

Abstract
Although imatinib showed high activity for advanced gastrointestinal stromal tumor (GIST) and improved the prognosis of GIST patients, resistance to the drug appears with prolonged use. Mechanisms of acquired resistance are still under investigation. In the present study, we carried out histologic and genetic analysis of 45 secondary resistant lesions obtained from 25 Japanese GIST patients treated with imatinib. All resistant lesions showed viable tumor cells expressing KIT protein, whereas imatinib‐sensitive lesions did not. All pre‐imatinib samples have KIT mutations either in exon 9 (n = 3) or exon 11 (n = 22), identified in the KIT gene of corresponding resistant tumors. In addition to primary mutations, 33 out of 45 tumors (73%) showed secondary KIT mutations in the kinase domain of the KIT gene. Secondary mutations are missense mutations and are mostly located in the kinase domains of the same allele to the primary mutations (cis‐position). Resistant lesions showed monoclonal development of tumor cells. Taken together, additional cis‐positioned mutations in the kinase domains are a major cause of secondary resistance to imatinib in Japanese GIST patients. (Cancer Sci 2008; 99: 799–804)
The great majority of gastrointestinal stromal tumors (GISTs) are accompanied with gain‐of‐function mutations in the KIT or platelet‐derived growth factor receptor alpha (PDGFRA) gene, which cause ligand‐independent constitutive activation of corresponding receptor tyrosine kinases and subsequent activation of downstream signal pathways.( 1 , 2 , 3 , 4 ) In the primary GIST, various types of KIT mutations are found in the juxtamembrane domain (exon 11), followed by the extracellular domain (exon 9), or rarely in kinase domains (exons 13 and 17).( 4 ) Similarly, 5–10% of GISTs have PDGFRA mutations either in the juxtamembrane domain (exon 12) or in the kinase domain (exons 14 and 18).( 5 ) Mutations in the KIT or PDGFRA genes are mutually exclusive, and the other approximately 5% of GISTs have no mutation in either KIT and PDGFRA. To date, primary and untreated GIST carries only a single mutation either in the KIT or the PDGFRA gene.
Imatinib mesylate (Glivec or Gleevec; Novartis Pharma, Basel, Switzerland), a selective tyrosine kinase inhibitor of BCR‐ABL, KIT and PDGFRA tyrosine kinases, has clinical activity on advanced and/or metastatic GIST with considerable tolerability.( 6 ) More than 40% of patients with advanced GISTs treated by imatinib show objective responses, and disease control is obtained for 85–90% of advanced GIST. Although imatinib improved the prognosis of advanced GIST patients with median progression‐free survival of 2 years and overall survival of 5 years,( 4 ) resistance to the drug appears with prolonged use and has become a serious problem in advanced GIST patients. Resistant lesions reportedly appear as a nodule in a mass, regrowth of a pre‐existing mass, or a new lesion in enhanced computed tomography (CT) scan.( 7 ) Several resistant mechanisms for molecular target agents, including target resistance with mutations or genomic as well as protein amplification, biological resistance, and functional resistance such as pharmacokinetic changes, are proposed.( 4 , 8 , 9 ) Previous studies have indicated clonal development, re‐expression as well as re‐activation of KIT tyrosine kinase, and acquired mutations in the kinase domain of the KIT gene, although each study had a limited number of the patients.( 8 , 9 , 10 , 11 , 12 , 13 , 14 ) The precise mechanisms of secondary resistance to imatinib, however, are still under investigation. In this study, we investigated clinicopathological and molecular features of secondary resistance to imatinib in Japanese patients with advanced GIST.
Materials and Methods
Patient demographics. The patients analyzed in this study had been diagnosed with GIST by histological examinations using either surgical or biopsy samples, of which spindle and/or epithelioid tumor cells were positive for KIT and/or CD34 by immunohistochemical examinations. The patients received 400 or 600 mg/day of imatinib for more than 180 days, although initial doses of imatinib varied from 200 to 600 mg/day. Tumor responses were determined by periodical enhanced CT scan. After progression under imatinib, samples were obtained at surgery, or by biopsy or autopsy from 25 patients (18 males and 7 females, median age 61 years old) at Osaka University Hospital (Osaka, Japan) or Niigata University Hospital (Niigata, Japan) (Table 1). Final prognostic analysis was done at the end of March 2007. A median follow‐up period from initial imatinib therapy is 43.5 months (range: 8.3–63 months). A median follow‐up period from initial diagnosis of GIST is 53.9 months (range: 8.3–124 months). Overall, 17 patients were alive with the disease and the the other eight patients died of the disease.
