

Abstract
It is reported that the agonistic antibodies against death receptors 4 and 5 (DR4, DR5) are cytotoxic to various cancer cells. In the present study, the sensitivity of five human lung cancer cell lines to previously reported AD5‐10 agonistic antibody against DR5 were investigated. Of these cell lines, A549 and small cell lung cancer showed a moderate sensitivity to AD5‐10 and three other cell lines were resistant. Cell line H460 is resistant to AD5‐10 despite a high level of cell‐surface DR5 expression. We demonstrated that the resistance of H460 cells to AD5‐10 was not related to the expression level of DR5, but the expression and cleavage of c‐FLIPL in the cells. Inhibition of endogenous c‐FLIPL expression by siRNA significantly enhanced AD5‐10‐induced cell death in these lung cancer cells. We further showed that this sensitizing effect was associated with decreased expression of Bcl‐2 family proteins Bid and Bcl‐XL, change of mitochondrial membrane potential, release of cytochrome c from mitochondria, and caspase activation. Therefore, these data provide evidence that c‐FLIPL is involved in the resistance of lung cancer cells to AD5‐10‐induced apoptosis. Moreover, immunohistochemistry on paraffin‐embedded tissue revealed that c‐FLIPL was expressed in 87.9% (29 of 33) of lung carcinoma tissues from the patients, but little in tissues from normal controls. This suggests that inhibition of c‐FLIPL expression might be a potential strategy for lung cancer therapy, especially for those lung cancers resistant to the agonistic antibody against death receptors. (Cancer Sci 2009; 100: 940–947)
Lung cancer is the leading cause of cancer‐related mortality in the world.( 1 ) Non‐small cell lung cancers (NSCLCs) account for about 75–80% of all types of lung cancers. Surgery offers substantial cure rates for those patients with NSCLC with early stage disease (stages I and II). However, 70% of lung cancer patients present at stage III/IV, and most of these patients continue to die of disease progression because of resistance to chemotherapeutic drugs or radiation therapy.( 2 ) Clearly, novel therapeutic strategies are necessary to improve lung cancer therapy.
Tumor necrosis factor (TNF)‐related apoptosis‐inducing ligand (TRAIL) is a member of the structurally related TNF family. To date, five TRAIL receptors have been identified: TRAIL‐R1 (DR4), TRAIL‐R2 (DR5), TRAIL‐R3 (DcR1, TRID), TRAIL‐R4 (DcR2, TRUNDD) and osteoprotegerin. Among these receptors, death receptors 4 and 5 (DR5 and DR4) are capable of recruiting adaptor proteins to trigger downstream caspase activation upon binding with TRAIL ligand.( 3 ) DR5‐mediated apoptosis is involved in the formation of a Death inducing signaling comp (DISC) consisting of Fas associated death domain protein (FADD), caspase‐8 and/or caspase‐10 to initiate a downstream caspase cascade and degradation of death substrates. In some cells, activation of the intrinsic pathway via Bcl‐2 family protein (Bid) cleavage occurs; this is required for apoptosis, in which Bid is cleaved by activated caspase‐8 and caspase‐10, and the truncated Bid promotes the release of cytochrome c and the second mitochondrial‐derived activator of caspase (Smac) from the mitochondria. The released cytochrome c binds to Apoptosis protease activating factor 1 (Apaf1) and pro‐caspase‐9 to activate caspase‐9 and subsequent effector caspases. It has been proved that both TRAIL ligand and agonistic antibodies against DR5 or DR4 are potential biological agents for cancer therapy in various tumor xenograft models and human clinical trials.( 4 , 5 ) However, some reports showed that certain versions of recombinant TRAIL may induce cell death in normal cells, for example, hepatocytes.( 6 , 7 ) As an alternative to TRAIL, agonistic antibodies to DR5 or DR4 are currently shouldering the hopes for the next generation of TNF‐related cancer therapeutics.( 8 )
We previously reported that a novel DR5 specific monoclonal antibody, AD5‐10, possessed a strong tumoricidal activity in various tumor cell lines in the absence of second cross‐linking in vitro and in vivo. Particularly, AD5‐10 does not compete with TRAIL when it binds with DR5 and induces cell death in caspase‐dependent and ‐independent manners.( 9 ) Moreover, the virally expressed scFv antibody fragment of AD5‐10 induced apoptosis in various human cancer cells and prevented tumor growth in mice.( 10 ) Therefore, AD5‐10 is one of most promising candidates for cancer therapy. However, some tumor cell lines are resistant to AD5‐10‐induced apoptosis despite high‐level expression of DR5 in these cells, and the molecular mechanism is not well understood.
Cellular Fas‐associated death domain‐like interleukin‐1 beta‐converting enzyme (FLICE) inhibitory protein (c‐FLIP) has been identified as a key inhibitor of TRAIL‐induced apoptotic signals, which has a high level of homology to caspase‐8 and caspase‐10, but without protease activity. Therefore, c‐FLIP interferes with the binding of caspase‐8 to FADD in the DISC, and thereby inhibits subsequent activation of the caspase cascade.( 11 ) Whether the resistant mechanism of TRAIL could be applied to the agonistic antibodies against DR4 or DR5 remains unknown. The c‐FLIP protein is expressed as three forms: c‐FLIPL, c‐FLIPS and c‐FLIPR.( 12 ) Studies by generation of c‐FLIP knockout mice showed the anticell death function against Fas ligand (FasL)‐ and TNF‐induced apoptosis and important roles in embryonic heart development.( 13 ) In addition, c‐FLIP is also reported as an important regulation factor in the life and death of T cells. Several studies indicated that c‐FLIP not only could interfere with the extrinsic apoptosis pathway, but also activates NF‐κB by recruiting Tumor necrosis factor receptor‐associated factor 2 (TRAF‐2) and receptor‐interacting protein‐1 (RIP1) into the DISC, therefore promoting cell survival and proliferation.( 14 , 15 ) However, controversial antiapoptotic( 11 ) and pro‐apoptotic( 16 ) effects of c‐FLIP have been reported.
In this study, we try to investigate the sensitivity of several lung cancer cell lines to AD5‐10‐induced apoptosis and evaluate the roles of cell‐surface DR5 expression and c‐FLIP in the resistance of lung cancer cells to AD5‐10. RNA interference was used to knockdown c‐FLIP expression in lung cancer cells, and the cell viabilities and apoptosis as well as possible signaling transduction pathways were further assessed. Then, the c‐FLIP expression in tissue samples was examined from patients with lung carcinoma.
Materials and Methods
Reagents. z‐IETD‐fmk (caspase‐8 inhibitor) was purchased from R & D Systems, Inc. (Minneapolis, MN, USA). Recombinant soluble TRAIL (rsTRAIL, a.a. 95–281, non‐tagged) was prepared as previously described by Shi et al.( 17 ) AD5‐10 was prepared as previously described by Guo et al.( 9 ) Anticaspase‐8 and anticaspase‐9 antibodies were purchased from Oncogene (La Jolla, CA, USA). Anticaspase‐3 and anticaspase‐10 antibodies were purchased from Cell Signaling (Beverly, MA, USA). Anti‐Poly ADP‐vibose polymerase (PARP) and anti‐Bcl‐2 antibodies were purchased from BD (San Jose, CA, USA). