

Abstract
Although hepatitis B virus (HBV) has been documented to cause hepatocellular carcinoma (HCC), the exact role of HBV in the development of HCC remains enigmatic. Several hypotheses have been proposed to explain the potential mechanism, including insertional mutagenesis of HBV genomes and transcriptional activators of HBV gene products such as hepatitis B x protein (HBx) and truncated middle S mutants. In the past few years, we have identified two types of large HBV surface antigens (LHBs) with deletions at the pre‐S1 (ΔS1‐LHBs) and pre‐S2 (ΔS2‐LHBs) regions in ground glass hepatocytes. The pre‐S mutant LHBs are retained in the endoplasmic reticulum (ER) and escape from immune attack. The pre‐S mutants, particularly ΔS2‐LHBs, are increasingly prevalent in patients with hepatitis B e antigen (HBeAg)‐positive chronic HBV infection, ranging from 6% before the 3rd decade to 35% in the 6th decade. In HCC patients, the two pre‐S mutants were detected in 60% of HCC patients, in the serum and in HCC tissues. Pre‐S mutant LHBs can initiate ER stress to induce oxidative DNA damage and genomic instability. Furthermore, pre‐S mutant LHBs can upregulate cyclooxygenase‐2 and cyclin A to induce cell cycle progression and proliferation of hepatocytes. In transgenic mice, the pre‐S mutants can induce dysplasia of hepatocytes and development of HCC. In a nested control study, the presence of pre‐S mutants carried a high risk of developing HCC in HBV carriers. In summary, the findings we describe in this review suggest a potential role for HBV pre‐S mutants in HBV‐related hepatocarcinogenesis, providing a model of viral carcinogenesis associated with ER stress. (Cancer Sci 2006; 97: 683–688)
Abbreviations:
- COX‐2
cyclooxygenase‐2
- ER
endoplasmic reticulum
- GGH
ground glass hepatocytes
- HBeAg
hepatitis B e antigen
- HBs
hepatitis B surface protein
- HBsAg
hepatitis B surface antigen
- HBV
hepatitis B virus
- HBx
hepatitis B x protein
- HCC
hepatocellular carcinoma
- JAB1
Jun activation domain‐binding protein 1
- LHBs
large surface protein
- NFκB
nuclear factor κB
- MAPK
mitogen‐activated protein kinase
- RB
retinoblastoma protein
- ΔS1‐LHBs
LHBs with a deletion at the pre‐S1 region
- ΔS2‐LHBs
LHB with a deletion at the pre‐S2 region
- UPR
unfolded protein response.
Hepatitis B virus is recognized as a major etiological factor in the development of HCC.( 1 , 2 ) Epidemiological studies have demonstrated an approximately 100‐fold increase in the relative risk of HCC among HBV carriers compared to non‐carriers.( 3 ) Although the relationship between chronic HBV infection and HCC has been well established, the exact role of HBV in the pathogenesis of HBV‐related hepatocarcinogenesis remains to be elucidated.
Hepatitis B virus is a partially double‐stranded DNA virus containing a genome of 3.2 kb in size, which contains four open reading frames encoding viral polymerase, the core and e antigen, the HBx protein and the pre‐S/S gene encoding the three surface antigens (i.e. the large [pre‐S1 + pre‐S2 + S], middle [pre‐S2 + S] and small [S only] surface proteins). In chronic HBV infection, HBV DNA can be integrated into the host genome. Almost all HCC harbor single or multiple copies of integrated HBV DNA.( 4 , 5 ) The integrated HBV DNA in tumors is usually rearranged and partially deleted. HBV DNA integration has been shown to be a random event and no specific cis‐effect has been observed on flanking cellular genes.( 6 , 7 ) Therefore, HBV DNA integration per se is not considered to be a general mechanism of HBV‐related hepatocarcinogenesis. Based on the observations obtained from several isolates of human HCC tissues, the integrated HBV DNA usually has truncated open reading frames coding for viral polymerase and the core antigen, and can only encode two gene products: the HBx and HBs proteins. Therefore, HBx and HBs proteins represent the two potential candidate proteins involved in HBV‐related hepatocarcinogenesis. Although still controversial, the HBx protein has been studied extensively. It has been shown to exhibit transactivating functions, and to activate JAK/STAT and the Ras–Raf–MAPK signal pathway.( 8 , 9 ) In the case of HBs protein, the LHBs has been demonstrated to exert a tumor promoter‐like function in the development of HCC.( 10 , 11 ) The C‐terminally truncated middle surface protein MHBst has been recognized as a transactivator and initiates c‐Raf‐1/MAPK‐2 signaling.( 12 , 13 )
In past years, we identified two types of LHBs with deletions at the pre‐S1 (ΔS1‐LHBs) and pre‐S2 (ΔS2‐LHBs) regions in GGH obtained from patients with advanced diseases of chronic HBV infection. These naturally occurring pre‐S mutant LHBs are increasingly prevalent in serum and liver tissues of patients with chronic HBV hepatitis and in HCC tissues. The pre‐S mutant LHBs are retained in the ER and induce ER stress signals. Furthermore, the pre‐S mutant LHBs can upregulate COX‐2 and cyclin A to induce cell cycle progression. In transgenic mice, ΔS2‐LHBs has been shown to induce dysplastic changes in hepatocytes and development of HCC. In this review, we describe the biological characteristics of HBV pre‐S mutants and their possible roles in the development of HCC based on the data mainly from our past studies.
Identification of pre‐S (LHBs) mutants with deletions at the pre‐S1 and pre‐S2 regions in GGH
Ground glass hepatocytes have been recognized as the histological hallmark of chronic HBV infection since 1973 when Hadziyannis et al. demonstrated the presence of HBV surface antigens in the ‘glassy’ cytoplasm of hepatocytes (Fig. 1a).( 14 , 15 ) Ultrastructurally, GGH are characterized by an abundance of ER in which HBsAg is accumulated.( 16 ) Several types of GGH have been recognized and correlated to the different replicative stages of chronic HBV infection.( 17 , 18 , 19 , 20 ) Of particular interest is the emergence of a so‐called ‘marginal type’ GGH (type II GGH) at advanced stages of chronic HBV infection.( 20 ) Marginal type GGH harbor HBsAg at the cell margin or periphery of hepatocytes, distinct from the ‘inclusion‐like’ accumulation of HBsAg in classic GGH (type I GGH; Fig. 1b). Furthermore, marginal type GGH consistently cluster in groups or nodules in liver tissues, suggesting a clonal proliferation or growth advantage of type II GGH.
Figure 1.
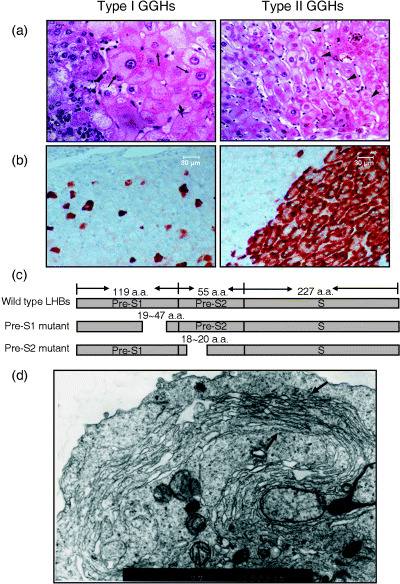
Histopathological characteristics of ground glass hepatocytes (GGH). (a) Hematoxylin–eosin staining of type I and type II GGH. Black arrows indicate hepatocytes containing ground glass substances in both type I (left) and type II GGH (right). (b) Immunostaining of surface proteins on livers of patients with chronic hepatitis B virus infection. Type I GGH (left) usually contain hepatitis B surface antigen (HBsAg) in a blot‐like or inclusion pattern. In contrast, HBsAg is usually expressed in the marginal region of type II GGH (right). (c) Mutation map of the identified pre‐S1 and pre‐S2 deletion mutants. Dotted lines indicate the deletion regions. (d) Electron microscopy of HuH‐7 cells expressing large surface protein (LHBs) with a deletion at the pre‐S2 region. Endoplasmic reticulum proliferation in the marginal region of the cell is indicated. (Original amplification: ×10 000.)
The differential expression patterns of HBsAg in GGH imply the possibility of the existence of different types of HBV surface antigens. By laser‐capture microdissection, we identified two major types of LHBs or pre‐S mutant LHBs with deletions at either the pre‐S1 (ΔS1‐LHBs) or pre‐S2 (ΔS2‐LHBs) region in type I and type II GGH, respectively (Fig. 1c).( 21 , 22 , 23 ) This interesting finding drives us to explore in depth the potential biological significance of GGH, particularly the role of type II GGH or ΔS2‐LHBs in the pathogenesis of HBV‐related tumorigenesis.
Pre‐S mutants represent immune selection variants and are increasingly prevalent during the course of chronic HBV infection
The presence of different types of viral mutants in different replicative stages of chronic HBV infection suggests the potential evolution of viral variants under immune pressure during HBV infection. Naturally occurring pre‐S mutants are detected frequently in serum obtained from patients with chronic HBV infection.( 24 ) The resulting mutants reveal shorter forms of LHBs proteins with internal deletions. By examining serum samples obtained from longitudinal studies of patients with chronic hepatitis B over 10 years, we observed the sequential appearance of a wide spectrum of LHBs mutants, with the first appearance of variants with point mutations at the S region, followed by the presence of ΔS1‐LHBs, and later the emergence of ΔS2‐LHBs after e seroconversion (after serum HBeAg status converted from positive to negative) (W. C. Chen and I. J. Su, unpublished data). The deletion site (nucleotides 4–57) of ΔS2‐LHBs has been recognized to correlate with the epitope of the CD8 T‐cell response and B‐cell neutralization.( 25 ) The pre‐S2 mutant ΔS2‐LHBs may therefore represent an immune escape mutant that usually appears at the later stage of chronic HBV infection. In liver biopsy tissues of chronic HBV infection, there is usually an absence of lymphocyte infiltration in regions of type II GGH, supporting the concept of an immune escape for pre‐S2 mutant ΔS2‐LHBs.
We have recently observed the prevalence of pre‐S mutants in patients with HBeAg‐positive chronic HBV infection who received antiviral therapy. The prevalence of pre‐S mutants in serum increases from around 6% at the early stage (2nd decade of age) of chronic HBV infection with high HBV viral load, to the peak level of 35% during the 5–6th decade with low titers of HBV DNA. In patients with HCC, the prevalence rate of pre‐S mutants increases up to 60% in both serum samples and liver tissues. Furthermore, the ΔS2‐LHBs appear to predominate over ΔS1‐LHBs in serum obtained from advanced diseases and in HCC tissues.
Pre‐S mutant proteins are accumulated in the ER and induce ER stress signals
Using double‐labeled immunofluorescence staining, we observed consistent colocalization of pre‐S mutant proteins and the ER protein calregulin, suggesting that these pre‐S mutant proteins, both the ΔS1‐LHBs and ΔS2‐LHBs, accumulate in the ER. Ultrastructurally, ΔS2‐LHBs could induce a significant proliferation of the ER to an extent that is similar to the characteristics of GGH in the liver (Fig. 1d). The accumulation of pre‐S mutants in ER may subsequently induce ER stress signals, leading to the induction of ER chaperones (GRP78 and GRP94).( 23 , 26 ) ER stress, also called the UPR in mammalian cells, is a cellular defense mechanism that responds to unfolded viral proteins or perturbed ER functions. Expression of viral gene products usually induces the UPR, which is sensed by two ER transmembrane kinases (IRE1 and PERK), and one ER transmembrane transcription factor (ATF‐6).( 27 ) These three UPR sensors are associated with ER chaperone GRP78/BiP when resting, and are dissociated from GRP78 upon ER stress. In our laboratory, we have demonstrated that the GRP78 gene is induced significantly in both types of GGH and in HCC, compared with non‐HBs‐expressing hepatocytes.( 23 , 28 ) It has been reported that the induction of GRP78 may prevent cells from death by forming a complex with caspase‐7 and caspase‐12, and ER stress‐regulated translation may increase tolerance to extreme hypoxia and promote tumor growth.( 29 , 30 ) The activation of ER‐stress downstream molecules such as ATF‐6, GRP78 and XBP‐1 is believed to be involved in hepatocarcinogenesis.( 31 ) These data suggest that ER stress may play a contributing role in cell transformation, especially in HCC.
Pre‐S mutant‐induced ER stress may induce genomic instability through oxidative stress, increased DNA damage and mutation frequency
The pre‐S mutants may induce oxidative DNA damage through ER stress signaling pathways. HuH‐7 cells carrying the pre‐S mutant LHBs exhibited enhanced levels of reactive oxygen species and oxidative DNA damage.( 32 ) Furthermore, pre‐S mutant LHBs can induce mutations on the X‐linked hprt gene. Oxidative DNA damage could also be observed in livers of transgenic mice carrying pre‐S mutant LHBs, as well as in GGH.( 32 ) Therefore, through ER stress signaling pathways, the pre‐S mutant LHBs can induce oxidative stress and lead to oxidative DNA damage of HBV‐infected hepatocytes. The oxidative DNA damage caused by pre‐S mutant LHBs may result in genomic instability and mutation of liver cells, and ultimately lead to HCC.
Recently, preliminary studies from our group revealed that ΔS2‐LHBs, but not wild‐type or ΔS1‐LHBs, could induce hyperphosphorylation of tumor suppressor RB. The ΔS2‐LHBs could interact directly with JAB1, dissociating JAB1 from the JAB1–IRE1 complex in the ER lumen and causing JAB1 to translocate to cell nuclei. JAB1 is an important factor for modulating the level of cyclin‐dependent kinase inhibitor p27Kip1, and targets nuclear p27Kip1 to the cytosolic 26S proteasome for degradation. The degradation of p27Kip1 activates the phosphorylation of RB. Therefore, the combined events of oxidative DNA damage and RB hyperphosphorylation may represent two independent events for hepatocellular carcinogenesis associated with the ER viral protein ΔS2‐LHBs.
Pre‐S mutants may upregulate COX‐2 through NFκB and p38 MAPK
Overexpression of COX‐2 has been detected in many types of cancer and is linked to disease progression and survival of candidate cells. One of the many roles of pre‐S mutant LHBs is to regulate the expression of COX‐2.( 33 ) Constitutive or inducible expression of pre‐S mutant LHBs induces the expression of COX‐2 and prostaglandin E2 in immortalized mouse liver ML‐1 cells and in transformed human HuH‐7 cells. Transgenic mice expressing pre‐S mutant LHBs express higher levels of COX‐2 protein in liver and kidney tissues. Similarly, increased expression of COX‐2 mRNA was observed in human HCC tissues expressing pre‐S mutant LHBs. COX‐2 induction is apparently important for the anchorage‐independent growth conferred by the expression of pre‐S mutant LHBs. Addition of etodolac, a specific COX‐2 inhibitor, can abolish the growth of ML‐1 cells expressing pre‐S mutant LHBs in soft agar.
The induction of COX‐2 is mediated mainly through transcriptional activation as actinomycin D (an inhibitor of RNA transcription) can attenuate the expression of COX‐2 significantly. The transcription factor NF‐κB is essential for transactivation of the COX‐2 promoter.( 34 ) Nuclear translocation of NF‐κB, especially the p65 subunit, is observed when cells are treated with tunicamycin and brefeldin A, two ER stress inducers. NF‐κB inhibitor can also block the induction of COX‐2 by either an artificial ER stress inducer such as tunicamycin or the expression of pre‐S mutant LHBs. Similar to our findings, NF‐κB has been proposed to play a major role in the progression of inflammation‐associated cancer.( 35 , 36 ) The activation of NF‐κB is not only involved in the traditional degradation of IκB but is also regulated by p38 MAPK. Inhibition of p38 MAPK does not affect the nuclear translocation of NF‐κB but does inhibit NF‐κB DNA binding activity( 33 ) and attenuate the induction of COX‐2.
Taken together, the above‐described role of NF‐κB and COX‐2 in the signal transduction of pre‐S mutant LHBs suggests that they may act as potential preventive or therapeutic targets for HBV‐related HCC. COX‐2 inhibitor has been verified for the chemoprevention of many human cancers.( 37 , 38 ) The chemoprevention of liver cancer with COX‐2 inhibitor may deserve future clinical trials.
Pre‐S2 mutant ΔS2‐LHBs can selectively upregulate cyclin A and induce cell cycle progression
Cyclin A is involved in both DNA synthesis and centrosome duplication during the cell cycle.( 39 ) It has been reported that both cyclin A and cyclin E are enhanced in HCC tissues and may be associated with tumor invasiveness and metastasis.( 40 , 41 ) Using cDNA microarray analysis, we observed that cyclin A, along with other cell cycle‐regulated genes, was induced by ΔS2‐LHBs.( 28 ) The induction of cyclin A was shown to be initiated via the specific transactivator function of ΔS2‐LHBs, independent of ER stress signals. The expression of ΔS2‐LHBs in hepatocytes led to cell cycle progression under strong ER stress conditions and enhanced 5‐bromo‐2‐deoxyuridine (BrdU)‐incorporation with a multinucleation phenotype. Histopathological examinations revealed that cyclin A expression was enhanced in GGH, HCC tissues and transgenic mouse livers.( 28 ) One interesting finding is the tremendous expression of cyclin A in the cytoplasm of hepatocytes induced by ΔS2‐LHBs. Although cyclin A functions predominantly in the nucleus, cytoplasmic expression of cyclin A may contribute to centrosome duplication.( 39 , 42 ) It is therefore interesting to clarify whether alteration of the subcellular localization of cyclin A could affect centrosome duplication that may subsequently contribute to cell aneuploidy.( 43 ) Recently, our study suggested that cytoplasmic localization of cyclin A is associated with its N‐terminal truncation by a calcium‐dependent protease, which is activated by ER stress (H. C. Wang and I. J. Su, unpublished data). These data suggest that cytoplasmic cyclin A may be initiated by ER stress and may contribute to aberrant centrosome duplication. The increased multinucleation and DNA aneuploidy observed in ΔS2‐LHBs transgenic mouse livers supports the potential role of ER stress and impaired cyclin A during the development of HCC.
