

Abstract
To identify additional alterations to c‐kit or platelet‐derived growth factor receptor α (PDGFRA) genes in gastrointestinal stromal tumors (GIST), we investigated the methylation status of nine known methylation‐sensitive CpG islands (p15, p16, p73, 0‐6‐methylguanine‐DNA methyltransferase, E‐cadherin, mutL homolog 1, colon cancer nonpolyposis type 2 (escherichia), methylated in tumors [MINT]1, MINT2, and MINT31), and compared the results with the malignant potential and gain‐of‐function mutation types of GIST. Thirty‐five GIST (c‐kit mutations in 25 cases, PDGFRA mutations in seven cases, and lacking either mutation in three cases) were subjected to methylation‐specific polymerase chain reaction to detect the methylation status of the nine methylation‐sensitive CpG islands. Aberrant DNA methylation of these loci was found in 94% of all GIST. The rates of DNA methylation at each locus were as follows: hMLH1, 60%; MINT2, 51%; MGMT, 49%; p73, 49%; p16, 20%; E‐cadherin, 14%; MINT1, 9%; p15, 6%; and MINT31, 0%. CpG islands methylator phenotype, which was defined as methylation involving more than three gene promoters, was found in 57% of GIST with c‐kit or PDGFRA gene mutations. According to the risk categories, CpG islands methylator phenotype was present in 55% of low‐risk GIST, and in 58% of high‐risk GIST. Our results suggested that in addition to c‐kit or PDGFRA mutations, the aberrant methylation of CpG islands, especially of mismatch‐repair genes, may have a role in the tumorigenesis of GIST. (Cancer Sci 2008; 99: 253–259)
Gastrointestinal stromal tumors (GIST) are thought to be the most common nonepithelial tumors in the gastrointestinal tract with an annual incidence estimated at 10–20 cases per million.( 1 ) During tumorigenesis of GIST, the most frequent changes are reported to be gain‐of‐function mutations in c‐kit protooncogene. These mutations can result in autophosphorylation, namely KIT ligand‐independent kinase activity.( 2 ) C‐kit mutations occur in up to 90% of GIST. Also, approximately 5% of GIST are characterized by a mutation in the related receptor tyrosine kinase platelet‐derived growth factor receptor α (PDGFRA) exons 12 and 18.( 3 , 4 ) Very recently, Lasota et al. reported PDGFRA exon 14 mutations in 11 of 200 GIST negative for mutations in c‐kit exons 9, 11, 13, and 17, and PDGFRA exons 12 and 18.( 5 ) However, approximately 5% of all GIST do not have a detectable mutation in either c‐kit or PDGFRA genes.
Although most GIST are characterized by a gene mutation in either c‐kit or PDGFRA, the clinical behavior of each GIST differs widely from benign to malignant with widespread metastasis. Therefore, we speculated that additional genetic alterations, other than the c‐kit and PDGFRA genes, were required for the progression of GIST.
We previously reported allelic losses of 1p, 14q, and 22q in both low‐ and high‐risk GIST.( 6 ) Moreover, additional chromosomal losses and microsatellite instability (MSI) were observed in high‐risk GIST. Fluorescence in situ hybridization (FISH) or comparative genomic hybridization (CGH) methods have also demonstrated the same chromosomal changes in high‐risk GIST.( 7 , 8 , 9 , 10 )
Recent advances in molecular biology have elucidated relevant epigenetic modifications in gene regulation, and have shown that epigenetic modifications play important roles in tumorigenesis and tumor progression as well as previously known genetic alterations in oncogenes and tumor‐suppressor genes. In particular, aberrant epigenetic hypermethylation of the gene‐promoter region and the subsequent loss of gene expression are closely related to the development and progression of several human cancers.( 11 ) For example, the hypermethylation of a mismatch‐repair gene closely correlates with the development of MSI‐positive colon cancer.( 12 , 13 ) The distinct methylation status of various genes has revealed characteristic differences among tumor phenotypes and clinical behaviors in several human cancers such as colon, breast,( 14 ) and liver cancers,( 15 ) and mucosa‐associated lymphoid tissue (MALT) lymphoma.( 16 ) The methylation status of various genes greatly influences the diagnosis and prognosis of several tumors.
However, only one report has analyzed the methylation status of GIST.( 17 ) Therefore, the present study aimed to identify the role of hypermethylation in the development and progression of GIST. We selected nine well‐known methylation‐sensitive CpG islands, evaluated the aberrant methylations in these loci, and compared the results of the mutational status of c‐kit and PDGFRA, and the malignant potential of GIST.
