

Abstract
We recently isolated a macrocyclic compound, versipelostatin (VST), that exerts in vivo antitumor activity. VST shows unique, selective cytotoxicity to glucose‐deprived tumor cells by preventing the unfolded protein response (UPR). Here we show that eukaryotic initiation factor 4E‐binding protein 1 (4E‐BP1), a negative regulator of eukaryotic initiation factor 4E‐mediated protein translation, plays a role in the UPR‐inhibitory action of VST. Indeed, 4E‐BP1 is aberrantly activated by VST. This activation occurs specifically during glucose deprivation and results in profound translation repression and prevents induction of the typical UPR markers glucose‐regulated protein (GRP) 78 and activating transcription factor (ATF) 4. Our overexpression and knockdown experiments showed that 4E‐BP1 can regulate GRP78 and ATF4 expression. These mechanisms appear to be specific for VST. By contrast, rapamycin, which activates 4E‐BP1 regardless of cellular glucose availability, has only marginal effects on the expression of GRP78 and ATF4. Our present findings demonstrate that aberrant 4E‐BP1 activation can contribute to UPR preventing by VST, possibly through a mechanism that does not operate in rapamycin‐treated cells. (Cancer Sci 2009; 100: 327–333)
Solid tumors have regions of low glucose and low oxygen (hypoxia) that arise from immature and irregular distribution of microvasculature.( 1 , 2 ) In this stressful microenvironment, tumor cells are thought to survive by activating adaptive response pathways.( 3 ) An important response for tumor development is the unfolded protein response (UPR), which can be activated in tumor cells during glucose deprivation as well as hypoxia.( 4 , 5 ) The UPR has also been associated with lowered chemosensitivity in breast cancer and gliomas.( 6 , 7 )
The UPR is a regulatory network that allows the cells to cope with stress that leads to the accumulation of misfolded or unfolded proteins in the endoplasmic reticulum (ER).( 8 ) The main signaling pathways are sensed and initiated by the ER‐localized transmembrane proteins Double‐standard RNA‐dependent Protein Kinase (PKR)‐like ER kinase (PERK), inositol‐requiring 1 (IRE1), and activating transcription factor (ATF) 6.( 8 ) These signaling pathways reduce global translation and produce several different active transcription factors to induce divergent UPR target genes, such as the ER‐resident molecular chaperones glucose‐regulated protein (GRP) 78 and GRP94.( 5 , 8 , 9 ) Thus, during the UPR, both translational and transcriptional control mechanisms operate to relieve ER stress to allow for cell survival.( 9 , 10 , 11 , 12 ) In the case of intolerable levels of ER stress, however, the sensor proteins can contribute to apoptosis.( 9 , 10 , 11 , 12 , 13 )
In the UPR translational control, PERK plays a major role by phosphorylating eukaryotic initiation factor (eIF) 2α at Ser51.( 12 , 14 ) Phosphorylation of eIF2α reduces global translation and, paradoxically, directs preferential translation of ATF4, a UPR transcription activator.( 15 ) The signaling pathway is further regulated by feedback eIF2α dephosphorylation that is mediated by ATF4‐directed Growth arrest and DNA damage (GADD)34 expression, thereby restoring translation for the UPR target transcripts.( 16 ) Both the PERK‐mediated translation repression and the subsequent GADD34‐mediated translation recovery have been shown to be important mechanisms that regulate UPR target gene expression and protect cells from ER stress.( 17 , 18 ) Thus, a delicately balanced translation control is required for cells to adapt to ER stress.
In general, translation control occurs mainly at the level of initiation for which eIF2α phosphorylation is a major regulatory mechanism. Translation initiation is also regulated at the point of eIF4F complex assembly, consisting of eIF4E, eIF4G, and eIF4A.( 19 ) Once assembled at the cap structure (7mGTP) of mRNA, the eIF4F complex recruits the 40S ribosomal subunit, GTP‐eIF2‐Met‐tRNA, and several other proteins for scanning toward the initiator AUG codon.( 20 ) In this system, formation of the GTP‐eIF2‐Met‐tRNA ternary complex is inhibited by eIF2α phosphorylation, whereas formation of the eIF4F complex is inhibited by eIF4E‐binding protein 1 (4E‐BP1).( 21 ) 4E‐BP1 is maintained in the inactive, hyperphosphorylated state by mammalian target of rapamycin (mTOR).( 22 ) When it is hypophosphorylated, 4E‐BP1 becomes activated and binds to eIF4E to inhibit eIF4F assembly by disturbing the association between eIF4E and eIF4G.( 20 , 23 )
Recently, the UPR has received considerable attention as a potential target for anticancer therapy.( 6 , 7 , 10 , 24 ) We have shown that versipelostatin (VST), a small molecule compound, can disrupt the UPR during glucose deprivation.( 25 ) Indeed, under that stressor, VST inhibits GRP78 and GRP94 induction and represses the production of the UPR transcription activators ATF4 and X‐box protein (XBP)1. VST shows highly selective cytotoxicity to glucose‐deprived tumor cells and exerts in vivo antitumor activity at well‐tolerated doses.( 25 ) However, it is not known how VST disrupts the UPR. It is interesting that VST induces profound repression of protein synthesis during glucose deprivation.( 25 ) From this observation, we have investigated the possibility that VST influences translation control mechanisms during the UPR. We demonstrate herein that 4E‐BP1 is activated by VST during glucose deprivation and can play an important role in the UPR inhibitory action of VST.