Pre‐S mutant LHBs can induce dysplastic changes and tumor formation in transgenic mice
Hepatitis B virus transgenic mice have been produced both with complete HBV genomes that can support viral replication and with selected viral genes, including three surface proteins (large, middle and small HBs), HBx, core and precore proteins.( 44 ) Transgenic mouse studies have revealed a contributing role for HBx in hepatocarcinogenesis but the importance of HBx remains debatable.( 45 , 46 ) The overexpression of LHBs protein in transgenic mice has been shown to be cytopathic, and could lead to liver injury, regenerative hyperplasia, chronic inflammation, oxidative DNA damage, hepatocyte aneuploidy and eventually progression to HCC.( 47 , 48 ) In our laboratory, we demonstrated that ΔS2‐LHBs can induce nodular proliferation and dysplasia of hepatocytes in transgenic mice,( 28 ) and tumor development was demonstrated recently (Fig. 2).
Figure 2.
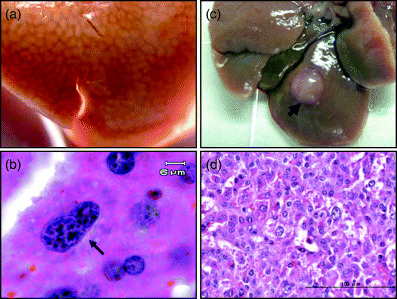
Large surface protein with a deletion at the pre‐S2 region (ΔS2‐LHBs) induces nodular proliferation, nuclear dysplastic changes and hepatocellular carcinoma on transgenic mice. (a) A transgenic mouse liver revealed nodular proliferation. (b) Nuclear dysplastic changes on transgenic liver. Black arrow indicates one affected hepatocyte with enlarged nucleus, hyperchromatism and nuclear pleomorphism. (c) Tumor formation on ΔS2‐LHBs transgenic mice. (d) Pathological finding of hepatocellular carcinoma on ΔS2‐LHBs transgenic mice.
Presence of pre‐S mutants carries a high risk of developing HCC and represents a potential prophylactic target to prevent HCC development
To test whether patients with HBV pre‐S mutants carry a higher risk of HCC development, a case‐control study nested in a cohort of 4155 HBsAg‐seropositive residents was conducted to assess the risk of developing HCC in HBV carriers with or without pre‐S mutants at enrolment. This nested case‐control analysis involved 68 men with newly diagnosed HCC and 132 matched controls. Among these cases, 14 men with HCC (20.6%) and 11 controls (8.3%) had detectable pre‐S mutants in serum samples at enrolment. After adjustment for age and HBeAg serostatus, the patients harboring pre‐S mutants at enrolment had a higher risk of developing HCC than those without pre‐S mutants during the 10‐year follow‐up period (OR = 3.2, 95% CI = 1.3–7.9, P = 0.01) (H. I. Yang and C. J. Chen, unpublished data). These data indicate that the emergence of pre‐S mutants may carry a high risk of developing HCC in patients with chronic HBV infection.
HBV pre‐S mutants may provide a model for viral carcinogenesis through latent ER stress signals
The potential role of HBV pre‐S mutants in the pathogenesis of HCC provides us with a model for viral carcinogenesis. The viral proteins may adopt a complex mechanism to escape from the host immune response by manipulating the machinery of cytoplasmic processing through the proteosome ubiquitination system. In this process, transport from the ER to the Golgi apparatus and cell membrane is manipulated meticulously by many viruses to escape from immune attack. In chronic HBV infection, a series of mutations under evolutionary cytotoxic T lymphocyte (CTL) immune pressure may result in the emergence of pre‐S mutants that accumulate in the ER.( 21 , 23 , 26 , 49 , 50 ) Retention of viral mutant proteins in the ER may induce ER stress and lead to apoptosis or, alternatively, the cells may try to survive through the activation of cell proliferation‐related signals such as COX‐2 and cyclin A to initiate cell proliferation (Fig. 3).( 28 , 32 , 33 ) In the presence of DNA damage and genomic instability, the proliferating cells may progress to tumor formation.