Materials and Methods
Patients and tumor tissues. We included 35 GIST patients who underwent potentially curative surgery without preoperative therapy at Gunma University Hospital and Jichi Medical School Hospital. The study was approved by the Institutional Review Board of the Gunma University Graduate School of Medicine and the Jichi Medical School.
The risk grade of the GIST was evaluated according to the methods of Fletcher et al.( 18 ) Tumors that were <2 cm in diameter with 0–4 mitoses/50 high‐power fields (HPF) were considered to be of very low risk, and tumors that were 2–5 cm in diameter with 0–4 mitoses/50 HPF were considered to be of low risk. Tumors <5 cm in diameter with 6–10 mitoses/50 HPF or tumors 5–10 cm in diameter with 0–4 mitoses/50 HPF were considered to be of intermediate risk. Tumors >5 cm in diameter with a mitotic count higher than 5/50 HPF, or tumors >10 cm in diameter with any mitotic rate, or tumors with >10 mitoses/50 HPF were classified as high‐risk tumors. Tumors with necrosis were also classified as high‐risk tumors.
Mutational analysis of c‐kit and PDGFRA. Genomic DNA was extracted from formalin‐fixed, paraffin‐embedded tumor tissues using standard proteinase K digestion and phenol/chloroform extraction methods, and was used for the following molecular analysis. To avoid contamination from normal tissues, only tumor tissues were scraped and obtained from each paraffin section, guided by serial hematoxylin–eosin stained sections.
Exons 9, 11, 13, and 17 of c‐kit, and exons 12, 14, and 18 of PDGFRA were amplified by polymerase chain reaction (PCR) using previously published primer sets (Table 1).( 5 , 19 ) Each of the amplified fragments was purified from a polyacrylamide gel, and direct sequencing was carried out using a BigDye Terminator v.3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) and an ABI PRISM 3100 DNA Sequencer (Applied Biosystems). All sequencing reactions were carried out in forward and reverse directions.
Table 1.
Sequences of primers used in the mutational analysis of c‐kit and PDGFRA genes
| Exon | Sequence |
|---|---|
| c‐kit | |
| Exon 9F | 5′‐ATGCTCTGCTTCTGTACTGCC‐3′ |
| Exon 9R | 5′‐CAGAGCCTAAACATCCCCTTA‐3′ |
| Exon 11F | 5′‐CCAGAGTGCTCTAATGACTG‐3′ |
| Exon 11R | 5′‐ACCCAAAAAGGTGACATGGA‐3′ |
| Exon 13F | 5′‐CATCAGTTTGCCAGTTGTGC‐3′ |
| Exon 13R | 5′‐ACACGGCTTTACCTCCAAATG‐3′ |
| Exon 17F | 5′‐TGTATTCACAGAGACTTGGC‐3′ |
| Exon 17R | 5′‐GGATTTACATTATGAAAGTCACAGG‐3′ |
| PDGFRA | |
| Exon 12F | 5′‐TCCAGTCACTGTGCTGCTTG‐3′ |
| Exon 12R | 5′‐GCAAGGGAAAAGGGAGTCTT‐3′ |
| Exon 14F | 5′‐GTAGCTCAGCTGGACTGATA‐3′ |
| Exon 14R | 5′‐AATCCTCACTCCAGGTCAGT‐3′ |
| Exon 18F | 5′‐ACCATGGATCAGCCAGTCTT‐3′ |
| Exon 18R | 5′‐AAGTGTGGGAGGATGAGCCTG‐3′ |
PDGFRA, platelet‐derived growth factor receptor α.
Methylation‐specific polymerase chain reaction. We selected nine methylation‐sensitive CpG islands as follows: tumor‐suppressor genes and related genes (p15, p16, p73, E‐cadherin), DNA repair genes (0‐6‐methylguanine‐DNA methyltransferase, mutL homolog 1, colon cancer nonpolyposis type 2 (escherichia)), and the three methylated clones methylated in tumors (MINT) 1, MINT2, and MINT31, originally recovered from a colorectal carcinoma cell line.( 20 ) The methylation status of the CpG islands in the promoter region of each gene was analyzed by the bisulfite modification technique using a CpGenome DNA modification kit (Chemicon, Temecula, CA, USA) according to the manufacturer's protocol. The primer sequences used in this methylation‐specific polymerase chain reaction (MSP) study are listed in Table 2, and the PCR conditions were as described previously.( 16 , 17 ) Each of the PCR products (10‐µL volume) was directly loaded onto 8% polyacrylamide gels, stained with ethidium bromide, and visualized directly under ultraviolet illumination (Fig. 1).