Materials and Methods
Cell culture and chemical treatment. Cells were maintained in either RPMI‐1640 (Wako Pure Chemical Industries, Osaka, Japan; HT1080 cells) or Dulbecco's Modified Eagle Medium (DMEM) (Wako Pure Chemical Industries; HeLa and 293T cells) supplemented with 10% fetal bovine serum and 100 µg/mL kanamycin and were cultured at 37°C in a humidified atmosphere of 5% CO2. To induce the UPR, cells were cultured for the indicated time periods in glucose‐containing medium in the presence of 10 mM 2‐deoxyglucose (2DG; Sigma, St Louis, MO, USA), 5 or 10 µg/mL tunicamycin (Nacalai Tesque, Kyoto, Japan), or 300 nM thapsigargin (Wako Pure Chemical Industries), or in glucose‐free medium (Sigma). To modulate the UPR, cells were treated with various concentrations of VST (1–10 µM) and of rapamycin (10–100 nM) (Sigma). These compounds were added to culture medium with the solvent being less than 0.5% of the medium's volume.
Plasmids and transfection. The 4E‐BP1 expression vector (pcHA‐4E‐BP1) was produced by inserting polymerase chain reaction‐amplified full‐length cDNA into pcHA, a derivative of pcDNA3 (Invitrogen, Carlsbad, CA, USA).( 26 ) The Δ4E‐BP1 expression vector (pcHA‐ Δ4E‐BP1) was produced by deleting the eIF4E binding site (amino acids 54–63) from the 4E‐BP1 expression vector using a QuikChange Site‐Directed Mutagenesis Kit (Stratagene, Cedar Creek, TX, USA).( 27 ) Plasmids were transiently transfected using FuGene6 Transfection Reagent (Roche Molecular Biochemicals, Indianapolis, IN, USA) for HT1080 cells or Lipofectamin 2000 (Invitrogen) for 293T cells, according to the manufacturers’ protocols.
RNA interference. Control short interfering RNA (siRNA) and Stealth siRNA against human 4E‐BP1 were purchased from Invitrogen. For knockdown analysis, HT1080 cells (0.5 × 105 cells/well in a 12‐well plate) were cultured overnight. The cells were transfected for 24 h with siRNA (40 pmol) using Lipofectamin RNAi MAX (Invitrogen) according to the manufacturer's protocol. After a further 24‐h incubation in fresh medium, the cells were used in the experiments.
Immunoblot analysis. Protein was extracted from cells as described previously.( 28 ) Equal amounts of lysate were electrophoresed in 4/20 or 10/20 sodium dodecylsulfate (SDS)–polyacrylamide gels (Daiichi Pure Chemicals, Tokyo, Japan) and transferred by electroblotting to a nitrocellulose membrane (Whatman, Springfield Mill, UK). Membranes were probed with the following antibodies: anti‐eIF2α (ABcam, Cambridge, MA, USA), antiphospho‐eIF2α, anti‐4EBP1, antiphospho‐4EBP1 (Ser65) (174A9), antiphospho‐4EBP1 (Thr70), anti‐eIF4E (Cell Signaling Technology, Danvers, MA, USA), anti‐KDEL (Stressgen, Victoria, BC, Canada), anti‐ATF4 (Santa Cruz Biotechnologies, Santa Cruz, CA, USA), and anti‐β‐actin (Sigma). The specific signals were detected using an enhanced chemiluminescence detection system (GE Healthcare Bio‐Sciences, Piscataway, NJ, USA).
Luciferase reporter assay. HT1080 cells (2 × 105 cells/well in a 12‐well plate) were cultured overnight under normal growth conditions. The cells were transfected with 200 ng empty (mock), pcHA‐4E‐BP1, or pcHA‐ Δ4E‐BP1 plasmid and 800 ng of the firefly luciferase reporter plasmid pGRP78pro160‐Luc or pGL3‐control together with 1.6 ng plasmid pRL‐CMV (Promega, Madison, WI, USA) (in which Renilla luciferase expression is under the control of the herpes simplex virus thymidine kinase promoter) as an internal control for 6 h. The medium was then replaced with fresh medium, and the cells were incubated under the same conditions for another 2 h. After passage into 96‐well plates, the cells were then treated for 18 h with UPR inducers. Relative firefly‐to‐Renilla luciferase activity was determined using the dual luciferase kit (Promega).( 25 , 28 )
7mGTP affinity purification. HeLa cells were lysed in lysis buffer (50 mM Tris‐HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% Triton X‐100, phosphatase inhibitor cocktails 1 and 2 [Sigma], and protease inhibitor [Sigma]) at 4°C for 30 min. Then, 500 µg of the cell extracts were incubated with 7mGTP‐conjugated sepharose beads (GE Healthcare Bio‐Sciences) in the lysis buffer at 4°C for 2 h.( 29 ) After three washings in the lysis buffer, the beads were boiled with 2× SDS sample loading buffer at 100°C. Each sample was analyzed by immunoblotting.
Measurement of protein synthesis. The rate of protein synthesis was assayed by measuring the incorporation of [35S]methionine/cysteine into HT1080 cells. Cells were treated with VST and UPR inducers for 2 h. After changing the culture medium to DMEM (plus 4.5 g/L d‐glucose, minus l‐glutamine, minus sodium pyruvate, minus l‐methionine, and minus l‐cysteine) (Sigma) supplemented with 10% fetal bovine serum, l‐glutamine, and 1% sodium pyruvate (Sigma), the cells were labeled with 1.85 MBq NEG‐772 Easy Tag Express (Perkin Elmer Life Sciences, Shelton, CT, USA) for 2 h. The cells were lysed in 1× SDS lysis buffer, and equal amounts of lysate were electrophoresed in a 4/20 SDS–polyacrylamide gel (Daiichi Pure Chemicals). After gel drying, the incorporated [35S]methionine/cysteine was visualized with Typhoon9410 (GE Healthcare).
Cell viability assay. HeLa cells were treated with VST or rapamycin in the presence or absence of 2DG for 18 h. The medium was then replaced with fresh growth medium, and cells were cultured for a further 15 h. 3‐(4,5‐dimethylthiazol‐2‐y1)‐2,5‐diphenyltetrazolium bromide (MTT) (Sigma) was then added to the culture medium. After 3 h, the absorbance of each well was determined as described previously.( 30 ) Relative cell survival was calculated by setting each control absorbance from untreated cells as 100%.
Results
Hypophosphorylation of 4E‐BP1 induced by VST. We initially carried out immunoblotting analysis to determine the eIF2α phosphorylation status. For this purpose, we treated HT1080 cells for 4 or 8 h with the hypoglycemia‐mimicking agent 2DG in the presence or absence of VST. Fig. 1(a) shows enhanced phosphorylation of eIF2α was seen in 2DG‐stressed HT1080 cells, regardless of whether VST is present or absent. In contrast, the same treatment with VST clearly suppressed 2DG‐induced GRP78 expression. Thus, VST had no effect on eIF2α phosphorylation status even at the dose that prevented GRP78 induction. Instead, we found that VST induced hypophosphorylation of 4E‐BP1 within 4 h (Fig. 1b) and in a dose‐dependent manner in 2DG‐stressed cells (Fig. 1c). The 4E‐BP1 hypophosphorylation was easily detected both by band shifts to lower molecular weights with the anti‐4E‐BP1 antibody and by signal decrease with each phophospecific anti‐4E‐BP1 antibody at Ser65 and Thr70.
Figure 1.
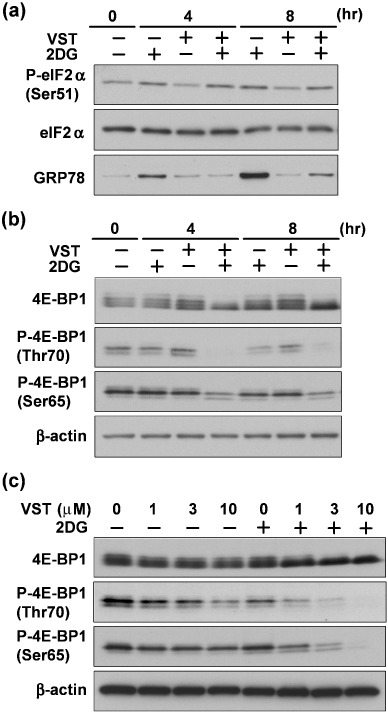
Effect of versipelostatin (VST) on translation initiation regulatory proteins. Immunoblotting analysis of eukaryotic initiation factor (eIF) 2α, phosphorylated eIF2α (P‐eIF2α), 4E‐BP1, phosphorylated 4E‐BP1 (P‐4E‐BP1) and GRP78. (a,b) HT1080 cells were treated with VST (3 µM) in the presence (+) or absence (–) of 2‐deoxyglucose (2DG) (10 mM) for the indicated times. (c) HT1080 cells were treated with the indicated concentrations of VST in the presence (+) or absence (–) of 2DG (10 mM) for 4 h.
Hypophosphorylation of 4E‐BP1 and prevention of GRP78 induction. To examine whether VST induces 4E‐BP1 hypophosphorylation under different types of ER stress, we treated HT1080 cells for 8 h with VST, together with 2DG, the ER Ca2+ pump inhibitor thapsigargin, or the N‐glycosylation inhibitor tunicamycin (Fig. 2a). As shown previously,( 25 ) VST prevented GRP78 induction in 2DG‐stressed cells but not in thapsigargin‐ or tunicamycin‐stressed cells, in spite of the observation that these chemical stressors induced GRP78 almost equally. Likewise, VST markedly induced 4E‐BP1 hypophosphorylation in the 2DG‐stressed cells. Effects similar to those of VST on 4E‐BP1 and GRP78 were also observed in HT1080 cells subjected to glucose withdrawal for 24 h and in HeLa cells stressed for 6 h with 2DG (Fig. 2b,c). Interestingly, the relatively long exposures (24 h) of HT1080 cells revealed that 4E‐BP1 hypophosphorylation was induced even by VST alone and by glucose withdrawal alone although it was much more profoundly induced by the combination (Fig. 2b).
Figure 2.
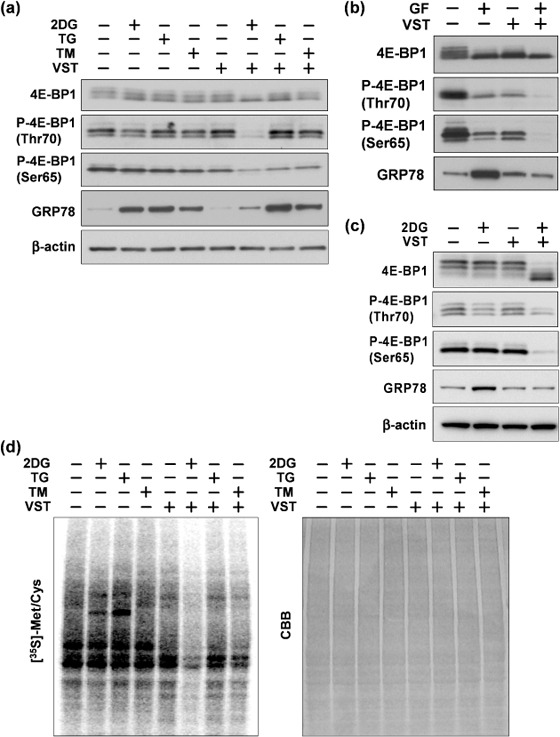
Effect of versipelostatin (VST) on 4E‐BP1 under different endoplasmic reticulum stress conditions. Immunoblotting analysis of 4E‐BP1, phosphorylated 4E‐BP1 (P‐4E‐BP1) and glucose‐regulated protein (GRP) 78. (a) HT1080 cells were treated with VST (3 µM) in the presence (+) or absence (–) of 2‐deoxyglucose (2DG) (10 mM), thapsigargin (TG) (300 nM), or tunicamycin (TM) (10 µg/mL) for 8 h. (b) HT1080 cells were treated with VST (3 µM) under glucose‐free (GF+) or normal (GF–) conditions for 24 h. (c) HeLa cells were treated with VST (3 µM) in the presence (+) or absence (–) of 2DG (10 mM) for 6 h. (d) Protein synthesis rates measured by the incorporation of [35S]methionine/cysteine into proteins during a 2‐h labeling. HT1080 cells were treated with VST (3 µM) in the presence (+) or absence (–) of 2DG (10 mM), thapsigargin (300 nM) or tunicamycin (10 µg/mL) for 4 h. Left panel, autoradiography; right panel, Coomassie brilliant blue (CBB)‐stained gel.
By measuring 35S‐labeled methionine incorporation into proteins, we estimated global protein synthesis activity in HT1080 cells treated for 4 h with VST, together with 2DG, thapsigargin, or tunicamycin (Fig. 2d). Like 4E‐BP1 hypophosphorylation, a strong inhibition of global protein synthesis was seen in cells treated with the combination of 2DG and VST, but not with each stressor or the other combinations. Taken together, prevention of GRP78 induction by VST correlated well with 4E‐BP1 hypophosphorylation as well as profound protein synthesis repression.
Regulation of GRP78 and ATF4 expression by 4E‐BP1. We next examined whether 4E‐BP1 influences the GRP78 promoter using pGRP78‐Luc, which contained a GRP78 promoter region (–160–+7) immediately upstream of firefly luciferase.( 25 ) Reporter activity was determined by cotransfecting a control plasmid that contained a Renilla luciferase gene and by calculating the ratios of the two luciferase activities to normalize alteration in protein synthesis activity as well as transfection efficiency. The effects of 4E‐BP1 on the GRP78 promoter reporter was determined by cotransfection of expression plasmids that contained empty (mock), full‐length 4E‐BP1, and eIF4E‐binding domain‐deleted mutant Δ4E‐BP1 (Fig. 3). In mock transfection cells, the GRP78 promoter reporter activity was increased approximately sixfold by each of 2DG and tunicamycin. Promoter activation was completely suppressed by VST in 2DG‐stressed cells, but not tunicamycin‐stressed cells. In the reporter system, we found that cotransfection of 4E‐BP1, but not Δ4E‐BP1, significantly attenuated the GRP78 promoter activity regardless of culture conditions (Fig. 3). Compared with a decrease in the net activity, the stressor inducibility of the GRP78 promoter was retained in the 4E‐BP1‐transfected cells. Meanwhile, cotransfection of mock, 4E‐BP1, and Δ4E‐BP1, as well as any drug treatments examined, had marginal effects on a control luciferase reporter that was driven by a simian virus 40 promoter and enhancer (data not shown).
