Immunodetection). In brief, a 96‐well flat bottom microplate was coated with 500 ng/well of CEA protein, diluted in PBS, and incubated overnight at 4°C. The plate was blocked with 200 µL PBS containing 1% bovine serum albumin (BSA) for 1 h at 37°C. Serum (1:10 dilution) in 1% PBS was added (50 µL) to each well and incubated for 30 min at 37°C. The plate was then incubated sequentially with 50 µL PBS for a blank column, one drop of biotinylated antibody (provided in the kit) for negative control, and one drop of subclass specific antibodies (IgG1, IgG2a, IgG2b, IgG3, Ig A, and IgM: provided in the kit) for 15 min at room temperature, with horseradish peroxidase (HRP)–streptavidin then added for 15 min at room temperature (RT). Finally, 100 µL of working substrate solution containing 2,2′‐azino‐bis(3‐ethylbenzthiazoline‐6‐sulphonic acid) (ABTS) substrate and hydrogen in citrate buffer (provided in the kit) was added. The absorbance was read at optical density (OD) 405 nm using an automated ELISA reader (Molecular Devices, Sunnyvale, CA, USA).
Tumor models. Mice were injected s.c. with 1 × 106 MC38/CEA2 tumor cells. At 10 days after tumor cell inoculation, mice were vaccinated s.c. with TAT‐CEA + CpG and CEA peptide/DC at weekly intervals for 5 weeks. Control groups of mice were vaccinated with either CEA, TAT‐CEA, CEA + CpG, or PBS. Seven mice were used for each treatment group.
Statistical analysis. Results are expressed as mean ± standard error of the mean. Statistical analysis was performed using an ANOVA test. The survival data was analyzed using the Kaplan–Meier test. Survival data were compared using a log‐rank test. Data were considered statistically significant at P < 0.05.
Results
Production of TAT‐CEA fusion protein. DNA fragments of the CEA domain 1 and the short TAT peptide insertion element, ‘YGRKKRRQRRR’, were subcloned into the corresponding restriction sites of the pET‐15b plasmid. The plasmid pET15b‐TAT‐CEA vector was purified and transformed into BL21 (Fig. 1a). Fusion protein expression was induced by adding IPTG to LB broth media, and the sonicated cell supernatants were purified using Ni2+ (Ni‐NTA superflow) affinity chromatography. CEA and TAT‐CEA spread to the expected sizes in western blotting analysis (Fig. 1b).
Figure 1.

Schematic map of the HIV TAT PTD domain and carcinoembryonic antigen (TAT‐CEA) and analysis of TAT‐CEA expression. (a) A schematic diagram of the plasmid encoding TAT‐CEA. (b) Expression of CEA and TAT‐CEA protein using western blotting analysis with 1 µg of purified protein from bacteria.
Enhancement of CTL activity. To test whether vaccination using TAT‐fusion protein and CpG‐ODN could enhance tumor‐specific CTL responses in vivo, mice were immunized with TAT‐CEA + CpG, CEA peptide/DC, CEA, TAT‐CEA, CEA + CpG, or PBS, and then re‐stimulated with 4% paraformaldehyde prefixed syngeneic MC38/CEA2 in vitro for 5 days. Cytotoxicity against syngeneic MC38/CEA2 target cells in the spleens of mice vaccinated with TAT‐CEA + CpG and CEA peptide/DC was significantly higher than those in the mice vaccinated in the other control groups (Fig. 2b). This CTL activity was CEA‐specific, in that none of these cells showed cytotoxic activities against the CEA‐negative MC38 or irrelevant GL26 (Fig. 2c,d). No killing activity was observed in effector cells vaccinated with the control group. In addition, the effector cells did not lyse the YAC‐1 target cell, indicating that the killing activity was not NK‐cell mediated (data not shown).
Figure 2.

Tumor‐specific cytotoxic T lymphocytes (CTL) activity. (a) Experiment schedule of vaccination for the in vitro assay. (b–d) Enhanced CTL activity using HIV TAT PTD domain and carcinoembryonic antigen (TAT‐CEA) + CpG. Splenocytes were harvested from mice vaccinated with TAT‐CEA + CpG, CEA peptide/dendritic cells (DC), TAT‐CEA, CEA + CpG, CEA, or phosphate‐buffered saline (PBS) (see Materials and Methods). The effector cells were generated by cocultivation of these splenocytes with 4% paraformaldehyde and prefixed syngeneic MC38/CEA2 cells for 5 days. The target cells (MC38/CEA2, MC38 or GL26) were labeled with 51Cr and incubated with the effector cells at the ratio indicated. *Statistically significant at P < 0.05 using anova compared with all other groups. Results are given as means ± SE, and are representative of two independent experiments. E/T, effector/target.