Table 1.
Characteristics of patients with secondary imatinib‐resistant gastrointestinal stromal tumor
| No. | Age | Gender | Primary location | Recurrent site | Initial imatinib dose (mg/day) | Best response | PFS (months) | Overall survival (months) | Present status | Resistant site | Treatments after imatinib‐resistance |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 67 | M | Small intestine | L + P | 400 | PR | 11.5 | 34.1 | AWD | P | Surgery then sunitinib |
| 2 | 61 | F | Colon | L + Lo | 400 | PR | 26.2 | 38.1 | DOD | Lo | Surgery then AMG706 |
| 3 | 43 | M | Stomach | L + P | 600 | PR | 29.2 | 57.9 | AWD | P | Surgery and RFA |
| 4 | 63 | M | Small intestine | L + P | 400 | PR | 26.8 | 47.0 | DOD | P | Surgery then sunitinib |
| P | |||||||||||
| 5 | 49 | M | Stomach | Lo + L | 400 | PR | 10.7 | 14.9 | AWD | Lo † | Surgery |
| P † | |||||||||||
| 6 | 66 | F | Stomach | L + P | 300 | PR | 25.2 | 32.0 | AWD | P | Surgery |
| 7 | 48 | M | Stomach | Lo + L | 300 | PR | 50.9 | 53.9 | AWD | L | Surgery |
| 8 | 54 | M | Small intestine | P | 400 | PR | 14.6 | 34.2 | DOD | P | Surgery then sunitinib |
| P | |||||||||||
| P | |||||||||||
| 9 | 72 | M | Stomach | L + P | 600 | PR | 24.9 | 56.3 | AWD | L | RFA, surgery, then sunitinib |
| L | |||||||||||
| L | |||||||||||
| 10 | 67 | M | Small intestine | P | 400 | SD | 19.1 | 25.2 | DOD | P | Surgery |
| 11 | 64 | M | Small intestine | P | 400 | CR | 33.4 | 46.8 | DOD | P | Sunitinib |
| 12 | 32 | F | Small intestine | L + P | 400 | PR | 49.4 | 59.3 | AWD | P † | Imatinib increase then surgery and RFA |
| P † | |||||||||||
| 13 | 66 | M | Stomach | L + P | 400 | PR | 9.0 | 19.5 | DOD | L | Surgery then imatinib increase |
| 14 | 45 | M | Small intestine | L + rib + skull | 400 | PR | 23.5 | 63.0 | AWD | skull | Surgery and TAE |
| L | |||||||||||
| rib | |||||||||||
| L | |||||||||||
| 15 | 56 | M | Small intestine | L | 600 | PR | 16.5 | 57.4 | AWD | L | Surgery and RFA |
| 16 | 46 | F | Small intestine | L + P | 400 | SD | 12.2 | 22.7 | DOD | P | Imatinib increase, surgery, then sunitinib |
| 17 | 49 | M | Stomach | L + P | 400 | PR | 24.6 | 40.7 | AWD | P | Surgery then sunitinib |
| P | |||||||||||
| 18 | 62 | M | Stomach | P + L | 400 | PR | 36.5 | 60.9 | AWD | Pl † | Surgery |
| P † | |||||||||||
| P | |||||||||||
| P | |||||||||||
| 19 | 71 | F | Small intestine | L + P | 400 | PR | 28.0 | 54.9 | AWD | P † | Imatinib increase, then surgery |
| P † | |||||||||||
| 20 | 49 | F | Small intestine | L + P | 200 | PR | 32.2 | 34.3 | AWD | P † | Eurgery |
| P † | |||||||||||
| 21 | 62 | M | Small intestine | L + P | 600 | PR | 23.4 | 54.7 | AWD | L † | Imatinib increase, then surgery |
| P † | |||||||||||
| 22 | 78 | M | Stomach | L + P + Lo | 400 | PR | 6.4 | 8.3 | AWD | Lo † | Surgery, then imatinib increase |
| P † | |||||||||||
| 23 | 55 | M | Small intestine | P | 600 | PR | 19.4 | 43.5 | DOD | P | Imatinib increase, then sunitinib |
| 24 | 49 | M | Small intestine | L + P | 300 | PR | 19.1 | 53.2 | AWD | L † | Surgery |
| P † | |||||||||||
| P | |||||||||||
| 25 | 70 | F | Small intestine | P | 300 | PR | 6.0 | 19.6 | AWD | P | Surgery |
Synchronous development of multiple resistant lesions. The others are metachronous. AWD, alive with disease; CR, complete response; DOD, dead of disease; F, female; L, liver; Lo, local; M, male; P, peritoneum; Pl, pleura; PR, partial response; RFA, radiofrequency ablation; SD, stable disease; TAE, transcatheter arterial chemoembolization.
