

Abstract
Cotylenin A, a plant growth regulator, and rapamycin, an inhibitor of mammalian target of rapamycin (mTOR), are potent inducers of differentiation of myeloid leukemia cells. Recently, we found that cotylenin A and rapamycin effectively inhibited the proliferation of several human breast cancer cell lines including MCF‐7. Herein, we demonstrate that cotylenin A and rapamycin rapidly and markedly induced the cyclin G2 gene expression in several cancer cells including MCF‐7 cells. The growth arrest of the MCF‐7 cells at the G1 phase, induced by the treatment with cotylenin A and rapamycin or the culture in low serum medium, markedly induced the cyclin G2 gene expression. Anticancer drugs including doxorubicin, etoposide and 5‐fluorouracil also induced cyclin G2 expression during induction of growth arrest of the MCF‐7 cell at the G1 phase or G2/M phase. Ectopically inducible cyclin G2 expression potently inhibited the proliferation of MCF‐7 cells. Furthermore, cyclin G2 knockdown induced by cyclin G2 small interfering RNA markedly reduced the potency of cotylenin A plus rapamycin to induce growth inhibition. Taken together, our results suggest that cotylenin A and rapamycin induce inhibition of cancer cell growth through the induction of cyclin G2. (Cancer Sci 2008; 99: 1693–1698)
Cotylenin A (CN‐A), which is a novel fusicoccane‐diterpene glycoside with a complex sugar moiety, was originally isolated as a plant growth regulator and has been shown to affect several physiological processes in higher plants.( 1 , 2 ) We reported previously that CN‐A has a potent differentiation‐inducing activity in several human and murine myeloid cell lines and in leukemia cells that were freshly isolated from patients with acute myelogenous leukemia.( 3 , 4 , 5 , 6 ) Furthermore, the administration of CN‐A also significantly prolonged the survival of mice with severe combined immunodeficiency that had been inoculated with cells of human promyelocytic leukemia cell line NB4.( 7 )
Rapamycin (Rapa), a macrocyclic antibiotic in complex with a 12‐kDa immunophilin (FKBP12) potently inhibits mammalian target of rapamycin (mTOR) signaling resulting in cytostatic or cytotoxic effects on cancer cells. Rapa and several analogs are currently undergoing clinical evaluation as anticancer agents.( 8 , 9 ) However, the level of sensitivity to Rapa with respect to growth inhibition differs markedly among various cancer cells, and Rapa shows promise against only a few cancers.( 10 ) To improve the therapeutic efficacy of Rapa against a broad range of human tumor cells, effective combination therapy of Rapa with other agents should be investigated. We also found previously that Rapa has a differentiation‐inducing activity in several human myeloid cell lines.( 11 )
Cyclin G2 (CG2) is an unconventional cyclin expressed at modest levels in proliferating cells, peaking during the late S/early G2‐phase, and is significantly upregulated as cells exit the cell cycle in response to DNA damage and receptor‐mediated negative signaling in B‐lymphocytes.( 12 , 13 , 14 ) Recent reports of cDNA microarray analyses consistently point to CG2 upregulation in parallel with cell cycle inhibition during the responses to diverse growth inhibitory signals, such as heat shock, oxidative stress and hypoxia.( 15 , 16 ) Ectopic expression of CG2 inhibits the proliferation of several cell types.( 16 , 17 ) Although these results support the hypothesis that CG2 has cell cycle inhibitory functions, the mechanism of CG2 regulation and cellular function in cancer cells is poorly understood.
While examining whether inducers of differentiation for leukemia can control the growth of solid tumors, we found recently that CN‐A and Rapa effectively inhibited the proliferation of human breast cancer cell line MCF‐7 cells in vitro and in vivo.( 18 ) This treatment induced growth arrest of the cells at the G1 phase, rather than apoptosis, and induced E‐cadherin and senescence‐associated β‐galactosidase activity. However, the mechanisms of the combined effects of these differentiation inducers are still unknown. Our previous results from cDNA microarray analysis showed that CG2 was markedly induced in MCF‐7 cells treated with CN‐A plus Rapa.( 18 ) In this report, we examine the role of CG2 in the CN‐A plus Rapa‐induced growth inhibition of breast cancer cells. We established an inducible CG2‐expressing cell line MCF7/CG2, in which CG2 is tightly regulated and induced only upon doxycycline (Dox) addition. Our results showed that CG2 exhibited an anticancer function by inducing growth arrest in human breast cancer cells. Furthermore, CG2 knockdown induced by cyclin G2 small interfering RNA (siRNA) reduced the potency of CN‐A plus Rapa to induce growth inhibition. These results suggest that the induction of CG2 in cancer cells treated with CN‐A plus Rapa is critical for inhibition of their growth.