Table 2.
Sequences of primers used in methylation‐specific polymerase chain reaction
| Gene | 5′ primer | 3′ primer |
|---|---|---|
| Methylated | ||
| p15 | GCGTTCGTATTTTGCGGTT | CGTACAATAACCGAACGACCGA |
| p16 | TTATTAGAGGGTGGGGCGGATCGC | GACCCCGAACCGCGACCGTAA |
| p73 | GGACGTAGCGAAATCGGGGTTC | CGTCGCAACCCCGAACATCG |
| MGMT | TTTCGACGTTCGTAGGTTTTCGC | GCACTCTTCCGAAAACGAAACG |
| E‐cadherin | TGTAGTTACGTATTTATTTTTAGTGGCGTC | CGAATACGATCGAATCGAACCG |
| hMLH1 | TTAATAGGAAGAGCGGATAGC | TCTATAAATTACTAAATCTCTTCG |
| MINT1 | AATTTTTTTATATATATTTTCGAAGC | AAAAACCTCAACCCCGCG |
| MINT2 | TTGTTAAAGTGTTGAGTTCGTC | AATAACGACGATTCCGTACG |
| MINT31 | TGTTGGGGAAGTGTTTTTCGGC | CGAAAACGAAACGCCGCG |
| Unmethylated | ||
| p15 | TGTGATGTGTTTGTATTTTGTGGTT | CCATACAATAACCAAACAACCAA |
| p16 | TTATTAGAGGGTGGGGTGGATTGT | CAACCCCAAACCACAACCATAA |
| p73 | AGGGGATGTAGTGAAATTGGGGTTT | ATCACAACCCCAAACATCAACATCCA |
| MGMT | TTTGTGTTTTGATGTTTGTAGGTTTTTGT | AACTCCACACTCTTCCAAAAACAAAACA |
| E‐cadherin | TGGTTGTAGTTATGTATTTATTTTTAGTGGTGTT | ACACCAAATACAATCAAATCAAACCAAA |
| hMLH1 | TTAATAGGAAGAGTGGATAGTG | TCTATAAATTACTAAATCTCTTCA |
| MINT1 | AATTTTTTTATATATATTTTTGAAGTGT | AACAAAAAACCTCAACCCCACA |
| MINT2 | GATTTTGTTAAAGTGTTGAGTTTGTT | CAAAATAATAACAACAATTCCATACA |
| MINT31 | TAGATGTTGGGGAAGTGTTTTTTGGT | TAAATACCCAAAAACAAAACACCACA |
hMLH, mutL homolog 1, colon cancer nonpolyposis type 2 (escherichia); MGMT, 0‐6‐methylguanine‐DNA methyltransferase; MINT, methylated in tumors.
Figure 1.
Representative methylation‐specific polymerase chain reaction (PCR) experiments for methylation analysis. PCR products amplified with unmethylated (U) and methylated (M) sequence‐specific primers.
Statistical analysis. Fisher's exact probability test, χ2‐test, or Student's t‐test were used to analyze correlations between the methylation status of CpG islands and the clinicopathological characteristics of GIST, including the mutation statuses. P‐values less than 0.05 were considered statistically significant.
Results
Clinicopathological characteristics. The clinicopathological characteristics of the patients are shown in Table 3. Tumor localizations were as follows: 26 GIST were in the stomach (74%), six were in the small intestine (17%), two were in the rectum (6%), and one was in the omentum (3%). The mean tumor size (maximum diameter) was 5.1 cm (range, 1.6–22 cm). Histopathologically, 28 GIST consisted of spindle tumor cells and the remaining seven were myxoid epithelioid GIST, a subtype of GIST closely correlated with PDGFRA gene mutations, as we reported previously.( 18 ) According to the risk categories, 22 cases were classified into the low‐risk group, one into the intermediate‐risk group and 12 into the high‐risk group. The median follow‐up time was 62 months (range, 5–125 months). Among the 35 patients, seven (20%) experienced tumor relapse, and two died of a recurrent tumor. The sites of recurrence were local (pelvic cavity and peritoneal dissemination, each in two cases), and distant metastasis (one each in liver, lung, and bone). The median tumor‐free time was 26 months (range, 5–63 months).
Table 3.