Frequency of IFN‐γ secreting T cells. CTL produces IFN‐γ in an antigen‐specific manner, so the levels of tumor‐specific CTL were measured using the IFN‐γ ELISPOT assay. Splenocytes from mice vaccinated with TAT‐CEA + CpG and CEA peptide/DC showed significantly higher numbers of IFN‐γ‐producing T cells (Fig. 3). In contrast, the effector cells of control mice produced negligible levels of IFN‐γ. This result suggests that vaccination of mice with TAT‐CEA + CpG enhanced the CEA‐specific immune response.
Figure 3.

Interferon (IFN)‐γ production by immunized splenocytes. IFN‐γ‐secreting splenocytes from the treated mice, were measured using the enzyme‐linked immunospot (ELISPOT) assay after re‐stimulation in vitro with prefixed MC38/carcinoembryonic antigen (CEA)2 cells for 5 days. Re‐stimulated splenocytes were stimulated in vitro with CEA RNA‐electroporated dendritic cells (DC) for 18 h. Results represent the mean number of IFN‐γ spots per 1 × 105 splenocytes from individually tested mice. *Statistically significant at P < 0.05 using ANOVA compared with all other groups. Results are given as means ± SE, and are representative of two independent experiments. PBS, phosphate‐buffered saline; TAT, HIV TAT PTD domain.
Isotypes of CEA‐specific antibodies. The authors investigated the in vivo effects of TAT‐CEA + CpG on the modulation of antigen‐specific antibody responses by favoring the development of Th1 versus Th2 cells. The levels of IgG subclasses were analyzed, as they provide an indication of the Th1 versus Th2 nature of the immune response (Fig. 4a). IgG1 is a Th2‐associated antibody, whereas IgG2a is a Th1‐associated antibody.( 22 ) IgG2a levels were significantly higher in the sera of TAT‐CEA + CpG‐vaccinated mice than in other groups (Fig. 4a). Mice vaccinated with TAT‐CEA + CpG presented a higher IgG2a/IgG1 ratio than did those in the other groups (Fig. 4b). Thus the CpG‐ODN enhances the TAT‐CEA‐induced, Th1 type immune response.
Figure 4.

Detection of serum antibodies. Each group of mice was vaccinated s.c. initially and 1 week later. (a) Mice were bled for detection of serum antigen‐specific immunoglobulin (Ig)G1 and IgG2a 1 week after the second injection. Sera were diluted to 1:10 and reacted with carcinoembryonic antigen (CEA) protein in enzyme‐linked immunosorbent assay (ELISA). Absorbance was measured at 405 nm. (b) The IgG2a/IgG1 ratio is shown. OD, optical density; PBS, phosphate‐buffered saline; TAT, HIV TAT PTD domain.
Antitumor effects. Because the TAT‐CEA + CpG combination induced tumor‐specific CTL, the authors also examined whether that vaccination had therapeutic antitumor immunity in a s.c. MC38/CEA2 tumor model. Mice were injected s.c. with 1 × 106 MC38/CEA2 tumor cells. At 10 days after tumor cell inoculation, mice were vaccinated s.c. with TAT‐CEA + CpG at weekly intervals for 5 weeks (shown schematically in Fig. 5a). Treatment with TAT‐CEA + CpG and CEA peptide/DC partially inhibited tumor growth compared with the control groups (Fig. 5b), and significantly prolonged survival compared with the TAT‐CEA or CEA + CpG (Fig. 5c).
Figure 5.
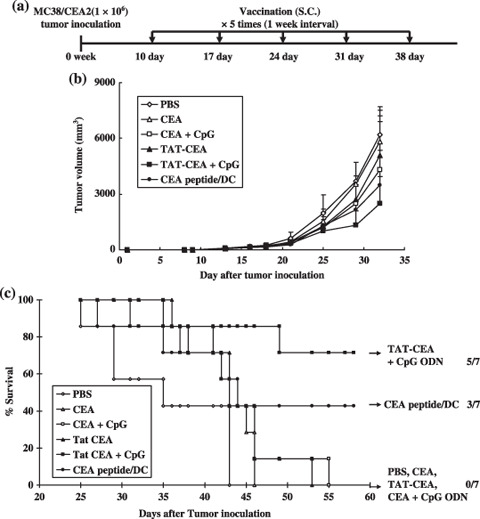
Antitumor therapeutic effect of the HIV TAT PTD domain and carcinoembryonic antigen (TAT‐CEA) and CpG. (a) Experiment schedule of vaccination for survival and tumor growth. C57BL/6 naive mice were inoculated s.c. with 1 × 106 MC38/CEA2 tumor cells on day 0 and subsequently immunized in the opposite flanks with TAT‐CEA + CpG or CEA peptide/dendritic cells (DC) at weekly intervals for 5 weeks from day 10 after tumor inoculation. (a) Tumor volume and (b) mice survival were monitored in groups of seven mice. (c) The numbers to the right indicate the number of surviving mice per total number of mice in each group. Significant differences (log‐rank test): TAT‐CEA + CpG versus TAT‐CEA, P = 0.032; TAT‐CEA + CpG versus CEA + CpG, P = 0.038; TAT‐CEA + CpG versus PBS, P = 0.003; TAT‐CEA + CpG versus CEA peptide/DC, P = 0.243. The survival advantage conferred by TAT‐CEA + CpG was statistically significant compared with either of the control groups (Kaplan–Meier test, P < 0.05).
Discussion
One priority for current vaccine research is the development of adjuvants that support the efficient priming of long‐lasting, antigen, effector/memory CD8+ T‐cell immunity. ODN with immune‐stimulating sequences containing CpG motifs facilitate the priming of MHC class I‐restricted CD8+ T‐cell responses to proteins or peptides. Similarly, synthetic ODN with CpG motifs (or immune‐stimulating sequences [ISS]) are potent adjuvants that facilitate priming of Th1 immunity.( 23 ) CpG‐ODN can modulate immune functions by activating dendritic cells, B cells, and NK cells,( 2 , 3 ) as well as the Th1/Th2 balance.( 24 ) The authors have previously reported that CpG‐ODN treatment in pulsing DC with E7 antigens significantly enhances antitumor immunity in an HPV 16 E6/E7‐associated cervical cancer animal model.( 25 ) In the present study, it was demonstrated that a CpG‐ODN adjuvant in a TAT fusion protein vaccine significantly enhanced antitumor activity. CpG‐ODN alone did not facilitate CD8+ T‐cell priming and did not affect tumor regression. This observation is in line with previous findings that co‐injection of plasmid‐based DNA and CpG‐ODN provides more protection from tumor challenges in a murine model.( 26 ) DNA‐based vaccines also contain nucleotide sequences with CpG motifs that stimulate innate host defense mechanisms by triggering signals from TLR9.( 8 ) Some cationic peptides, such as HIV TAT, polylysines, and polyarginines, can enhance antigen immunogenicity.( 27 , 28 ) Enhanced peptide‐specific immune responses have been induced by a synthetic vaccine composed of antigenic peptides (T‐cell epitopes), ODN with CpG motifs, and poly L‐arginine.( 29 ) Cationic HIV TAT peptides efficiently translocate through plasma membranes and can be used as molecular transporters to deliver drugs, proteins, or DNA that are (covalently or non‐covalently) linked to it into cells.( 30 , 31 , 32 ) Furthermore, chemical coupling of synthetic polylysines, polyarginines, or natural cationic domains from bacterial or viral proteins to heterologous protein domains or peptides enhances their cellular uptake and stimulates antigen‐specific T‐cell responses in vivo.( 33 , 34 ) The present data indicate that targeting of the CpG‐ODN adjuvant by the HIV TAT peptide is essential for potentiating the induction of T‐cell immunity.
Vaccination with TAT‐CEA + CpG significantly increased IgG2a production, which is consistent with findings that coadministration of modified tumor antigen with CpG ODN increased IgG2a or IgG2b.( 25 , 34 ) This increase in IgG2a is consistent with the induction of Th1 cells induced by CpG‐ODN. This result suggests that vaccination with TAT‐CEA + CpG improved vaccine efficacy through a Th‐1 cell response.
In conclusion, it was observed that covaccination with a TAT‐CEA fusion protein and CpG could induce significant antitumor immunity in a CEA tumor model through induction of tumor‐specific CTL.
Acknowledgments
This study was supported in part by a grant from the Ministry of Commerce, Industry and Energy (10016766‐2005‐12).