Materials and Methods
Cell culture. Human breast carcinoma cell line MCF‐7 cells and human promyelocytic leukemia NB4 cells were cultured in RPMI‐1640 supplemented with 10% fetal bovine serum at 37°C in a humidified atmosphere of 5% carbon dioxide in air.
Materials. Rapamycin was purchased from Sigma Chemical (St. Louis, MO, USA). CN‐A was purified from a stock ethyl acetate extract obtained from the culture filtrate of Cladosporium fungus sp. 501‐7 W by flash chromatography on silica gel with more than 99% purity.( 1 , 2 )
Assay of cell growth. The cells were seeded at 1–3 × 104/mL in a 24‐well multidish. After culture with or without the test compounds for the indicated times, viable cells were examined using either the trypan blue dye exclusion test or a modified MTT (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide) assay.( 18 )
Gene expression analysis by reverse transcription polymerase chain reaction. Total RNA was extracted using Isogen (Nippon Gene, Toyama, Japan) according to the manufacturer's instructions. Total RNA (1 µg) from tumor cells was converted to first‐strand cDNA primed with random nonamer in a reaction volume of 20 µL using TaKaRa RNA PCR kit (TaKaRa Bio, Otsu, Japan), and 4 µL of this reaction was used as a template in the polymerase chain reaction. The oligonucleotides used in polymerase chain reaction (PCR) amplification were as follows: sense strand, 5′‐AGCACTTGGCAGGTCATGAA‐3′ and antisense strand, 5′‐CAACTATTCTAGCAGCCAGC‐3′ for cyclin G2; sense strand, 5′‐GGTCGGAGTCAACGGATTTG‐3′ and antisense strand, 5′‐ATGAGCCCCAGCCTTCTCCAT‐3′ for glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH); sense strand, 5′‐TGACCTCCATAGAAGACACC‐3′ and antisense strand, 5′‐CAACTATTCTAGCAGCCAGC‐3′ for transfected cyclin G2 (Exo. CG2). The sequence of the sense strand primer for Exo. CG2 is located in the plasmid pTRE2hyg‐CG2. PCR consisted of 22 cycles for cyclin G2 and 17 cycles for GAPDH with denaturing at 94°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s as described previously.( 18 ) Under these conditions, the amounts of PCR products increased linearly up to 0.4 µg total RNA.
Cell cycle analysis. Cell cycle analysis was carried out by staining DNA with propidium iodide in preparation for flow cytometry as described previously.( 18 )
Establishment of inducible CG2 stable clones in MCF‐7 cells (MCF‐7/CG2). Tetracycline/Dox inducible expression of CG2 was established in MCF‐7 cells using the Tet‐On Gene Expression System according to the manufacturer's instructions (Clontech, Palo Alta, CA, USA). Stable Tet‐On MCF‐7 cells were first established by the introduction of pTet‐On vector DNA (Clontech, Palo Alta, CA, USA) into the cells followed by selection for growth in the presence of the antibiotic G418. G418‐resistant clones were tested for the expression of Dox‐responsive luciferase by transient transfection assay with pTET2hyg‐Luc (Clontech). CG2 cDNA was prepared from MCF‐7 cells treated with CN‐A plus Rapa by reverse transcription (RT)‐PCR using LA‐Taq (Takara Bio) and then subcloned into the SalI and EcoRV restriction sites of expression vector pTRE2hyg (Clontech) to form plasmid pTRE2hyg‐CG2. The sequence of the cDNA insert of the plasmid was confirmed by sequencing. MCF‐7/CG2 cells and MCF‐7/empty cells were established by the introduction of pTRE2hyg‐CG2 plasmid DNA or pTRE2hyg plasmid DNA, respectively, into the Tet‐On MCF‐7 cells followed by selection for growth in the presence of G418 and hygromycin.