Clinicopathological characteristics of 35 gastrointestinal stromal tumors
| Variable | Value |
|---|---|
| Age (years, mean ± SD) | 61.9 ± 12.0 |
| Sex | |
| Male | 17 (49%) |
| Female | 18 (51%) |
| Tumor location | |
| Stomach | 26 (74%) |
| Small intestine | 6 (17%) |
| Rectum | 2 (6%) |
| Omentum | 1 (3%) |
| Tumor size (cm, average ± SD) | 5.09 ± 3.90 |
| Grade | |
| High | 12 (34%) |
| Intermediate | 1 (3%) |
| Low | 22 (63%) |
| Histological type | |
| Spindle | 28 (80%) |
| Myxoid epithelioid | 7 (20%) |
| Recurrence | |
| Absent | 28 (80%) |
| Present | 7 (20%) |
| Mutation status | |
| c‐kit | 25 (71%) |
| PDGFRA | 7 (20%) |
| No mutation | 3 (9%) |
PDGFRA, platelet‐derived growth factor receptor α.
Mutation analysis of the c‐kit and PDGFRA genes. As shown in Table 3 and Figure 2a, 25 of 35 GIST showed mutations in c‐kit: one (case 22) in exon 9 (duplication) and 24 in exon 11 (10 point mutations, nine in‐frame deletions, and five internal tandem duplications). No mutations in exons 13 or 17 were found in any of the tumors. As reported previously,( 19 ) all seven myxoid epithelioid GIST had mutations in PDGFRA exon 18 (four in‐frame deletions and three point mutations).( 19 ) The three remaining GIST showed no mutations in either the c‐kit or PDGFRA genes.
Figure 2.
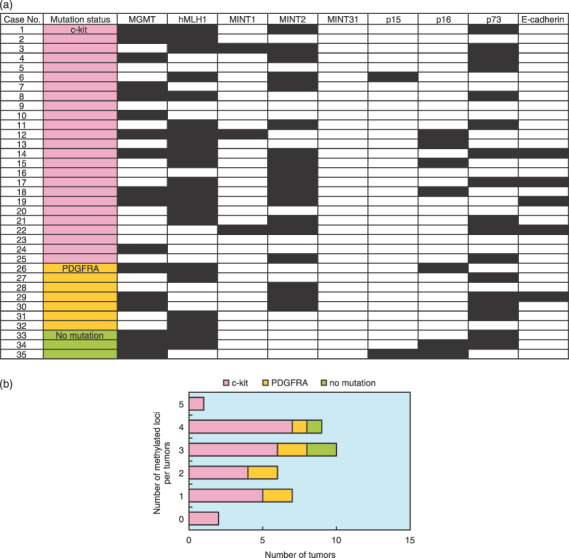
(a) Methylation profiles of nine CpG islands and mutation statuses of c‐kit and platelet‐derived growth factor receptor α (PDGFRA) genes. The black grid squares represent positive methylation. The open squares denote no detectable methylation. (b) Correlation between the number of methylated loci and the mutational status of gastrointestinal stromal tumors (GIST). Although GIST lacking either mutation showed relatively higher rates of multigene methylation, there were no statistical differences between GIST with c‐kit or PDGFRA mutations or those without those mutations.
Methylation analysis. The results of MSP analysis are summarized in Figure 2a. Unmethylated alleles of each locus were amplified successfully in all samples examined in the present study, confirming the accuracy of bisulfate modification in the MSP protocol.
The frequency rates for hypermethylation of the nine CpG islands are listed in Table 4. High frequency rates were observed in hMLH1 (60%), MGMT (49%), p73 (49%), and MINT2 (51%), whereas low frequency rates were observed in p15 (5.7%), MINT1 (8.6%), and MINT31 (0%). The average number of methylated genes per tumor was 2.6. The majority of GIST (33 of 35, 94.3%) showed aberrant DNA methylation of at least one CpG island. Two GIST (cases 9 and 23) showed no methylation of the CpG islands examined. On average, 2.6 (0–5) loci were methylated in GIST with c‐kit gene mutations and 2.3 (1–4) loci were methylated in those with PDGFRA mutations (Fig. 2b).
Table 4.
Frequency rates of aberrant methylation in each CpG island
| CpG island | Frequency (%) |
|---|---|
| hMLH1 | 60 |
| MINT2 | 51 |
| MGMT | 49 |
| p73 | 49 |
| p16 | 20 |
| E‐cadherin | 14 |
| MINT1 | 9 |
| p15 | 6 |
| MINT31 | 0 |
No statistical difference was found in the frequency rate of hypermethylation between GIST with c‐kit and PDGFRA gene mutations. Comparing GIST with c‐kit or PDGFRA gene mutations, the three GIST without any mutations showed higher frequency rates of methylation (3–4, average 3.3), although there was no statistical difference (Fig. 2b). The results of MSP in each risk grade are summarized in Figure 3a. On average, 2.5 (0–5) loci were methylated in low‐risk GIST, whereas 2.7 (1–4) loci were methylated in high‐risk GIST (Fig. 3b). No statistical difference was also found among the risk categories.
Figure 3.
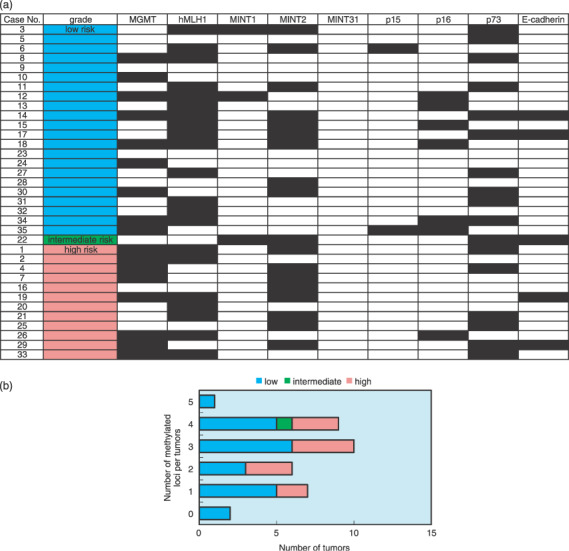
(a) Methylation profiles for the risk categories of gastrointestinal stromal tumors (GIST) by Fletcher et al. The black grid squares represent positive methylation. The open squares denote no detectable methylation. (b) Correlation between the number of methylated loci and risk categories of GIST. There were no statistical differences among the risk categories.
The hypermethylated phenotype (CpG islands methylator phenotype; CIMP), which was methylated at three or more of the nine loci, as described previously,( 21 ) was also analyzed. CIMP‐positive tumors were found in 20 of 35 (57%) GIST. We analyzed the relationship between CIMP status and clinicopathological data (age, sex, tumor location, tumor size, histological type, mitotic score, mindbomb homolog 1 labeling index, risk grade, presence of recurrence, and mutation status) (Table 5). For risk grade, CIMP was present in 12 (55%) of the low‐risk GIST, and in seven (58%) of the high‐risk GIST (Fig. 3b). For the mutation status, 14 (56%) of 25 GIST with c‐kit mutations, three (43%) of seven with PDGFRA mutations, and all three (100%) of three GIST with no mutation were CIMP positive. However, there was no significant correlation between CIMP status and any clinicopathological characteristic.
Table 5.
Comparison of clinicopathological variables between CpG islands methylator phenotype (CIMP)‐positive and ‐negative gastrointestinal stromal tumors
| Variable | CIMP | ||
|---|---|---|---|
| Positive (n = 20) | Negative (n = 15) | P‐value | |
| Age (years, mean ± SD) | 60.9 ± 10.4 | 63.3 ± 14.0 | 0.551 |
| Sex (male:female) | 9:11 | 8:7 | 0.738 |
| Tumor location (St/SI/R/Ome) | 15/3/1/1 | 11/3/1/0 | 0.922 |
| Tumor size (cm, average ± SD) | 4.98 ± 2.78 | 4.18 ± 2.27 | 0.370 |
| Histological type (spindle/epithelioid) | 17/3 | 11/4 | 0.430 |
| Mitotic score | 4.75 ± 4.99 | 7.00 ± 10.32 | 0.399 |
| MIB‐1 labeling index | 5.14 ± 4.13 | 6.65 ± 5.37 | 0.353 |
| Grade (high/intermediate/low) | 7/1/12 | 5/0/10 | 0.655 |
| Recurrence (absent/present) | 18/2 | 10/5 | 0.088 |
| Mutation (c‐kit/PDGFRA/negative) | 14/3/3 | 11/4/0 | 0.241 |
Ome, omentum; PDGFRA, platelet‐derived growth factor receptor α; R, rectum; SI, small intestine; St, stomach.