References
- 1. Sparwasser T, Koch ES, Vabulas RM et al . Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur J Immunol 1998; 28: 2045–54. [DOI] [PubMed] [Google Scholar]
- 2. Ballas ZK, Rasmussen WL, Krieg AM. Induction of NK activity in murine and human cells by CpG motifs in oligodeoxynucleotides and bacterial DNA. J Immunol 1996; 157: 1840–5. [PubMed] [Google Scholar]
- 3. Sparwasser T, Vabulas RM, Villmow B et al . Bacterial CpG‐DNA activates dendritic cells in vivo: T helper cell‐independent cytotoxic T cell responses to soluble proteins. Eur J Immunol 2000; 30: 3591–7. [DOI] [PubMed] [Google Scholar]
- 4. Bohle B, Jahn‐Schmid B, Maurer D et al . Oligodeoxynucleotides containing CpG motifs induce IL‐12, IL‐18 and IFN‐γ production in cells from allergic individuals and inhibit IgE synthesis in vitro . Eur J Immunol 1999; 29: 53. [DOI] [PubMed] [Google Scholar]
- 5. Davis HL, Weeranta R, Waldschmidt TJ et al . CpG DNA is a potent enhancer of specific immunity in mice immunized with recombinant hepatitis B surface antigen. J Immunol 1998; 160: 870–6. [PubMed] [Google Scholar]
- 6. Warren TL, Bhatia SK, Acosta AM et al . APC stimulated by CpG oligodeoxynucleotide enhance activation of MHC class I‐restricted T cells. J Immunol 2000; 165: 6244–51. [DOI] [PubMed] [Google Scholar]
- 7. Chaung HC. CpG oligodeoxynucleotides as DNA adjuvants in vertebrates and their applications in immunotherapy. Int Immunopharmacol 2006; 6: 1586–96. [DOI] [PubMed] [Google Scholar]
- 8. Spies B, Hochrein H, Vabulas M et al . Vaccination with plasmid DNA activates dendritic cells via Toll‐like receptor 9 (TLR9) but functions in TLR9‐deficient mice. J Immunol 2003; 171: 5908–12. [DOI] [PubMed] [Google Scholar]
- 9. Heit A, Huster KM, Schmitz F et al . CpG‐DNA aided cross‐priming by cross‐presenting B cells. J Immunol 2004; 172: 1501–7. [DOI] [PubMed] [Google Scholar]
- 10. Schirmbeck R, Riedl P, Zurbriggen R et al . Antigenic epitopes fused to cationic peptide bound to oligonucleotides facilitate Toll‐like receptor 9‐dependent, but CD4+ T cell help‐independent, priming of CD8+ T cells. J Immunol 2003; 171: 5198–207. [DOI] [PubMed] [Google Scholar]
- 11. Krieg AM. Therapeutic potential of Toll‐like receptor 9 activation. Nat Rev Drug Discov 2006; 5: 471–84. [DOI] [PubMed] [Google Scholar]
- 12. Kim DT, Mitchell DJ, Brockstedt DG et al . Introduction of soluble proteins into the MHC class I pathway by conjugation to an HIV tat peptide. J Immunol 1997; 159: 1666–8. [PubMed] [Google Scholar]
- 13. Riedl P, Reimann J, Schirmbeck R. Peptides containing antigenic and cationic domains have enhanced, multivalent immunogenicity when bound to DNA vaccines. J Mol Med 2004; 82: 144–52. [DOI] [PubMed] [Google Scholar]
- 14. Thompson JA, Grunert F, Zimmermann W. Carcinoembryonic antigen gene family: molecular biology and clinical perspectives. J Clin Laboratory Anal 1991; 5: 344–66. [DOI] [PubMed] [Google Scholar]
- 15. Oh ST, Kim CH, Park MY et al . Dendritic cells transduced with recombinant adenoviruses induce more efficient anti‐tumor immunity than dendritic cells pulsed with peptide. Vaccine 2006; 24: 2860–8. [DOI] [PubMed] [Google Scholar]
- 16. Aldrich WA, Ren C, White AF et al . Enhanced transduction of mouse bone marrow‐derived dendritic cells by repetitive infection with self‐complementary adeno‐associated virus 6 combined with immunostimulatory ligands. Gene Ther 2006; 13: 29–39. [DOI] [PubMed] [Google Scholar]
- 17. Hörig H, Medina FA, Conkright WA et al . Strategies for cancer therapy using carcinoembryonic antigen vaccines. Expert Rev Mol Med 2000; 2: 1–24. [DOI] [PubMed] [Google Scholar]