Western blot analysis. Cells were packed after washing with cold phosphate‐buffered saline, and then lysed at a concentration of 1.5 × 107 cells/mL in sample buffer (63 mM Tris‐HCl [pH 6.8], 15% glycerol, 2% sodium dodecyl sulfate [SDS], 5% 2‐mercaptethanol and 0.005% bromophenol blue). The resultant lysates were resolved on 10–20% SDS‐polyacrylamide gels. The proteins were transferred electrically from gel to an Immobilon‐P membrane (Millipore, Bedford, MA, USA) and immunoblotted with anti‐CG2 antibody (rabbit monoclonal IgG) (Epitomics, Long Beach, CA, USA) (1:5000 dilution) and anti‐α‐tubulin antibody (TU‐02) (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Horseradish peroxidase (HRP)‐conjugated antirabbit or antimouse immunoglobulin G antibody (Cell Signaling Technology, CA, USA) was used as a secondary antibody (1:2000 dilution). The bands were developed by treatment with the Immune‐Star HRP chemiluminescent (Bio‐Rad Laboratories, Danvers, MA, USA) for 5 min at room temperature, and detected using a Fuji Lumino Image Analyzer LAS‐1000 system (Fuji Film, Tokyo, Japan).
Transfection with siRNA. Duplexes of siRNA corresponding to the nucleotide sequence from CG2 (siCG2) (5′‐CUAGAAGCUCAGCUGAAAGTT‐3′) and a negative control siRNA (5′‐AAUUCACAGGCGCUGAAAGTT‐3′), were transfected using TransIT‐TKO (Mirus, Madison, WI, USA) according to the manufacturer's instructions. The siCG2 and the negative control siRNA were prepared using the siRNA Design Support System (TaKaRa Bio). The sequence of the negative control siRNA was a scrambled sequence of the siCG2 and its specificity was confirmed by BLAST (National Institutes of Health; http://www.ncbi.nlm.nih.gov/blast/Blast.cgi).
Results
CG2 expression is markedly increased in cancer cells by treatment with CN‐A plus Rapa. As described in our previous report,( 18 ) CN‐A and Rapa synergistically inhibited the proliferation of breast cancer carcinoma cell line MCF‐7 cells (Fig. 1a). CG2 was initially identified by cDNA microarray analysis as one of the genes that are markedly upregulated by treatment with CN‐A plus Rapa.( 18 ) Fig. 1(b) shows the detailed time‐course of CG2 gene expression in MCF‐7 cells after treatment with CN‐A and Rapa. The treatment with CN‐A or Rapa was capable of rapidly increasing CG2 expression as early as 3 h in MCF‐7 cells. The combinatorial treatment of CN‐A plus Rapa induced greater expression of CG2 than did either agent alone. CN‐A and Rapa also synergistically inhibited the proliferation of human promyelocytic leukemia NB4 cells (Fig. 1c) and also induced greater expression of CG2 than did either agent alone (Fig. 1d). We also observed similar growth inhibition and induction of CG2 expression in two other breast cancer cell lines (T‐47D and MDA‐MB‐231 cells) and a human lung cancer cell line A549 when they were treated with CN‐A plus Rapa (data not shown).
Figure 1.
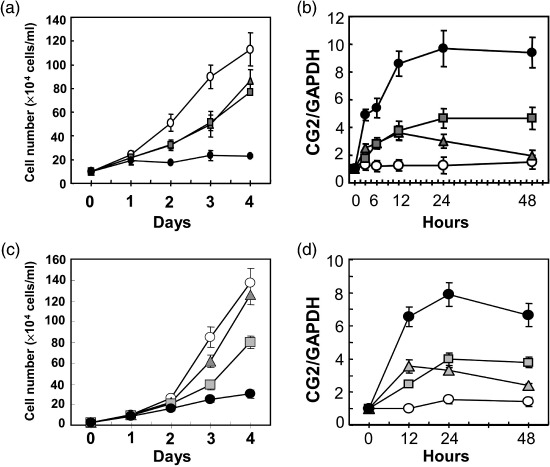
Combined treatment of cotylenin A (CN‐A) and rapamycin (Rapa) markedly induced (a,c) growth inhibition and (b,d) CG2 expression. (a,b) MCF‐7 cells (3 × 104 cells/mL) or (c,d) NB4 cells (1 × 104 cells/mL) were cultured without ( ) or with 5 µg/mL CN‐A (
) or with 5 µg/mL CN‐A ( ), 0.5 ng/mL Rapa (
), 0.5 ng/mL Rapa ( ), or 5 µg/mL CN‐A plus 0.5 ng/mL Rapa (
), or 5 µg/mL CN‐A plus 0.5 ng/mL Rapa ( ) for the indicated number of days. The values are expressed as mean ± standard deviation of three determinations.
) for the indicated number of days. The values are expressed as mean ± standard deviation of three determinations.