Discussion
In an earlier report, we demonstrated the presence of loss of heterozygosity (LOH) and MSI preferentially in high‐risk GIST at a rate of 11–40%.( 6 ) Similar results are reported in other studies using FISH or CGH.( 7 , 8 , 9 , 10 ) Moreover, MSI was observed in 50% of GIST. In low‐risk GIST, MSI was detected at a single locus in two of nine tumors. In contrast, it was observed in 69% of high‐risk GIST; two at a single locus, five at two loci, one at three loci, and one at five loci.( 6 ) It is known that the silencing of tumor‐related genes by promoter hypermethylation at CpG islands results in gene alterations.( 6 ) Therefore, these results indicated that a higher frequency of LOH and the accumulation of MSI might relate to the progression of GIST from low to high risk.
In the present study, to elucidate additional molecular mechanisms that participate in the development or malignant conversion of GIST from low to high risk, we characterized the methylation status of GIST using a panel of nine independent CpG islands. In many previous studies analyzing hypermethylation of human malignancies,( 11 ) the target genes inactivated by DNA methylation were diverse and the list of such genes continues to grow. Therefore, a standard list of CpG islands that should be analyzed when profiling hypermethylation is not settled. Here, we selected mismatch repair genes, tumor suppressor genes, and related genes that, when hypermethylated, have been reported to occur in many kinds of tumors. As well as these, we analyzed the hypermethylation of MINT1, MINT2, and MINT31. These three regions of MINT are CpG islands that were observed in colorectal carcinoma by Toyota et al. in 1999.( 20 , 21 ) In those studies, Toyota et al. subclassified colorectal carcinoma into CIMP‐positive and ‐negative tumors and found a close correlation between CIMP and MSI. Subsequently, CIMP has been reported in various tumors, such as hepatocellular carcinoma,( 15 ) pancreatic adenocarcinoma,( 22 ) and gastric adenocarcinoma.( 23 ) Moreover, we reported a distinct methylation pattern in Helicobacter pylori‐dependent and ‐independent gastric MALT lymphoma.( 16 ) In these studies,( 15 , 16 , 20 , 21 , 22 , 23 ) MINT1, MINT2, and MINT31 were methylated in various tumors, and were thought to be useful markers of CIMP. However, no study has analyzed the methylation status of MINT and CIMP in mesenchymal tumors, including GIST. Thus we selected MINT1, MINT2, and MINT31 for the present study.
We demonstrated that hMLH1 and MGMT had the highest rates of hypermethylation of the mismatch‐repair genes. As the silencing of hMLH1 by promoter hypermethylation at a CpG island resulted in MSI, these results seem to be consistent with our previous report,( 6 ) in which MSI was observed frequently in high‐risk GIST.
Interestingly, there were no correlations between methylation status and any clinicopathological characteristics, including the mutational status of c‐kit and PDGFRA. In a previous study using an oligonucleotide microarray,( 24 ) GIST with either c‐kit or PDGFRA gene mutation showed different gene expression profiles, which might indicate that GIST with different gene mutations progress by different additional mechanisms. However, FISH analysis revealed a similar cytogenetic profile for GIST with PDGFRA gene mutations and GIST with c‐kit gene mutations, and losses of 1p, 14q, and 22q were seen in both types of GIST.( 25 ) The similar methylation status of GIST with different mutations in the present study supports the latter result of FISH analysis, which might indicate that GIST, irrespective of mutation status, have common pathways for tumor progression.
However, GIST without any mutation, although a minority, were all CIMP positive. This suggests that hypermethylation may play a more important role in tumorigenesis of GIST without mutation than those with c‐kit or PDGFRA mutations.
To date, only one study has reported the methylation status of GIST. House et al. revealed hypermethylation of at least 1 of 11 candidate gene regions in 84% of the 38 KIT‐positive GIST.( 17 ) In that study, they indicated that the presence of methylated E‐cadherin alleles or the absence of methylated hMLH1 alleles correlated with increased early tumor recurrence for GIST. However, in our study, hypermethylation of hMLH1 and E‐cadherin was found at different rates to those of House et al. (hMLH1, 60 vs 34%; E‐cadherin, 37 vs 14%), and no correlation was found between the hypermethylation of hMLH1 or E‐cadherin and the risk categories of GIST. Although we could not explain the reason for these differences, the early tumor recurrence of GIST as shown by House et al. might not necessarily be consistent with the high‐risk GIST investigated in the present study. Longer follow‐up periods will be required to clarify the exact correlation between the aberrant methylation status of GIST and the prognosis of patients.