Figure 3.

Effect of 4E‐BP1 overexpression on glucose‐regulated protein (GRP) 78 promoter. HT1080 cells were transfected with empty vector (mock), pcHA‐4E‐BP1, or pcHA‐Δ4E‐BP1, together with pGRP78‐Luc (firefly luciferase) and pRL‐CMV (Renilla luciferase). The transfected cells were treated with versipelostatin (VST) (3 µM) in the presence (+) or absence (–) of 2‐deoxyglucose (2DG) (10 mM) and tunicamycin (TM) (10 µg/mL) for 18 h. Relative firefly‐to‐Renilla luciferase activity was determined. Data shown are mean values and standard deviations (bar) of quadruplicate samples. Right panel, immunoblotting analysis of Hemagglutinin (HA)‐tagged 4E‐BP1 proteins (4E‐BP1 and Δ4E‐BP1) in HT1080 cells that were transfected with pcHA‐4E‐BP1 and pcHA‐ Δ4E‐BP1, respectively. TM, tunicamycin.
Immunoblotting analysis of lysates from 293T cells revealed that 4E‐BP1 overexpression significantly attenuated endogenous GRP78 and ATF4 induction under 2DG stress conditions (Fig. 4a). In contrast to the above‐mentioned reporter assays, 4E‐BP1 overexpression did not affect the basal expression levels of GRP78 and ATF4. We also found that siRNA‐mediated silencing of 4E‐BP1 in HT1080 cells, compared with non‐silenced cells, enhanced the production of endogenous GRP78 and ATF4 under 2DG stress conditions (Fig. 4b). Essentially the same results were obtained using two independent siRNA targeting 4E‐BP1 (Fig. 4b; data not shown). In the 4E‐BP1‐silenced cells, VST was still able to suppress 2DG‐induced GRP78 and ATF4, but close examination of the data suggested that the inhibitory activity was somewhat weakened, as seen with VST at 1 µM being less effective in 4E‐BP1‐silenced cells than in non‐silenced cells.
Figure 4.
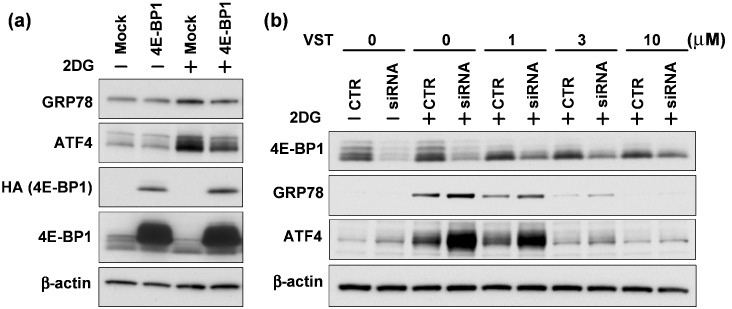
Regulation of glucose‐regulated protein (GRP) 78 and activating transcription factor (ATF) 4 expression by 4E‐BP1. Immunoblotting analysis of 4E‐BP1, Hemagglutinin (HA) (exogenous 4E‐BP1), GRP78, and ATF4. (a) 293T cells were transfected with empty vector (mock) or 4E‐BP1 and treated with 2‐deoxyglucose (2DG) (10 mM) for 4 h. (b) Control short interfering RNA (siRNA) (CTR) or siRNA against 4E‐BP1 (siRNA) were transfected into HT1080 cells. The cells were treated with 2DG (10 mM) and the indicated concentrations of versipelostatin (VST) for 18 h.
Differences between the effects of VST and rapamycin on stress response. We next compared the actions of VST and the mTOR inhibitor rapamycin.( 22 ) Immunoblotting analysis of lysates from HT1080 cells revealed that a 4‐h treatment with 100 nM rapamycin led to hypophosphorylation of 4E‐BP1 under both non‐stress and 2DG‐stress conditions (Fig. 5a). Thus, unlike VST, rapamycin induced 4E‐BP1 hypophosphorylation regardless of cell culture conditions (Fig. 5a). Under 2DG‐stress conditions, VST tended to induce 4E‐BP1 hypophosphorylation more profoundly than rapamycin. Essentially the same results were obtained in HeLa cells (Fig. 5b). Consistent with the 4E‐BP1 hypophosphorylation levels, more 4E‐BP1 proteins were coprecipitated with eIF4E in an mRNA cap structure 7mGTP‐binding assay using VST‐ and 2DG‐treated cells compared with rapamycin‐treated cells (Fig. 5b). Conversely, fewer eIF4G proteins were found in the eIF4E‐containing initiation complex of the VST‐treated, 2DG‐stressed cells than in the rapamycin‐treated cells (Fig. 5b).