Induction of cell arrest correlates the induction of CG2 expression. The treatment of CN‐A plus Rapa induced growth arrest of MCF‐7 cells at G1 phase (Fig. 2a‐ii) and induced CG2 expression (Fig. 2b). Low serum concentration (0.1%) also induced growth arrest at G1 phase and also induced CG2 gene expression (Fig. 2a‐iii,b). To examine whether the induction of CG2 expression associates with cell cycle arrest at a specific phase, we induced cell cycle arrest in MCF‐7 cells with chemotherapeutic drugs (Fig. 2a‐iv–vi and Fig. 2b). 5‐Fluorouracil (5‐FU) induced growth arrest at the G1 phase and markedly induced CG2 gene expression. On the other hand, daunomycin (DNR) and etoposide induced growth arrest at G2/M phase, but they also could induce significant increases in CG2 gene expression. These results suggest that the induction of CG2 expression is associated with cell cycle arrest in any phase rather than cell cycle arrest in a specific phase.
Figure 2.
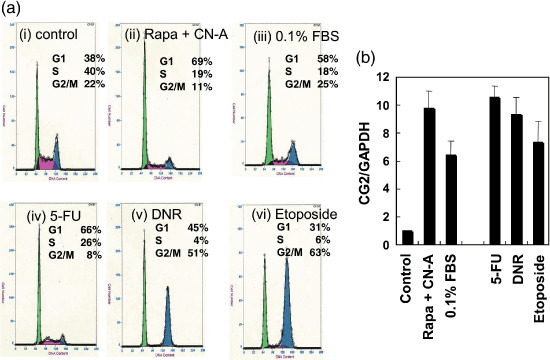
Effect of cell cycle arrest on CG2 expression. (a) MCF‐7 cells were cultured (a‐i) without or (a‐ii) with 5 µg/mL cotylenin A (CN‐A) plus 0.5 ng/mL rapamycin (Rapa) (a‐iii) low serum (0.1%) (a‐iv) 48 µM 5‐fluorouracil (5‐FU) (a‐v) 67 ng/mL daunomycin (DNR), or (a‐vi) 10 µM etoposide for 2 days. Then, the cells were subjected to cell cycle analysis. (b) Using an aliquot of the cells, expression of CG2 mRNA was examined by reverse transcription polymerase chain reaction analysis. Bars show the mean ± standard deviation of three determinations. FBS, fetal bovine serum.
Ectopic expression of CG2 inhibited proliferation of MCF‐7 cells. Stable clones that express Dox‐inducible CG2 under the control of a tetracycline response element‐driven promoter in MCF‐7 cells were established as described in Materials and Methods. The transfected cells were incubated in the presence and absence of 1 µg/mL Dox for 2 days. The results showed that MCF7/CG2 cells, but not MCF7/empty cells, were induced to express CG2 mRNA (Fig. 3a) and CG2 protein (Fig. 3b). Cell growth in MCF7/CG2 cells (Fig. 3d) was progressively inhibited in the presence of Dox, compared to that in Dox‐treated MCF7/empty cells (Fig. 3c). The inhibition began to appear on day 4 after Dox addition and became much more significant from that point in MCF7/CG2 cells. Another clone of MCF7/CG2 cells showed similar results (data not shown). These results suggest that CG2 expression induces growth arrest of MCF‐7 cells.
Figure 3.
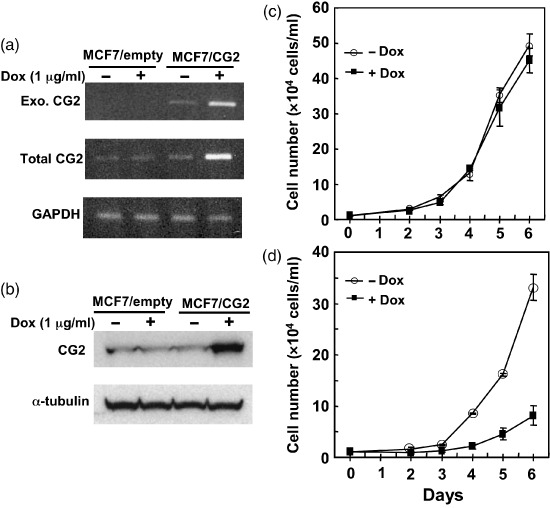
Ectopic expression of CG2 inhibited proliferation of MCF‐7 cells. Stable clones of MCF‐7/CG2 cells (clone 6) and MCF‐7/empty cells (clone 10) were cultured in the absence or presence of 1 µg/mL doxycycline (Dox) for 2 days. Cells were then collected for total RNA extraction and cell lysate. (a) Exogenously expressed CG2 (Exo. CG2) mRNA, total CG2 mRNA, and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) mRNA were measured by reverse transcription polymerase chain reaction. (b) Levels of CG2 and α‐tubulin proteins were measured by Western blotting. (c) MCF7/empty cells or (d) MCF7/CG2 cells were seeded at 1 × 104 cells/mL and cultured without () or with () 0.5 µg/mL Dox for the indicated number of days. The values are expressed as mean ± standard deviation of three determinations.