- 18. Cho HI, Kim HJ, Oh ST et al . In vitro induction of carcinoembryonic antigen (CEA) ‐specific cytotoxic T lymphocytes by dendritic cells transduced with recombinant adenoviruses. Vaccine 2003; 22: 224–36. [DOI] [PubMed] [Google Scholar]
- 19. Park MY, Kim CH, Sohn HJ et al . The optimal interval for dendritic cell vaccination following adoptive T cell transfer is important for boosting potent anti‐tumor immunity. Vaccine 2007; 25: 7322–30. [DOI] [PubMed] [Google Scholar]
- 20. Romani N, Gruner S, Brang D et al . Proliferating dendritic cell progenitors in human blood. J Exp Med 1994; 180: 83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim CH, Hong MJ, Park SD et al . Enhancement of anti‐tumor immunity specific to murine glioma by vaccination with tumor cell lysate‐pulsed dendritic cells engineered to produce interleukin‐12. Cancer Immunol Immunother 2006; 55: 1309–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Finkelman FD, Holmes J, Katona IM et al . Lymphokine control of in vivo immuno‐globulin isotype selection. Annu Rev Immunol 1990; 8: 303–33. [DOI] [PubMed] [Google Scholar]
- 23. Shen Y, Ichino M, Nakazawa M et al . CpG oligodeoxynucleotides prevent the development of scleroderma–like syndrome in tight‐skin mice by stimulating a Th1 immune response. J Invest Dermatol 2005; 124: 1141–8. [DOI] [PubMed] [Google Scholar]
- 24. Liu N, Ohnishi N, Ni L, Akira S et al . CpG directly induces T‐bet expression and inhibits IgG1 and IgE switching in B cells. Nat Immunol 2003; 4: 687–93. [DOI] [PubMed] [Google Scholar]
- 25. Kim TG, Kim CH, Won EH et al . CpG‐ODN‐stimulated dendritic cells act as a potent adjuvant for E7 protein delivery to induce antigen‐specific antitumour immunity in a HPV 16, E7‐associated animal tumour model. Immunology 2004; 112: 117–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schneeberger A, Wagner C, Zemann A et al . CpG motifs are efficient adjuvants for DNA cancer vaccines. J Invest Dermatol 2004; 123: 371–9. [DOI] [PubMed] [Google Scholar]
- 27. Luhrs P, Schmidt W, Kutil R et al . Induction of specific immune responses by polycation‐based vaccines. J Immunol 2002; 169: 5217–26. [DOI] [PubMed] [Google Scholar]
- 28. Mattner F, Fleitmann JK, Lingnau K et al . Vaccination with poly‐L‐arginine as immunostimulant for peptide vaccines: induction of potent and long‐lasting T‐cell responses against cancer antigens. Cancer Res 2002; 62: 1477–80. [PubMed] [Google Scholar]
- 29. Lingnau K, Egyed A, Schellack C et al . Poly‐L‐arginine synergizes with oligodeoxynucleotides containing CpG‐motifs (CpG‐ODN) for enhanced and prolonged immune responses and prevents the CpG‐ODN‐induced systemic release of pro‐inflammatory cytokines. Vaccine 2002; 20: 3498–508. [DOI] [PubMed] [Google Scholar]
- 30. Brooks H, Lebleu B, Vives E. Tat peptide‐mediated cellular delivery: back to basics. Adv Drug Deliv Rev 2005; 57: 559–77. [DOI] [PubMed] [Google Scholar]
- 31. Morris MC, Depollier J, Mery J et al . peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat Biotechnol 2001; 19: 1173–6. [DOI] [PubMed] [Google Scholar]
- 32. Snyder EL, Dowdy SF. Protein/peptide transduction domains: potential to deliver large DNA molecules into cells. Curr Opin Mol Ther 2001; 3: 147–52. [PubMed] [Google Scholar]
- 33. Mitsui H, Inozume T, Kitamura R et al . Polyarginine‐mediated protein delivery to dendritic cells presents antigen more efficiently onto MHC class I and class II and elicits superior antitumor immunity. J Invest Dermatol 2006; 126: 1804–12. [DOI] [PubMed] [Google Scholar]
- 34. Salucci V, Mennuni C, Calvaruso F et al . CD8+ T‐cell tolerance can be broken by an adenoviral vaccine while CD4+ T‐cell tolerance is broken by additional co‐administration of a Toll‐like receptor ligand. Scand J Immunol 2006; 63: 35–41. [DOI] [PubMed] [Google Scholar]