Radiographic evaluation of resistance. Clinical responses were assessed by contrast‐enhanced multidetector CT scan. Target lesions were evaluated every 3 months. When necessary, 18F‐fluoro‐2‐deoxy‐d‐glucose‐positron emission tomography scan was carried out. Tumor responses and progression were assessed according to the Response to Treatment in Solid Tumors criteria as well as recently proposed criteria.( 7 ) Radiological appearance of resistance was classified into a nodule within a mass, enlargement or regrowth of a pre‐existing mass, or appearance of a new lesion. The secondary resistance in this study is defined as tumor progression under imatinib therapy for more than 180 days, and the primary resistance progression within 180 days.( 8 , 9 )
Histologic examinations. Pre‐imatinib, imatinib‐sensitive, and imatinib‐resistant samples were examined by hematoxylin–eosin (HE) staining and KIT as well as CD34 immunohistochemistry using paraffin‐embedded sections (3 µm) of formalin‐fixed tissues. Proliferative activity of the imatinib‐resistant lesions was evaluated by immunohistochemistry of Ki‐67 antigen. Immunohistochemistry was carried out using the EnVision+ kit horseradish peroxidase (3,3′‐diaminobenzidine‐tetrachloride) system (Dako Cytomation, Kyoto, Japan) with rabbit polyclonal antibody against human KIT (A4502; Dako Cytomation), mouse monoclonal antibody against human CD34 (QBend10; Novocastra Laboratories, Newcastle, UK), or mouse monoclonal antibody against human Ki‐67 antigen (MIB‐1, Dako Cytomation) as the primary antibodies as described previously.( 15 )
RNA extraction, reverse transcription–polymerase chain reaction (RT‐PCR) and sequence analysis. All the fresh samples were snap‐frozen in liquid nitrogen at the time of surgical resection or biopsy and were kept at –80°C until RNA extraction. Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Valencia, CA). cDNA was synthesized using reverse transcriptase (Superscript II; Gibco BRL, Grand Island, NY) and KIT or PDGFRA cDNA was amplified by RT‐PCR as previously described.( 16 ) Adequate cDNA for mutational analysis was obtained in 45 resistant tumor nodules from 25 patients. All the samples were tested including the known sites of KIT (exons 9, 11, 13, 14, 16, and 17) and PDGFRA (exons 12, 14, and 18) mutations. Sequencing was carried out as previously described.( 16 ) Fresh samples of primary lesions were only available in four cases. In the other 21 cases, genomic DNA was extracted from formalin‐fixed, paraffin‐embedded specimens of pre‐imatinib samples (10 µm thick) using DEXPAT (TaKaRa, Kyoto, Japan) and used for direct sequencing as described previously.( 15 ) When mutations were detected at two or more sites, amplified cDNA, including the mutational sites, was subcloned into pT7‐blue plasmid and sequencing of 10–20 independent cloned cDNAs was carried out to examine whether the mutations were present on the same allele.
The study was carried out under the authors’ institutional guidelines and written informed consent for the present analysis was obtained from all patients and their relatives.
Results
Clinical characteristics. The study included 25 patients with a median age of 61 years at the start of imatinib treatment (range: 32–78 years). Nine primary GISTs were located in the stomach, 15 in the small intestine, and one in the colon. Initial imatinib‐target lesions included 20 peritoneal disseminations, 19 liver metastases, four local recurrences, one pleural metastasis, and one bone metastasis (duplicated number) (Table 1). Patients received either 400 mg/day (20 patients) or 600 mg/day (five patients) of imatinib for more than 180 days during the therapy, although initial doses of imatinib varied from 200 to 600 mg/day. Best responses to imatinib included one complete response, 22 partial responses, and two stable disease.