CG2 knockdown induced by CG2 siRNA reduced the potency of CN‐A plus Rapa to induce growth inhibition. To determine the role of CG2 gene expression in CN‐A plus Rapa‐induced growth inhibition, a siRNA corresponding to the nucleotide sequence from CG2 (siCG2) and a negative control siRNA were prepared and transfected transiently into MCF‐7 cells. The CG2 siRNA reduced the induction of CG2 mRNA (Fig. 4a) and CG2 protein (Fig. 4b) in MCF‐7 cells treated with CN‐A plus Rapa compared to the control siRNA. The basal level of CG2 mRNA in CG2 siRNA‐transfected MCF‐7 cells was also reduced compared to that in control siRNA‐transfected MCF‐7 cells (Fig. 4a). Although the growth of CG2 siRNA‐transfected MCF‐7 cells without CN‐A plus Rapa treatment was not significantly different from that of control siRNA‐transfected cells without the combination treatment as shown in Fig. 4(c), the growth of CG2 siRNA‐transfected MCF‐7 cells without treatment slightly but significantly increased compared with the control siRNA‐transfected MCF‐7 cells without treatment when inoculum sizes of cells were increased (>0.75 × 104 cells/well) (Fig. 5 and data not shown). Similar to the results in non‐transfected MCF‐7 cells as mentioned above, CN‐A plus Rapa markedly inhibited the proliferation of the control siRNA‐transfected MCF‐7 cells. In contrast, the potency of CN‐A plus Rapa to induce growth inhibition was almost impaired in CG2 siRNA‐transfected MCF‐7 cells (Fig. 4c). We also obtained similar results using one other CG2 siRNA prepared from a different nucleotide sequence from CG2 (data not shown). These results suggest that CN‐A plus Rapa induced inhibition of cancer cell growth through induction of CG2. The potency of DNR or etoposide to induce growth inhibition was also markedly impaired in CG2 siRNA‐transfected MCF‐7 cells (Fig. 5a,b), whereas the levels of 5‐FU‐induced growth inhibition were almost same in CG2 siRNA‐transfected MCF‐7 cells and control siRNA‐transfected MCF‐7 cells (Fig. 5c).
Figure 4.
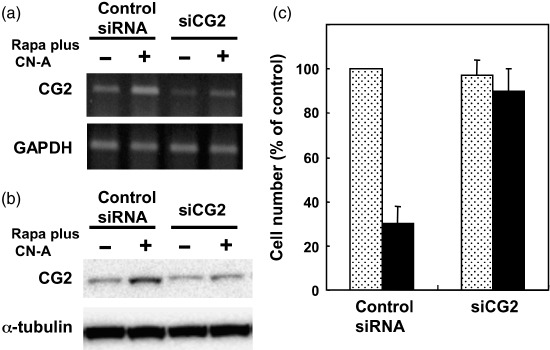
CG2 knockdown induced by CG2 small interfering RNA (siRNA) reduced the potency of cotylenin A (CN‐A) plus rapamycin (Rapa) to induce growth inhibition. (a) CG2‐specific siRNA (siCG2) (20 nM) but not control siRNA (20 nM) reduced the expression of CG2 mRNA in both untreated MCF‐7 cells and CN‐A (2.5 µg/mL) plus Rapa (0.5 ng/mL)‐treated MCF‐7 cells. The expression of (a) CG2 mRNA and levels of (b) CG2 protein and α‐tubulin protein were measured after 2 days. Similar results were obtained in two additional experiments. (c) siCG2‐ or control siRNA‐transfected MCF‐7 cells (0.5 × 104 cells/well in a 24‐well dish) were cultured without ( ) or with () CN‐A (2.5 µg/mL) plus Rapa (0.5 ng/mL) for 4 days. Then, the cell number was determined by MTT assay. The values are expressed as mean ± standard deviation of three determinations. GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
) or with () CN‐A (2.5 µg/mL) plus Rapa (0.5 ng/mL) for 4 days. Then, the cell number was determined by MTT assay. The values are expressed as mean ± standard deviation of three determinations. GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
Figure 5.