Figure 5.
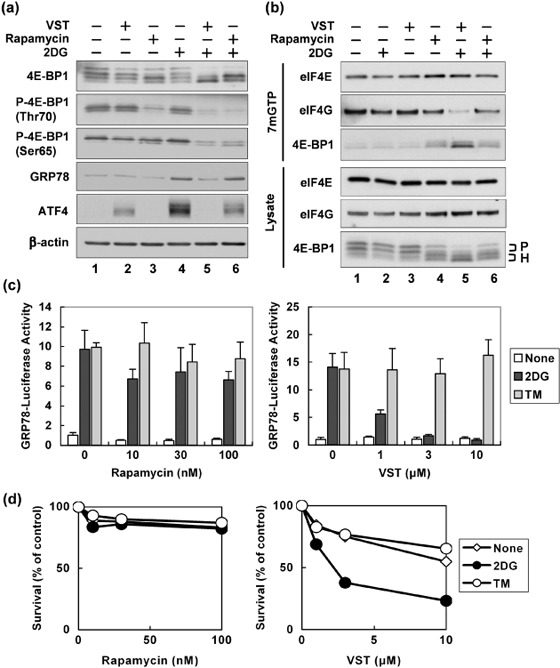
Comparison of versipelostatin (VST) and rapamycin. (a) Immunoblotting analysis of 4E‐BP1, phosphorylated 4E‐BP1 (P‐4E‐BP1), activating transcription factor (ATF) 4, and glucose‐regulated protein (GRP) 78. HT1080 cells were treated with VST (3 µM) or rapamycin (100 nM) in the presence (+) or absence (–) of 2‐deoxyglucose (2DG) (10 mM) for 4 h. (b) Immunoblotting analysis of eukaryotic initiation factor (eIF) 4E, eIF4G, 4E‐BP1, phosphorylated 4E‐BP1 (P‐4E‐BP1), ATF4, and GRP78. HeLa cells were treated with VST (3 µM) or rapamycin (100 nM) in the presence (+) or absence (–) of 2DG (10 mM) for 4 h. The cell lysates were affinity purified with 7mGTP‐conjugated sepharose beads. The purified samples and the corresponding lysates underwent immunoblotting analysis. (c) HT1080 cells were transfected with pGRP78‐Luc together with pRL‐CMV and treated with the indicated concentrations of VST (right panel) or rapamycin (left panel) in the presence (+) or absence (–) of 2DG (10 mM) and tunicamycin (TM) (5 µg/mL) for 18 h. Relative firefly‐to‐Renilla luciferase activity was determined. Data shown are mean values and standard deviations of triplicate samples. (d) assay of HeLa cells treated with the indicated concentrations of VST (right panel) or rapamycin (left panel) in the presence (+) or absence (–) of 2DG (10 mM) and tunicamycin (5 µg/mL) for 18 h. Data shown are mean values and standard deviations of triplicate samples. H, hypophosphoryalted 4E‐BP1; P, phosphorylated 4E‐BP1; TM, tunicamycin.
We also found that rapamycin had little effect on 2DG‐induced expression of ATF4 and GRP78 in HT1080 cells (Fig. 5a). In the absence of 2DG, VST paradoxically induced ATF4 protein accumulation, but rapamycin induced neither ATF4 nor GRP78. Rapamycin had marginal effects on GRP78 promoter reporter activity, in sharp contrast to VST, which suppressed the promoter activity induced by 2DG but not tunicamycin in a dose‐dependent manner (Fig. 5c). In agreement with these results, VST caused selective and massive killing of 2DG‐stressed HeLa cells whereas rapamycin did not (Fig. 5d).
Discussion
In the present study, we have shown that 4E‐BP1 can play an important role in the UPR inhibitory action of VST. Indeed, VST induced hypophosphorylation of 4E‐BP1, which was closely associated with suppression of ATF4, UPR transcriptional activator, and GRP78 expression (1, 2, 5). It is important to note that overexpression of 4E‐BP1 attenuated ATF4 production, whereas knockdown of 4E‐BP1 enhanced ATF4 and GRP78 expression during glucose deprivation (Fig. 4). Thus, 4E‐BP1 has the ability to regulate the UPR. Furthermore, this activity of 4E‐BP1 appears to depend on its translation repressor activity because Δ4E‐BP1, which lacks the eIF4E binding site, loses UPR‐regulating activity (Fig. 3).