Secondary resistance appeared in peritoneal lesions (29), hepatic metastases (10), local lesions (3), bone (2), or pleural metastasis (1) as shown in Table 1. Median progression‐free survival of patients in this study was 23.5 months (range: 6–50.9 months). In radiological examinations, secondary resistance mainly appeared as an enlargement of pre‐existing mass in 26 lesions of 15 patients, and 16 lesions of 10 patients showed a nodule in a mass appearance (Table 2). New lesions were observed in three patients. These three new lesions appeared at the same time as, or after the appearance of, the other patterns of secondary resistance.
Table 2.
Summary of mutations in imatinib‐resistant GIST
| No. | Timing | Radiological appearance | Viable tumor cells | KIT | Primary mutation | Secondary mutation | Allelic location | ||
|---|---|---|---|---|---|---|---|---|---|
| 1 | Enlargement | + | + | ex11 † | del 554–570 | ex13 | V654A | cis | |
| 2 | Nodule in a mass | + | + | ex11 † | del 557–558 | ex13 | V654A | ||
| 3 | Enlargement | + | + | ex11 † | del 557–558 | ex13 | V654A | ||
| 4 | Metachronous | Enlargement | + | + | ex11 † | del 556–557 | ex13 | V654A | cis |
| Metachronous | Enlargement | + | + | ex13 | V654A | ||||
| 5 | Synchronous | Enlargement | + | + | ex11 | del 557–561 | ex13 | V654A | |
| Synchronous | New lesion | + | + | ex13 | V654A | ||||
| 6 | Enlargement | + | + | ex11 † | del 552–572 | ex13 | V654A | ||
| 7 | Enlargement | + | + | ex11† | del 552–572 | ex13 | V654A | ||
| 8 | Metachronous | Nodule in a mass | + | + | ex11 | V560D | ex13 | V654A | cis |
| Metachronous | Nodule in a mass | + | + | Absent | |||||
| Metachronous | Nodule in a mass | + | + | Absent | |||||
| 9 | Metachronous | Nodule in a mass | + | + | ex11 † | del 557–558 | ex14 | T670I | cis |
| Metachronous | Nodule in a mass | + | + | ex14 | T670I | ||||
| Metachronous | New lesion | + | + | ex14 | T670I | ||||
| 10 | Enlargement | + | + | ex11 † | del 557 + 558 | ex16 + ex17 | K786N + D816H | all cis | |
| 11 | Enlargement | + | + | ex11 | del 560 | ex17 | N822K | cis | |
| 12 | Synchronous | Nodule in a mass | + | + | ex11 † | del 555–572 | ex17 | N822K | cis |
| Synchronous | Nodule in a mass | + | + | ex17 | N822Y | cis | |||
| 13 | Nodule in a mass | + | + | ex11 † | P551I and del 552–559 | ex17 | Y823D | ||
| 14 | Metachronous | Enlargement | + | + | ex9 † | dup 502 + 503 | ex17 | C809G | |
| Metachronous | Enlargement | + | + | ex17 | N822K | ||||
| Metachronous | Enlargement | + | + | ex17 | D816E | ||||
| Metachronous | New lesion | + | + | ex17 | N822K | cis | |||
| 15 | Nodule in a mass | + | + | ex11 † | del 560–572 | ex17 | A829P | ||
| 16 | Nodule in a mass | + | + | ex11 † | del 557–558 | ex17 | D816H | cis | |
| 17 | Metachronous | Nodule in a mass | + | + | ex11 † | del 557–558 | ex17 | Y823D | cis |
| Metachronous | Nodule in a mass | + | + | n.e. | n.e. | ||||
| 18 | Synchronous | Enlargement | + | + | ex11 † | del 557–558 | ex13 | V654A | cis |
| Synchronous | Enlargement | + | + | ex17 | D816E, D820V | each cis | |||
| Metachronous | Enlargement | + | + | ex13 | V654A | cis | |||
| Metachronous | Enlargement | + | + | ex13 | Y823D | cis | |||
| 19 | Synchronous | Enlargement | + | + | ex11 † | del 559–574 | ex17 | V654A | cis |
| Synchronous | Enlargement | + | + | ex13 | N822K | cis | |||
| 20 | Synchronous | Enlargement | + | + | ex11 † | del 557–561 | ex13 | V654A | |
| Synchronous | Enlargement | + | + | ex17 | N822D | ||||
| 21 | Synchronous | Enlargement | + | + | ex11 † | K558N del 559 | Absent | ||
| Synchronous | Enlargement | + | + | Absent | |||||
| 22 | Synchronous | Nodule in a mass | + | + | ex11 | V558S, V559S | Absent | ||
| Synchronous | Nodule in a mass | + | + | Absent | |||||
| 23 | Nodule in a mass | + | + | ex9 † | dup 502 + 503 | Absent | |||
| 24 | Synchronous | Enlargement | + | + | ex11 † | del 570–578 | Absent | ||
| Synchronous | Enlargement | + | + | Absent | |||||
| Metachronous | Enlargement | + | + | Absent | |||||
| 25 | Enlargement | + | + | ex9 † | dup 502 +503 | Absent | |||
Primary mutation was confirmed using genomic DNA extracted from formalin‐fixed, paraffin‐embedded sections. +, positive; del, deletion; dup, duplication; ex, exon; n.e., not examined.
Histologic examinations. Next, we examined the microscopic viability of tumor cells in specimens obtained after imatinib treatment including resistant and sensitive lesions. HE and KIT immunostaining showed regrowth of viable spindle and/or epithelioid tumor cells with significant mitotic figures in imatinib‐resistant lesions, whereas a few viable cells, some of which were KIT‐positive, were scattered in the background of hyaline degeneration in imatinib‐sensitive lesions (Fig. 1). In the former, tumor cells strongly and uniformly expressed KIT protein, and showed substantial numbers of mitotic figures and Ki‐67‐positive tumor cells, suggesting proliferating activities (Fig. 1). In the latter, KIT expression was attenuated. As shown in Table 2, all imatinib‐resistant lesions consisted of viable tumor cells in HE, in which KIT protein was strongly expressed as pre‐imatinib samples. Importantly, all resistant lesions strongly express KIT protein regardless of secondary mutations (Table 2).
Figure 1.
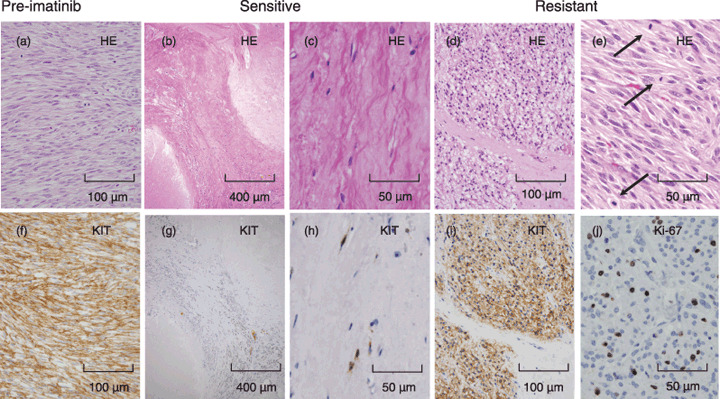
Pathological findings of pre‐imatinib, imatinib‐sensitive, and imatinib‐resistant lesions. Representatives of pre‐imatinib (a and f), imatinib‐sensitive (b, c, g, and h), and imatinib‐resistant (d, e, i and j) lesions are shown. Upper panels (a–e) show hematoxylin–eosin (HE) staining and lower panels show immunohistochemistry ([f–i], KIT; [j], Ki‐67 antigen). Arrows in (e) indicate mitotic figures. Imatinib‐sensitive lesions show a few KIT‐positive spindle cells in a background of hyaline degeneration. Imatinib‐resistant lesions show a lump of KIT‐positive spindle tumor cells that contains many mitotic figures and Ki‐67‐potitive cells.
Additional cis mutation in the kinase domains. We further examined the sequences of the KIT and PDGFRA genes of samples obtained before and after imatinib therapy (Table 2). The analysis using pre‐imatinib samples revealed that all tumors had mutations either in exon 9 (n = 3) or exon 11 (n = 22) of the KIT gene. No primary mutation was found in the PDGFRA gene. Forty‐five imatinib‐resistant tumors, obtained from 25 patients, were analyzed for full sequence of the KIT and PDGFRA genes. All mutations found in primary GISTs were identified in the KIT gene of corresponding resistant tumors. In addition to primary mutations, 33 out of 45 tumors (73%) showed additional (or secondary) KIT mutations in the kinase domain of the KIT gene. This corresponds to the fact that secondary mutation was found in 20 of 25 patients (80%) (Table 2). No additional mutation was found in the PDGFRA gene. Of interest, all secondary mutations detected were missense mutations, and all secondary mutations examined for allelic distribution were located on the same allele to their primary mutations (cis‐position), even two secondary hits occurred at the same time (Table 2, case no. 10). The other allele of the KIT gene was still wild‐type. No trans‐positioned secondary mutation was found. Secondary mutations were found either in kinase domain I or II of the KIT gene. In kinase domain I (exons 13 and 14), mutations are limited to codon 654 (Val to Ala; 14 lesions) and codon 670 (Thr to Ile; 3 lesions). In the kinase domain II (exon 16 and 17), various types of missense mutations have been found in several codons (809, 816, 820, 822, 823, 829, and 786+816; 1, 3, 1, 7, 3, 1, and 1 lesion, respectively).