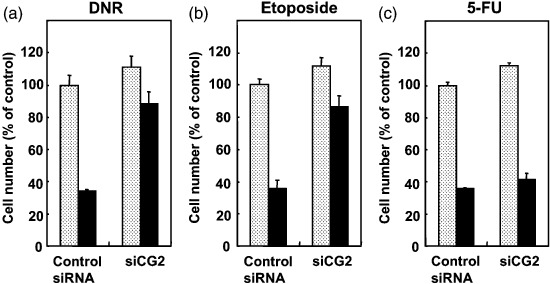
Effect of CG2 small interfering RNA (siRNA) on the chemotherapeutic drug‐induced growth inhibition of MCF‐7 cells. siCG2‐ or control siRNA‐transfected MCF‐7 cells (0.75 × 104 cells/well in 24‐well dish) were cultured without () or with () (a) 50 ng/mL daunomycin (DNR), (b) 7.5 µM etoposide or (c) 40 µM 5‐fluorouracil (5‐FU) for 4 days. Then, the cell number was determined by MTT assay. The values are expressed as mean ± standard deviation of three determinations.
Discussion
In this study, we found that the induction of CG2 gene expression was very important for the induction of CN‐A plus Rapa‐induced growth inhibition of mammary cancer cells and leukemia cells. The effect of CN‐A, Rapa, or CN‐A plus Rapa on MCF‐7 and NB4 cell proliferation was almost the same (Fig. 1). CN‐A moderately suppressed the growth of both cell lines. Rapa also suppressed the growth of the both cell lines until 3 days after treatment, but thereafter its growth inhibiting activities appeared to decrease. The kinetic patterns of CG2 expression between CN‐A‐, Rapa‐, and CN‐A plus Rapa‐treated MCF‐7 cells and CN‐A‐, Rapa‐, and CN‐A plus Rapa‐treated NB4 cells were also quite similar (Fig. 1). In both cell lines the induction levels of CG2 expression were well correlated with the levels of growth inhibition. These results suggest that both induction of growth inhibition and induction of CG2 expression are observed not only in CN‐A plus Rapa‐treated MCF‐7 cells but also in other cancer cells.
Treatment with CN‐A plus Rapa also induced phenotypic changes such as the induction of E‐cadherin and senescence‐associated β‐galactosidase activity in MCF‐7 cells.( 18 ) Because the induction of CG2 could also occur when growth arrest of the cells was induced by various agents or low serum conditions (Fig. 2), the induction of CG2 may be correlated with the induction of growth arrest rather than the induction of phenotypic changes. Although CN‐A, retinoic acid and 1α,25‐dihydroxyvitamin D3 can induce differentiation of human promyelocytic leukemia HL‐60 cells,( 19 ) CN‐A and retinoic acid, but not 1α,25‐dihydroxyvitamin D3, could induce significant CG2 expression (data not shown). CN‐A and retinoic acid showed strong induction of differentiation with marked growth suppression, whereas 1α,25‐dihydroxyvitamin D3 induced strong induction of differentiation without marked growth suppression.( 19 ) These results also support our suggestion that CN‐A plus Rapa‐induced CG2 induction is correlated with the induction of growth arrest rather than the induction of phenotypic changes.
Because ectopic expression of CG2 is expected to suppress the proliferation of MCF‐7 cells, we anticipated that the establishment of a stable clone of MCF‐7 cells expressing exogenous CG2 would be difficult. We then first established stable clones of Tet‐On MCF‐7 cells by introduction of pTet‐On vector DNA into the MCF‐7 cells. Next, we generated an inducible human CG2‐expressing cell line MCF‐7/CG2 using human breast cancer Tet‐On MCF‐7 cells by the introduction of the pTRE2hyg‐CG2 plasmid. This cell line presented the advantage of not having been directly selected for CG2 expression during its establishment. As such, it had not been preselected with particular features such as reduced proliferation or differentiation/apoptotic potential prior to the analysis of CG2 expression. In MCF7/CG2 cells, CG2 was tightly regulated and induced only upon Dox addition. By using the cell line, we examined the effect of CG2 in breast cancer cells. Our results clearly showed that the proliferation of MCF7/CG2 cells was arrested upon CG2 induction. Other reports show that the expression levels of CG2 are downregulated in human oral cancers,( 16 ) and in papillary carcinomas of the thyroid.( 20 ) Therefore, our results suggest that CG2 may play antagonistic roles with regard to breast cancer development.