It is likely that VST‐induced 4E‐BP1 activation disrupts the translation control mechanisms that are regulated by the PERK–eIF2α signaling pathway. This pathway contains two important regulatory steps: PERK‐mediated eIF2α phosphorylation for translation repression, and ATF4‐directed GADD34 expression for translation recovery.( 8 ) Although VST had no effect on eIF2α phosphorylation (Fig. 1), cells treated with VST during glucose deprivation have features similar to cells that lack functional GADD34.( 17 , 18 ) Indeed, GADD34‐mutated cells have reduced survival in response to ER stress and show persistent repression of protein synthesis and impaired induction of ATF4 and GRP78.( 17 , 18 ) The consistent observations between GADD34‐mutated and VST‐treated cells, together with the present findings of UPR‐inhibitory activity of 4E‐BP1, suggest that VST‐induced 4E‐BP1 activation may affect the translational recovery process of the PERK–eIF2α pathway.
Recently, 4E‐BP1 has been shown to be a direct target gene of ATF4 and to be induced during the UPR of murine pancreatic β cells.( 31 ) Although induction of 4E‐BP1 expression did not occur in the human tumor cell lines we used here, activating hypophosphorylation of 4E‐BP1 occurred during prolonged exposure to glucose withdrawal as well as to the chemical stressors 2DG, thapsigargin, and tunicamycin (Fig. 2b; data not shown). Furthermore, prolonged hypoxia also induces hypophosphorylation of 4E‐BP1.( 29 , 32 ) These observations indicate that translational control during the UPR can be a biphasic mechanism involving the PERK–eIF2α pathway, mainly at the early stage, and the 4E‐BP1‐mediated pathway, mainly at the late stage. In this context, VST could cause activation of the translation inhibition late‐stage mechanism during the early phase of response to glucose deprivation, thereby disrupting the UPR translation control. Thus, future studies on the VST action would be helpful to explain the complex mechanisms of translational control during the UPR.
We also showed that 4E‐BP1 activation by VST during glucose deprivation is not equivalent to that seen in rapamycin‐treated cells. Not limited to 4E‐BP1, VST and rapamycin also affected ribosomal protein S6 phosphorylation in essentially the same manner (data not shown). VST too can cause 4E‐BP1 hypophosphorylation, even under normal growth conditions, although longer exposure periods are required (Fig. 2b). Thus, the actions of VST, especially during glucose deprivation, overlap considerably with those of rapamycin. Nevertheless, the effect of rapamycin on UPR activation was marginal. As a possible explanation of the difference between VST and rapamycin, we found a tendency during glucose deprivation for VST to cause 4E‐BP1 hypophosphorylation more profoundly than rapamycin does (Fig. 5). This tendency was also confirmed by a functional 7mGTP‐binding assay. Another simple explanation is also possible, in addition to 4E‐BP1 activation: each compound has other different effects on glucose‐deprived cells, which result in different effects on the UPR. In this context, it is noteworthy that VST and rapamycin also showed different activities regarding ATF4 induction under normal growth conditions (Fig. 5a), although the mechanisms behind this difference are unclear at present.
In summary, we have shown that 4E‐BP1 activation plays an important role in VST mechanisms that prevent the UPR during glucose deprivation. Our findings also demonstrated that under glucose deprivation conditions, VST exerts many activities similar to those of rapamycin. Recently, CCI‐779 (temsirolims), a rapamycin analog, was approved for treatment of renal cell carcinoma in the USA, and another mTOR inhibitor, RAD001, has shown promise.( 33 , 34 , 35 ) Therefore, VST may be interesting not only as an antitumor UPR inhibitor but also as a different type of antitumor agent that modulates the mTOR signaling pathway.
Acknowledgments
This work was supported in part by a Grant‐in‐Aid for Cancer Research (15‐2) from the Ministry of Health, Labor, and Welfare (to A.T.), and a Grant‐in‐Aid for scientific research on priority areas for cancer from the Ministry of Education, Culture, Sport, Science, and Technology of Japan (to T.T.).