The standard therapy for primary GIST is complete surgical resection, which is carried out in approximately 85% of patients with this as a primary disease.( 26 ) Nevertheless, tumor recurrence is frequent, and 5‐year survival after removal of the primary localized GIST is approximately 50%.( 26 , 27 ) Imatinib mesylate (STI571, Gleevec; Novartis Pharmaceuticals, Basel, Switzerland) is an oral agent that selectively inhibits specific tyrosine kinases including KIT, ABL, BCR‐ABL, and PDGFR,( 28 ) and can induce a partial response or stable disease in up to 80% of patients with metastatic or unresectable GIST.( 29 , 30 ) However, GIST without mutations in c‐kit and PDGFRA do not respond to imatinib.( 31 ) Moreover, acquired resistance to imatinib in GIST with mutations has been reported by several groups and is often caused by secondary c‐kit mutations.( 32 , 33 , 34 ) It is now clear that many patients subsequently develop resistance to this agent. The frequent aberrant methylation found in GIST in our study might indicate that some therapeutic strategies using demethylating drugs could be effective against GIST, especially those without c‐kit or PDGFRA gene mutations.
In conclusion, we reported aberrant methylation status using nine methylation‐sensitive CpG islands in GIST with and without mutations. The frequencies of methylation for each locus were similar in GIST with c‐kit or with PDGFRA mutations. However, CIMP was present in 100% of GIST without mutation, although there was no statistical difference between GIST with or without gene mutations. From these results, we hypothesized that aberrant methylation of multiple gene promoters, as well as the c‐kit and PDGFRA genes, plays a role in the tumorigenesis of GIST.
Acknowledgments
This work was supported by a Grant‐in‐Aid for Scientific Research (C), Scientific Research on Priority Areas and Initiatives for Attractive Education in Graduate Schools from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1. Nilsson B, Bumming P, Meis‐Kindblom JM et al . Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era: a population‐based study in western Sweden. Cancer 2005; 103: 821–9. [DOI] [PubMed] [Google Scholar]
- 2. Hirota S, Isozaki K, Moriyama Y et al . Gain‐of‐function mutations of c‐kit in human gastrointestinal stromal tumors. Science 1998; 279: 577–80. [DOI] [PubMed] [Google Scholar]
- 3. Kitamura Y, Hirota S. Kit as a human oncogenic tyrosine kinase. Cell Mol Life Sci 2004; 61: 2924–31. [DOI] [PubMed] [Google Scholar]
- 4. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol 2004; 22: 3813–25. [DOI] [PubMed] [Google Scholar]
- 5. Lasota J, Stachura J, Miettinen M. GISTS with PDGFRA exon 14 mutations represent a subset of clinically favorable gastric tumors with epithelioid morphology. Laboratory Invest 2006; 86: 94–100. [DOI] [PubMed] [Google Scholar]
- 6. Fukasawa T, Chong JM, Sakurai S et al . Allelic loss of 14q and 22q, NF2 mutation, and genetic instability occur independently of c‐kit mutation in gastrointestinal stromal tumor. Jpn J Cancer Res 2000; 91: 1241–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Breiner JA, Meis‐Kindblom J, Kindblom LG et al . Loss of 14q and 22q in gastrointestinal stromal tumors (pacemaker cell tumors). Cancer Genet Cytogenet 2000; 120: 111–16. [DOI] [PubMed] [Google Scholar]
- 8. Kim NG, Kim JJ, Ahn JY et al . Putative chromosomal deletions on 9P, 9Q and 22Q occur preferentially in malignant gastrointestinal stromal tumors. Int J Cancer 2000; 85: 633–8. [DOI] [PubMed] [Google Scholar]
- 9. Assamaki R, Sarlomo‐Rikala M, Lopez‐Guerrero JA et al . Array comparative genomic hybridization analysis of chromosomal imbalances and their target genes in gastrointestinal stromal tumors. Genes Chromosomes Cancer 2007; 46: 564–76. [DOI] [PubMed] [Google Scholar]
- 10. Meza‐Zepeda LA, Kresse SH, Barragan‐Polania AH et al . Array comparative genomic hybridization reveals distinct DNA copy number differences between gastrointestinal stromal tumors and leiomyosarcomas. Cancer Res 2006; 66: 8984–93. [DOI] [PubMed] [Google Scholar]
- 11. Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res 2001; 61: 3225–9. [PubMed] [Google Scholar]
- 12. Kana MF, Loda M, Gaida GM et al . Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair‐defective human tumor cell lines. Cancer Res 1997; 57: 808–11. [PubMed] [Google Scholar]