Although evidence suggests that CG2 modulates cell cycle arrest responses, little is known about the effects of elevated CG2 on cellular physiology. Recently, Don et al.( 17 ) reported that ectopic and endogenous CG2 localized at centrosomes and co‐purified along with protein phosphatase 2 A (PP2A) in centrosome‐enriched subcellular fractions. Because alteration of centrosomal components that results in inhibition of cytokinesis and centrosome duplication has been shown to induce cell cycle arrest,( 21 , 22 , 23 ) high CG2 expression may modulate the cell cycle and cellular division processes through modulation of PP2A and centrosomal associated activities.
We showed finally that CG2 knockdown induced by CG2 siRNA reduced the potency of CN‐A plus Rapa to induce growth inhibition of MCF‐7 cells. These results suggest that the induction of CG2 in cancer cells treated with CN‐A plus Rapa is critical for inhibition of their growth. Recently Le et al.( 24 ) reported that CG2 expression was increased in breast cancer cells that overexpress human epidermal growth factor receptor 2 (HER2) by treatment with anti‐HER2 antibody. Our and these results suggest that treatment with agents targeted at the induction of CG2 could be a therapeutically effective method for human cancer including breast cancer. The anti‐HER2 antibody cannot exert its biological and therapeutic effects in cancer cells that do not express HER2 or in cancer cells displaying forms of the receptor that lacks the ectodomains of HER2. It is possible that treatment with CN‐A plus Rapa may induce CG2 expression and suppresses the proliferation of these anti‐HER2 antibody‐unresponsive cancer cells. Furthermore, we found that CN‐A plus Rapa also markedly induced CG2 expression and significantly inhibited the growth of Dox‐resistant MCF‐7 cells (data not shown). Therefore, treatment with CN‐A plus Rapa also might be effective against antibody therapy‐ and chemotherapy‐resistant breast cancer cells through induction of the CG2 gene. Additional studies of the mechanisms of induction of CG2 by CN‐A plus Rapa are required.
Acknowledgments
This study was supported partly by a grant from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1. Sassa T, Tojyo T, Munakata K. Isolation of new plant growth substance with cytokinin‐like activity. Nature 1970; 227: 379. [DOI] [PubMed] [Google Scholar]
- 2. Sassa T, Ooi T, Nukina M, Ikeda M, Kato N. Structural confirmation of cotylenin A, novel fusicoccane‐diterpene glycoside with potent plant growth‐regulating activity from Cladosporium fungus sp. 501‐7W. Biosci Biotechnol Biochem 1998; 62: 1815–8. [DOI] [PubMed] [Google Scholar]
- 3. Asahi K, Honma Y, Hazeki K et al . Cotylenin A, a plant‐growth regulator, induces differentiation in murine and human myeloid leukemia cells. Biochem Biophys Res Commun 1997; 238: 758–63. [DOI] [PubMed] [Google Scholar]
- 4. Yamamoto‐Yamaguchi Y, Yamada K, Ishii Y, Asahi K, Tomoyasu S, Honma Y. Induction of monocytic differentiation of myeloid leukaemia cells by cotylenin A, a plant growth regulator. Br J Haematol 2001; 112: 697–705. [DOI] [PubMed] [Google Scholar]
- 5. Honma Y. Cotylenin A – a plant growth regulator as a differentiation‐inducing agent against myeloid leukemia. Leuk Lymphoma 2002; 43: 1169–78. [DOI] [PubMed] [Google Scholar]
- 6. Yamada K, Honma Y, Asahi K, Sassa T, Hino K, Tomoyasu S. Differentiation of human acute myeloid leukaemia cells in primary culture in response to cotylenin A, a plant growth regulator. Br J Haematol 2001; 114: 814–21. [DOI] [PubMed] [Google Scholar]
- 7. Honma Y, Ishii Y, Sassa T, Asahi K. Treatment of human promyelocytic leukemia in the SCID mouse model with cotylenin A, an inducer of myelomonocytic differentiation of leukemia cells. Leukemia Res 2003; 27: 1019–25. [DOI] [PubMed] [Google Scholar]