Figure 3.
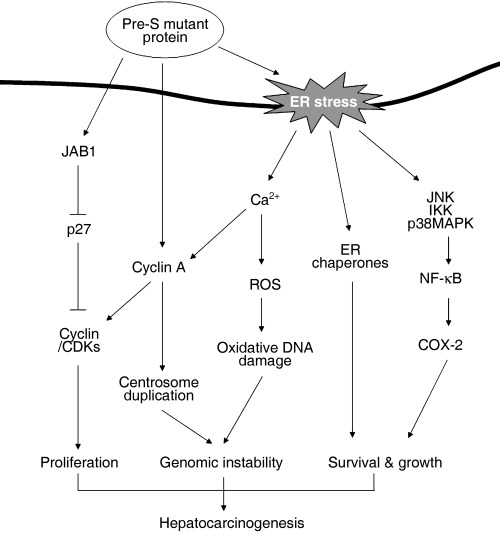
Oncogenic pathway of hepatitis B virus pre‐S mutant proteins. CDK, cyclin dependent kinase; COX‐2, cyclooxygenase‐2; ER, endoplasmic reticulum; IKK, Ikappa B kinase; JAB1, Jun activation domain‐binding protein 1; JNK, Jun N‐terminal kinase; MAPK, mitogen‐activated protein kinase; NKκB, nuclear factor κB; ROS, reactive oxygen species.
Acknowledgments
This research was supported by grants from the National Health Research Institutes and the Program to Promote the Excellence of Universities from the Department of Education, Taiwan (Dr Su).
References
- 1. Beasley RP, Hwang LY. Hepatocellular carcinoma and hepatitis B virus. Semin Liver Dis 1984; 4: 113–21. [DOI] [PubMed] [Google Scholar]
- 2. Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet 1981; 2: 1129–33. [DOI] [PubMed] [Google Scholar]
- 3. Beasley RP. Hepatitis B virus. The major etiology of hepatocellular carcinoma. Cancer 1988; 61: 1942–56. [DOI] [PubMed] [Google Scholar]
- 4. Brechot C, Hadchouel M, Scotto J et al. State of hepatitis B virus DNA in hepatocytes of patients with hepatitis B surface antigen‐positive and ‐negative liver diseases. Proc Natl Acad Sci USA 1981; 78: 3906–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Imazeki F, Omata M, Yokosuka O, Okuda K. Integration of hepatitis B virus DNA in hepatocellular carcinoma. Cancer 1986; 58: 1055–60. [DOI] [PubMed] [Google Scholar]
- 6. Hsu T, Moroy T, Etiemble J et al. Activation of c‐myc by woodchuck hepatitis virus insertion in hepatocellular carcinoma. Cell 1988; 55: 627–35. [DOI] [PubMed] [Google Scholar]
- 7. Robinson WS. Molecular events in the pathogenesis of hepadnavirus‐associated hepatocellular carcinoma. Annu Rev Med 1994; 45: 297–323. [DOI] [PubMed] [Google Scholar]
- 8. Diao J, Garces R, Richardson CD. X protein of hepatitis B virus modulates cytokine and growth factor related signal transduction pathways during the course of viral infections and hepatocarcinogenesis. Cytokine Growth Factor Rev 2001; 12: 189–205. [DOI] [PubMed] [Google Scholar]
- 9. Kim H, Lee YH, Won J, Yun Y. Through induction of juxtaposition and tyrosine kinase activity of Jak1, X‐gene product of hepatitis B virus stimulates Ras and the transcriptional activation through AP‐1, NF‐κB, and SRE enhancers. Biochem Biophys Res Commun 2001; 286: 886–94. [DOI] [PubMed] [Google Scholar]
- 10. Hildt E, Hofschneider PH. The PreS2 activators of the hepatitis B virus: activators of tumour promoter pathways. Recent Results Cancer Res 1998; 154: 315–29. [DOI] [PubMed] [Google Scholar]
- 11. Hildt E, Saher G, Bruss V, Hofschneider PH. The hepatitis B virus large surface protein (LHBs) is a transcriptional activator. Virology 1996; 225: 235–9. [DOI] [PubMed] [Google Scholar]
- 12. Hildt E, Munz B, Saher G, Reifenberg K, Hofschneider PH. The PreS2 activator MHBs (t) of hepatitis B virus activates c‐raf‐1/Erk2 signaling in transgenic mice. EMBO J 2002; 21: 525–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hildt E, Urban S, Hofschneider PH. Characterization of essential domains for the functionality of the MHBst transcriptional activator and identification of a minimal MHBst activator. Oncogene 1995; 11: 2055–66. [PubMed] [Google Scholar]
- 14. Hadziyannis S, Gerber MA, Vissoulis C, Popper H. Cytoplasmic hepatitis B antigen in ‘ground‐glass’ hepatocytes of carriers. Arch Pathol 1973; 96: 327–30. [PubMed] [Google Scholar]
- 15. Hadziyannis S, Moussouros A, Vissoulis C, Afroudakis A. Cytoplasmic localisation of Australia antigen in the liver. Lancet 1972; 1: 976–9. [DOI] [PubMed] [Google Scholar]