- 13. Salahshor S, Koelble K, Rubio C, Lindblom A. Microsatellite instability and hMLH1 and hMSH2 expression analysis in familial and sporadic colorectal cancer. Laboratory Invest 2001; 81: 535–41. [DOI] [PubMed] [Google Scholar]
- 14. Bae YK, Brown A, Garrett E et al . Hypermethylation in histologically distinct classes of breast cancer. Clin Cancer Res 2004; 10: 5998–6005. [DOI] [PubMed] [Google Scholar]
- 15. Shen L, Ahuja N, Shen Y et al . DNA methylation and environmental exposures in human hepatocellular carcinoma. J Natl Cancer Inst 2002; 94: 755–61. [DOI] [PubMed] [Google Scholar]
- 16. Kaneko Y, Sakurai S, Hironaka M et al . Distinct methylated profiles in Helicobacter pylori dependent and independent gastric MALT lymphomas. Gut 2003; 52: 641–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. House MD, Guo MZ, Efron DT et al . Tumor suppressor gene hypermethylation as a predictor of gastric stromal tumor behavior. J Gastrointest Surg 2003; 7: 1004–14. [DOI] [PubMed] [Google Scholar]
- 18. Fletcher CD, Berman JJ, Corless C et al . Diagnosis of gastrointestinal stromal tumors: a consensus approach. Hum Pathol 2002; 33: 459–65. [DOI] [PubMed] [Google Scholar]
- 19. Sakurai S, Hasegawa T, Sakuma Y et al . Myxoid epithelioid gastrointestinal stromal tumor (GIST) with mast cell infiltrations: a subtype of GIST with mutations of platelet‐derived growth factor receptor alpha gene. Hum Pathol 2004; 35: 1223–30. [DOI] [PubMed] [Google Scholar]
- 20. Toyota M, Ho C, Ahuja N et al . Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res 1999; 59: 2307–12. [PubMed] [Google Scholar]
- 21. Toyota M, Ahuja N, Ohe‐Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 1999; 96: 8681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ueki T, Toyota M, Sohn T et al . Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res 2000; 60: 1835–9. [PubMed] [Google Scholar]
- 23. Toyota M, Ahuja N, Suzuki H et al . Aberrant methylation in gastric cancer associated with the CpG island methylator phenotype. Cancer Res 1999; 59: 5438–42. [PubMed] [Google Scholar]
- 24. Kang HJ, Nam SW, Kim H et al . Correlation of KIT and platelet‐derived growth factor receptor alpha mutations with gene activation and expression profiles in gastrointestinal stromal tumors. Oncogene 2005; 24: 1066–74. [DOI] [PubMed] [Google Scholar]
- 25. Debiec‐Rychter M, Wasag B, Stul M et al . Gastrointestinal stromal tumours (GISTS) negative for KIT (CD117 antigen) immunoreactivity. J Pathol 2004; 202: 430–8. [DOI] [PubMed] [Google Scholar]
- 26. DeMatteo RP, Lewis JJ, Leung D, Mudan SS, Woodruff JM, Brennan MF. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg 2000; 231: 51–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ng EH, Pollock RE, Munsell MF, Atkinson EN, Romsdahl MM. Prognostic factors influencing survival in gastrointestinal leiomyosarcomas. Implications for surgical management and staging. Ann Surg 1992; 215: 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Buchdunger E, Cioffi CL, Law N et al . Abl protein‐tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c‐kit and platelet‐derived growth factor receptors. J Pharmacol Exp Ther 2000; 295: 139–45. [PubMed] [Google Scholar]
- 29. Joensuu H, Robers PJ, Sarlomo‐Rikala M et al . Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 2001; 344: 1052–6. [DOI] [PubMed] [Google Scholar]
- 30. Demetri GD, Von Mehren M, Blanke CD et al . Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002; 347: 472–80. [DOI] [PubMed] [Google Scholar]
- 31. Heinrich MC, Corless CL, Demetri GD et al . Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003; 21: 4342–9. [DOI] [PubMed] [Google Scholar]
- 32. Fletcher JA, Corless CL, Dimitrijevic S et al . Mechanisms of resistance to imatinib mesylate (IM) in advanced gastrointestinal stromal tumor (GIST). Proc Am Soc Clin Oncol 2003; 22: 815S. [Google Scholar]
- 33. Chen LL, Trent JC, Wu EF et al . A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res 2004; 64: 5913–19. [DOI] [PubMed] [Google Scholar]
- 34. Debiec‐Rychter M, Cools J, Dumez H et al . Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib‐resistant mutants. Gastroenterology 2005; 128: 270–9. [DOI] [PubMed] [Google Scholar]