Some resistant lesions showed different genotypes even in the same individuals (case no. 8, 12, 14, 18, 19, and 20), suggesting monoclonal development of resistance. Although radiographic findings (nodule in mass, enlargement, or new lesion) did not correlate with genotype, three new lesions found in case no. 5, 9, and 14 appeared simultaneously with, or after, the other patterns, and had the same mutations to resistant lesions previously or simultaneously found.
Eleven imatinib‐resistant lesions in six patients had viable tumor cells expressing KIT protein without secondary mutations. In these lesions, including case no. 21–25, resistant tumor cells carry a KIT gene with a primary mutation in one allele and a wild‐type KIT gene in the other allele. The presence or absence of secondary mutation as well as secondary mutation loci were independent to the primary location, best response, primary mutation type, radiographic appearance of resistance, resistant site, time from imatinib start to resistance, cell types (spindle or epithelioid), and the prognosis.
Discussion
Since the first successful case report in 2001,( 17 ) imatinib mesylate has been established as a first‐line therapy for unresectable, metastatic, and/or recurrent GIST.( 4 , 6 ) Although imatinib improves the prognosis of advanced GIST patients with a median progression‐free survival and overall survival of 2 and 5 years, respectively, resistance to imatinib eventually occurs in these patients.( 9 , 10 , 11 , 12 , 13 , 14 ) Efficacy of imatinib depends on the genotype of GIST, and primary resistant GIST includes GIST without mutations in the KIT and PDGFRA genes or GIST with resistant mutations in their kinase domain.( 18 , 19 ) Most GISTs with mutations in either exon 11 or 9 of the KIT gene are usually sensitive to imatinib, however, subsequent resistance to imatinib was reported during treatment.( 9 , 10 , 11 , 12 , 13 , 14 ) Although the mechanisms of secondary resistance to the drug have not been completely elucidated, postulated imatinib resistance mechanisms in GIST include: (i) target resistance, such as amplification of target gene or overexpression of target kinase, and mutations in target genes; (ii) functional resistance, including pharmacokinetic changes in imatinib and expression of drug‐resistant transport genes; and (iii) biological resistance with KIT‐independent progression sometimes associated with disappearance of KIT protein. The analysis of imatinib‐resistant mechanisms has been undertaken in chronic myeloid leukemia.( 20 , 21 ) In this disease, target resistance with target mutation or over‐expression of the target kinase appears to be a main cause.( 22 ) In GIST, recent reports have shown various degrees of secondary mutations in the kinase domain of the target genes concomitant with re‐activation of the corresponding kinase, even in the presence of imatinib.( 9 , 10 , 11 , 12 , 13 , 14 ) Frequency of secondary mutation in resistant lesions is 46–100%, although the numbers of reported cases are limited. In this study, 73% of resistant lesions had secondary mutations in the kinase domain of the KIT gene, in another way, 80% of patients with imatinib‐resistant GIST had GIST secondary mutations.