- 16. Gerber MA, Hadziyannis S, Vissoulis C, Schaffner F, Paronetto F, Popper H. Electron microscopy and immunoelectronmicroscopy of cytoplasmic hepatitis B antigen in hepatocytes. Am J Pathol 1974; 75: 489–502. [PMC free article] [PubMed] [Google Scholar]
- 17. Popper H. The ground glass hepatocyte as a diagnostic hint. Hum Pathol 1975; 6: 517–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hsu HC, Lai MY, Su IJ et al. Correlation of hepatocyte HBsAg expression with virus replication and liver pathology. Hepatology 1988; 8: 749–54. [DOI] [PubMed] [Google Scholar]
- 19. Su IJ, Kuo TT, Liaw YF. Hepatocyte hepatitis B surface antigen. Diagnostic evaluation of patients with clinically acute hepatitis B surface antigen‐positive hepatitis. Arch Pathol Laboratory Med 1985; 109: 400–2. [PubMed] [Google Scholar]
- 20. Su IJ, Lai MY, Hsu HC et al. Diverse virological, histopathological and prognostic implications of seroconversion from hepatitis B e antigen to anti‐HBe in chronic hepatitis B virus infection. J Hepatol 1986; 3: 182–9. [DOI] [PubMed] [Google Scholar]
- 21. Fan YF, Lu CC, Chang YC et al. Identification of a pre‐S2 mutant in hepatocytes expressing a novel marginal pattern of surface antigen in advanced diseases of chronic hepatitis B virus infection. J Gastroenterol Hepatol 2000; 15: 519–28. [DOI] [PubMed] [Google Scholar]
- 22. Fan YF, Lu CC, Chen WC et al. Prevalence and significance of hepatitis B virus (HBV) pre‐S mutants in serum and liver at different replicative stages of chronic HBV infection. Hepatology 2001; 33: 277–86. [DOI] [PubMed] [Google Scholar]
- 23. Wang HC, Wu HC, Chen CF, Fausto N, Lei HY, Su IJ. Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre‐S mutants that may induce endoplasmic reticulum stress. Am J Pathol 2003; 163: 2441–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gunther S, Fischer L, Pult I, Sterneck M, Will H. Naturally occurring variants of hepatitis B virus. Adv Virus Res 1999; 52: 25–137. [DOI] [PubMed] [Google Scholar]
- 25. Chisari FV, Ferrari C. Hepatitis B virus immunopathogenesis. Annu Rev Immunol 1995; 13: 29–60. [DOI] [PubMed] [Google Scholar]
- 26. Xu Z, Yen TS. Intracellular retention of surface protein by a hepatitis B virus mutant that releases virion particles. J Virol 1996; 70: 133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kadowaki H, Nishitoh H, Ichijo H. Survival and apoptosis signals in ER stress: the role of protein kinases. J Chem Neuroanat 2004; 28: 93–100. [DOI] [PubMed] [Google Scholar]
- 28. Wang HC, Chang WT, Chang WW et al. Hepatitis B virus pre‐S2 mutant upregulates cyclin A expression and induces nodular proliferation of hepatocytes. Hepatology 2005; 41: 761–70. [DOI] [PubMed] [Google Scholar]
- 29. Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase‐7 activation. J Biol Chem 2003; 278: 20 915–24. [DOI] [PubMed] [Google Scholar]
- 30. Bi M, Naczki C, Koritzinsky M et al. ER stress‐regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J 2005; 24: 3470–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shuda M, Kondoh N, Imazeki N et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J Hepatol 2003; 38: 605–14. [DOI] [PubMed] [Google Scholar]
- 32. Hsieh YH, Su IJ, Wang HC et al. Pre‐S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis 2004; 25: 2023–32. [DOI] [PubMed] [Google Scholar]