- 8. Huang S, Houghton PJ. Inhibitors of mammalian target of rapamycin as novel antitumor agents. Curr Opin Invest Drugs 2002; 3: 295–304. [PubMed] [Google Scholar]
- 9. Huang S, Houghton P. Targeting mTOR signaling for cancer therapy. Curr Opin Pharmacol 2003; 3: 371–7. [DOI] [PubMed] [Google Scholar]
- 10. Noh W‐C, Mondesire WH, Peng J et al . Determinants of rapamycin sensitivity in breast cancer cells. Clin Cancer Res 2004; 10: 1013–23. [DOI] [PubMed] [Google Scholar]
- 11. Yamamoto‐Yamaguchi Y, Okabe‐Kado J, Kasukabe T, Honma Y. Induction of differentiation of human myeloid leukemia cells by immunosuppressant macrolides (rapamycin and FK506) and calcium/calmodulin‐dependent kinase inhibitors. Exp Hematol 2001; 29: 582–8. [DOI] [PubMed] [Google Scholar]
- 12. Horne MC, Goolsby GL, Donaldson KL, Tran D, Neubauer M, Wahl AF. Cyclin G1 and cyclin G2 comprise a new family of cyclins with contrasting tissue‐specific and cell cycle‐regulated expressions. J Biol Chem 1996; 271: 6050–61. [DOI] [PubMed] [Google Scholar]
- 13. Horne MC, Donaldson KL, Goolsby GL et al . Cyclin G2 is up‐regulated during growth inhibition and B cell antigen receptor‐mediated cell cycle arrest. J Biol Chem 1997; 272: 12650–61. [DOI] [PubMed] [Google Scholar]
- 14. Bates S, Rowan S, Vousden KH. Characterization of human cyclin G1 and G2: DNA damage inducible genes. Oncogene 1996; 13: 1103–9. [PubMed] [Google Scholar]
- 15. Murray JI, Whitfield ML, Trinklein ND, Myers RM, Brown PO, Botstein D. Diverse and specific gene expression responses to stresses in cultured human cells. Mol Biol Cell 2004; 15: 2361–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim Y, Shintani S, Kohno Y, Zhang R, Wong DT. Cyclin G2 dysregulation in human oral cancer. Cancer Res 2004; 64: 8980–6. [DOI] [PubMed] [Google Scholar]
- 17. Don AS, Dallapiazza RF. Bennin DA, Brake T, Cowan CE, Horne MC. Cyclin G2 is a centrosome‐associated nucleoplasmic shuttling protein that influences microtubule stability and induces a p53‐dependent cell cycle arrest. Exp Cell Res 2006; 312: 4181–8204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kasukabe T, Okabe‐Kado J, Kato N, Sassa T, Honma Y. Effects of combined treatment with rapamycin and cotylenin A, a novel differentiation‐inducing agents, on human breast carcinoma MCF‐7 cells and xenografts. Breast Cancer Res 2005; 7: R1097–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ishii Y, Kasukabe T, Honma Y. Immediate up‐regulation of the calcium‐binding protein S100P and its involvement in the cytokinin‐induced differentiation of human myeloid leukemia cells. Biochim Biophys Acta 2005; 1745: 156–65. [DOI] [PubMed] [Google Scholar]
- 20. Ito Y, Yoshida H, Uruno T et al . Decreased expression of cyclin G2 is significantly linked to the malignant transformation of papillary carcinoma of thyroid. Anticancer Res 2003; 23: 2335–8. [PubMed] [Google Scholar]
- 21. Khodjakov A, Rieder CL. Centrosomes enhance the fidelity of cytokinesis in vertebrate and are required for cell cycle progression. J Cell Biol 2001; 153: 237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Keryer G, Witczak O, Delouvee A et al . Dissociating the centrosomal matrix protein AKAP450 from centrioles impairs centriol duplication and cell cycle progression. Mol Biol Cell 2003; 14: 2436–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gromley A, Jurczyk A, Sillibourne J et al . A novel human protein of the maternal centriole is required for the final stages of cytokinesis and entry into S phase. J Cell Biol 2003; 161: 535–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Le X‐F, Arachchige‐Don AS, Mao W, Horne MC, Bast Jr. RC. Roles of human epidermal growth factor receptor 2, c‐jun NH2‐terminal kinases, phosphoinositide 3‐kinase, and p70, S6 kinase pathways in regulation of cyclin G2 expression in human breast cancer cells. Mol Cancer Ther 2007; 6: 2843–57. [DOI] [PubMed] [Google Scholar]