References
- 1. Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res 1998; 58: 1408–16. [PubMed] [Google Scholar]
- 2. Scrivaen P, Brown NJ, Pockley AG, Wyld L. The unfolded protein response and cancer: a brighter future unfolding? J Mol Med 2007; 85: 331–41. [DOI] [PubMed] [Google Scholar]
- 3. Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Curr Opin Cell Biol 2006; 18: 1–9. [DOI] [PubMed] [Google Scholar]
- 4. Ma Y, Hendershot LM. The role of the unfolded protein response in tumor development: friend or foe? Nat Rev Cancer 2004; 4: 966–77. [DOI] [PubMed] [Google Scholar]
- 5. Lee AS. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res 2007; 67: 3496–9. [DOI] [PubMed] [Google Scholar]
- 6. Lee E, Nichols P, Spicer D, Groshen S, Yu MC, Lee AS. GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res 2007; 66: 7849–53. [DOI] [PubMed] [Google Scholar]
- 7. Pyrko P, Schonthal AH, Hofman FM, Chen TC, Lee AS. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res 2007; 67: 9809–16. [DOI] [PubMed] [Google Scholar]
- 8. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8: 519–29. [DOI] [PubMed] [Google Scholar]
- 9. Marciniak SJ, Ron D. Endoplasmic reticulum stress signalling in disease. Physiol Rev 2006; 86: 1133–49. [DOI] [PubMed] [Google Scholar]
- 10. Dong D, Ni M, Li J et al . Critical role in the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene‐induced mammary tumor development. Cancer Res 2008; 68: 498–505. [DOI] [PubMed] [Google Scholar]
- 11. Bi M, Naczki C, Koritzinsky M et al . ER stress‐regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J 2005; 24: 3470–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 2000; 5: 897–04. [DOI] [PubMed] [Google Scholar]
- 13. Marciniak SJ, Yun CY, Oyadomari S et al . CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 2004; 18: 3066–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic reticulum‐resident kinase. Nature 1999; 397: 271–4. [DOI] [PubMed] [Google Scholar]
- 15. Harding HP, Novoa I, Zhang Y et al . Regulated translation initiation controls stress‐induced gene expression in mammalian cells. Mol Cell 2000; 6: 1099–08. [DOI] [PubMed] [Google Scholar]
- 16. Ma Y, Hendershot LM. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J Biol Chem 2003; 278: 34 864–73. [DOI] [PubMed] [Google Scholar]
- 17. Kojima E, Takeuchi A, Haneda M et al . The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress‐elucidation by GADD34‐deficient mice. FASEB J 2003; 17: 1573–5. [DOI] [PubMed] [Google Scholar]
- 18. Novoa I, Zhang Y, Zeng H, Jungreis R, Harding HP, Ron D. Stress‐induce gene expression requires programmed recovery from translational repression. EMBO J 2003; 22: 1180–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Graff JR, Konicek BW, Carter JH, Marcusson EG. Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer Res 2008; 68: 631–4. [DOI] [PubMed] [Google Scholar]
- 20. Polunovsky VA, Bitterman PB. The cap‐dependent translation apparatus integrates and amplifies cancer pathway. RNA Biol 2006; 3: 10–17. [DOI] [PubMed] [Google Scholar]
- 21. Dever TE. Gene‐specific regulation by general translation factors. Cell 2002; 108: 545–56. [DOI] [PubMed] [Google Scholar]
- 22. Gingras A, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev 2001; 15: 807–26. [DOI] [PubMed] [Google Scholar]
- 23. Gingras A, Gygi SP, Raught B et al . Regulation of 4E‐BP1 phosphorylation: a novel two‐step mechanism. Genes Dev 1999; 13: 1422–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fels DR, Koumenis C. The PERK/eIF2α/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol Ther 2006; 5: 723–8. [DOI] [PubMed] [Google Scholar]
- 25. Park H, Tomida A, Sato S et al . Effect on tumor cells of blocking survival response to glucose deprivation. J Natl Cancer Inst 2004; 96: 1300–10. [DOI] [PubMed] [Google Scholar]
- 26. Horie K, Tomida A, Sugimoto Y et al . SUMO‐1 conjugation to intact DNA topoisomerase I amplifies cleavable complex formation induced by camptothecin. Oncogene 2002; 21: 7913–22. [DOI] [PubMed] [Google Scholar]
- 27. Polunovsky VA, Gingras A, Sonenberg N et al . Translational control of the antiapoptotic function of Ras. J Biol Chem 2000; 275: 24 776–80. [DOI] [PubMed] [Google Scholar]
- 28. Tsukumo Y, Akihiro T, Kitahara O. Nucleobindin 1 controls the unfolded protein response by inhibiting ATF6 activation. J Biol Chem 2007; 282: 29 264–72. [DOI] [PubMed] [Google Scholar]
- 29. Arsham AM, Howell JJ, Simon MC. A novel hypoxia‐inducible factor‐independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem 2003; 32: 29 655–60. [DOI] [PubMed] [Google Scholar]
- 30. Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Meth 1986; 89: 271–7. [DOI] [PubMed] [Google Scholar]
- 31. Yamaguchi Y, Ishihara H, Yamada T et al . ATF4‐mediated induction of 4E‐BP1 contributes to pancreatic β cell survival under endoplasmic reticulum stress. Cell Metab 2008; 7: 269–76. [DOI] [PubMed] [Google Scholar]
- 32. Koritzinsky M, Magagnin MG, Beucken T et al . Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J 2006; 25: 1114–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hudes G, Carducci M, Tomczak P et al . Temsirolimus, interferon alfa, or both for advanced renal‐cell carcinoma. N Engl J Med 2007; 356: 2271–81. [DOI] [PubMed] [Google Scholar]
- 34. Motzer R, Escudier B, Oudard S et al . Efficiency of everolimus in advanced renal cell carcinoma: a double‐blind, randomized, placebo‐controlled phase III trial. Lancet 2008; 372: 449–56. [DOI] [PubMed] [Google Scholar]
- 35. Gridelli C, Maione P, Rossi A. The potential role of mTOR inhibitors in non‐small cell lung cancer. Oncologist 2007; 13: 139–47. [DOI] [PubMed] [Google Scholar]