- 33. Hung JH, Su IJ, Lei HY et al. Endoplasmic reticulum stress stimulates the expression of cyclooxygenase‐2 through activation of NF‐κB and pp38 mitogen‐activated protein kinase. J Biol Chem 2004; 279: 46 384–92. [DOI] [PubMed] [Google Scholar]
- 34. Wu D, Marko M, Claycombe K, Paulson KE, Meydani SN. Ceramide‐induced and age‐associated increase in macrophage COX‐2 expression is mediated through up‐regulation of NF‐κB activity. J Biol Chem 2003; 278: 10 983–92. [DOI] [PubMed] [Google Scholar]
- 35. Karin M, Greten FR. NF‐κB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 2005; 5: 749–59. [DOI] [PubMed] [Google Scholar]
- 36. Pikarsky E, Porat RM, Stein I et al. NF‐κB functions as a tumour promoter in inflammation‐associated cancer. Nature 2004; 431: 461–6. [DOI] [PubMed] [Google Scholar]
- 37. Wang X, Ju W, Renouard J, Aden J, Belinsky SA, Lin Y. 17‐Allylamino‐17‐demethoxygeldanamycin synergistically potentiates tumor necrosis factor‐induced lung cancer cell death by blocking the nuclear factor‐κB pathway. Cancer Res 2006; 66: 1089–95. [DOI] [PubMed] [Google Scholar]
- 38. Mazhar D, Ang R, Waxman J. COX inhibitors and breast cancer. Br J Cancer 2006; 94: 346–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA. Centrosome duplication in mammalian somatic cells requires E2F and Cdk2‐cyclin A. Nat Cell Biol 1999; 1: 88–93. [DOI] [PubMed] [Google Scholar]
- 40. Ohashi R, Gao C, Miyazaki M et al. Enhanced expression of cyclin E and cyclin A in human hepatocellular carcinomas. Anticancer Res 2001; 21: 657–62. [PubMed] [Google Scholar]
- 41. Zhou Q, He Q, Liang LJ. Expression of p27, cyclin E and cyclin A in hepatocellular carcinoma and its clinical significance. World J Gastroenterol 2003; 9: 2450–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jackman M, Kubota Y, Den Elzen N, Hagting A, Pines J. Cyclin A– and cyclin E–Cdk complexes shuttle between the nucleus and the cytoplasm. Mol Biol Cell 2002; 13: 1030–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Faivre J, Frank‐Vaillant M, Poulhe R et al. Centrosome overduplication, increased ploidy and transformation in cells expressing endoplasmic reticulum‐associated cyclin A2. Oncogene 2002; 21: 1493–500. [DOI] [PubMed] [Google Scholar]
- 44. Akbar SK, Onji M. Hepatitis B virus (HBV)‐transgenic mice as an investigative tool to study immunopathology during HBV infection. Int J Exp Pathol 1998; 79: 279–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991; 351: 317–20. [DOI] [PubMed] [Google Scholar]
- 46. Lee TH, Finegold MJ, Shen RF, DeMayo JL, Woo SL, Butel JS. Hepatitis B virus transactivator X protein is not tumorigenic in transgenic mice. J Virol 1990; 64: 5939–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang SN, Chisari FV. Strong, sustained hepatocellular proliferation precedes hepatocarcinogenesis in hepatitis B surface antigen transgenic mice. Hepatology 1995; 21: 620–6. [PubMed] [Google Scholar]
- 48. Hagen TM, Huang S, Curnutte J et al. Extensive oxidative DNA damage in hepatocytes of transgenic mice with chronic active hepatitis destined to develop hepatocellular carcinoma. Proc Natl Acad Sci USA 1994; 91: 12 808–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Protzer U, Schaller H. Immune escape by hepatitis B viruses. Virus Genes 2000; 21: 27–37. [PubMed] [Google Scholar]
- 50. Yamamoto K, Horikita M, Tsuda F et al. Naturally occurring escape mutants of hepatitis B virus with various mutations in the S gene in carriers seropositive for antibody to hepatitis B surface antigen. J Virol 1994; 68: 2671–6. [DOI] [PMC free article] [PubMed] [Google Scholar]