From our results and previous reports, secondary mutations occur to the kinase domain.( 9 , 10 , 11 , 12 , 13 , 14 ) KIT and PDGFRA are type III tyrosine kinases in which the kinase domain splits into domains I and II. The former is mainly composed of exons 13 and 14, which form the ATP‐binding domain, and the latter is composed of exons 16 and 17 forming an activation loop. Secondary mutations observed in the ATP‐binding domain are limited to V654A and T670I. These mutated loci are different from primary mutations in the KIT gene found in untreated primary GIST, that is, K642E, E643K, N655K. The substitution of these residues induces substantial modifications in the conformation of the kinase domain. In addition, V654 is indicated to interact with the diaminophenyl ring of imatinib( 23 ) and changes in this amino acid (V654A) resulted in decrease in the binding affinity. T670I, called the gate‐keeper mutation, also causes steric hindrance for imatinib binding.( 8 , 9 , 11 , 24 ) Mutations in the activation loop are rather variable compared to the ATP‐binding domain. Mutated codons include S709, D716, K786, C809, D816, D820, N822, Y823, and A829, a part of which is similar to mutated amino acids found in the primary GIST (D816, D820, and N822) and the others have not been seen in the primary GIST.( 8 , 9 ) Mutations in the activation loop might be considered to destabilize the inactive conformation by introducing charged side chains into the pocket.( 8 ) These secondary mutations are shown to confer resistance to imatinib, in vitro. ( 9 ) Interestingly, the preliminary in vitro study shows that secondary mutations in the ATP‐binding domain are sensitive to sunitinib malate, a new drug recently approved by the US Food and Drug Administration for imatinib‐resistant GIST, whereas secondary mutations in the activation loop are still resistant to sunitinib.( 25 )
Another interesting finding in this study is that, in all secondary mutations examined, their allelic distribution was detected on the same allele to the primary mutations. In the present study, we carried out RT‐PCR, subcloning, and full sequencing of the KIT and PDGFRA genes from fresh frozen samples of resistant lesions. All primary mutations, detected either by using frozen specimens and RT‐PCR or by paraffin‐embedded sections and PCR, were confirmed in secondary resistant lesions. No additional mutation was found in the PDGFRA gene. A previous preliminary report suggests that seven out of eight secondary mutations occurred in the same allele to the primary mutation (cis‐location) and one mutation was trans‐positioned.( 9 ) Our results are consistent with this result, however, reasons of mono‐allelic distribution of multiple mutations in resistant GIST have not been elucidated. In conjunction with these results, evolutional mutations are also clustered to the same allele.( 26 )
Resistant lesions are composed of tumor cells strongly re‐expressing KIT protein whereas sensitive lesions show few viable cells with faint KIT immunoreactivity. All resistant lesions examined in this study expressed KIT protein with or without secondary mutations. Variable KIT activation is also indicated both in resistant nodules with and without secondary mutations by measuring phospho‐KIT.( 8 , 9 ) In these studies, KIT amplification was not identified in any tumors. These results suggest that activation of KIT protein and the downstream signaling pathway might still play an important role in resistant GIST even if secondary mutations are absent, and that biological resistance of KIT‐independent progression with disappearance of KIT expression seems to be rare.
Imatinib resistance can be clinically diagnosed before any increase in size. Typical representations of resistance include a nodule in a mass, regrowth of pre‐existing lesion, and an appearance of a new lesion.( 7 ) In this study, new lesions did not appear independently, and are usually followed after the appearance of other patterns of secondary resistance. The genotype of new lesions that appeared in three patients was the same as that observed in the other resistant patterns of the patients (Table 2). In particular, new lesions in cases 9 and 14 emerged after previous lesions with other patterns of secondary resistance, which carry the same KIT mutations to corresponding new lesions. In case 5, a new resistant lesion in the liver coincided with local resistance, and both lesions carry the same KIT mutation. These results suggest that a new lesion could spread from the other secondary resistant lesions, and that secondary resistance might be clonal development in pre‐existing masses treated by imatinib.
In summary, we analyzed Japanese patients with GISTs who had acquired resistance to imatinib. Secondary cis‐mutations in the kinase domain were observed in about 75% of patients with resistant lesions or those with imatinib‐resistant GIST. Multiple resistant lesions show different mutations in the KIT gene, suggesting clonal development of resistance.
Acknowledgments
Supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- 1. Hitrota S, Isozaki K, Moriyama Y et al . Gain‐of‐function mutations of c‐kit in human gastrointestinal stromal tumors. Science 1998; 279: 577–80. [DOI] [PubMed] [Google Scholar]
- 2. Heinrich MC, Corless CL, Duencing A et al . PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003; 299: 708–10. [DOI] [PubMed] [Google Scholar]
- 3. Hirota S, Nishida T, Isozaki K et al . Gain‐of‐function mutation at the extracellular domain of KIT in gastrointestinal stromal tumours. J Pathol 2001; 193: 505–10. [DOI] [PubMed] [Google Scholar]
- 4. Rubin BP, Heinrich MC, Corless CL. Gastrointestinal stromal tumor. Lancet 2007; 369: 1731–41. [DOI] [PubMed] [Google Scholar]
- 5. Corless CL, Schroeder A, Griffith D et al . PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol 2005; 23: 5357–64. [DOI] [PubMed] [Google Scholar]
- 6. Demetri GD, Von Mehren M, Blanke CD et al . Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Eng J Med 2002; 347: 472–80. [DOI] [PubMed] [Google Scholar]
- 7. Shankar S, Van Sonnenberg E, Desai J et al . Gastrointestinal stromal tumor: new nodule‐within‐a mass pattern of recurrence after partial response to imatinib mesylate. Radiology 2005; 235: 892–8. [DOI] [PubMed] [Google Scholar]
- 8. Antonescu CA, Besmar P, Tianhua G et al . Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res 2005; 11: 4182–90. [DOI] [PubMed] [Google Scholar]
- 9. Heinrich MC, Corless CL, Blanke CD et al . Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol 2006; 24: 4764–74. [DOI] [PubMed] [Google Scholar]
- 10. Wakai T, Kanda T, Hirota S et al . Late resistance to imatinib therapy in a metastatic gastrointestinal stromal tumour is associated with a second KIT mutation. Br. J Cancer 2004; 90: 2059–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tamborini E, Bonadiman L, Greco A et al . A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology 2004; 127: 294–9. [DOI] [PubMed] [Google Scholar]
- 12. Chen LL, Trent JC, Wu EF et al . A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res 2004; 64: 5913–19. [DOI] [PubMed] [Google Scholar]
- 13. Debiec‐Rychter M, Cools J, Dumez H et al . Mechanism of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib‐resistant mutants. Gastroenterology 2005; 128: 270–9. [DOI] [PubMed] [Google Scholar]
- 14. Wardelmann E, Merkelbach‐Bruse S, Pauls K et al . Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res 2006; 12: 1743–9. [DOI] [PubMed] [Google Scholar]
- 15. Nishitani A, Hirota S, Nishida T et al . Different expression of connexin 43 in gastrointestinal stromal tumours between gastric and small intestinal origin. J Pathol 2005; 206: 377–82. [DOI] [PubMed] [Google Scholar]
- 16. Hirota S, Ohashi A, Nishida T et al . Gain‐of‐function mutations of platelet‐derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology 2003; 125: 660–7. [DOI] [PubMed] [Google Scholar]
- 17. Joensuu H, Roberts PJ, Sarlomo‐Rikala M et al . Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 2001; 344: 1052–6. [DOI] [PubMed] [Google Scholar]
- 18. Heinrich MC, Corless CL, Demetri GD et al . Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003; 21: 4342–9. [DOI] [PubMed] [Google Scholar]
- 19. Debiec‐Rychter M, Sciot R, Cesne AL et al . KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 2006; 42: 1093–103. [DOI] [PubMed] [Google Scholar]
- 20. Gorre ME, Mohammed M, Ellwood K et al . Clinical resistance to STI‐571 cancer therapy caused by BCR‐ABL gene mutation or amplification. Science 2001; 293: 876–80. [DOI] [PubMed] [Google Scholar]
- 21. Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition and STI‐571/imatinib resistance revealed by mutagenesis of BCR‐ABL . Cell 2003; 112: 831–43. [DOI] [PubMed] [Google Scholar]
- 22. Ohno R. Treatment of chronic myeloid leukemia with imatinib mesylate. Int J Clin Oncol 2006; 11: 176–83. [DOI] [PubMed] [Google Scholar]
- 23. McLean SR, Gana‐Weisz M, Hartzoulakis B et al . Imatinib binding and cKIT inhibition is abrogated by the cKIT kinase domain I missense mutation Val654Ala. Mol Cancer Ther 2005; 4: 2008–15. [DOI] [PubMed] [Google Scholar]
- 24. Tamborini E, Pricl S, Negri T et al . Functional analyses and molecular modeling of two c‐Kit mutations responsible for imatinib secondary resistance in GIST patients. Oncogene 2006; 25: 6140–6. [DOI] [PubMed] [Google Scholar]
- 25. Prenen H, Cools J, Mentens N et al . Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin Cancer Res 2006; 12: 2622–7. [DOI] [PubMed] [Google Scholar]
- 26. McGregor AP, Orgogozo V, Delon I et al . Morphological evolution through multiple cis‐regulatory mutations at a single gene. Nature 2007; 448: 587–90. [DOI] [PubMed] [Google Scholar]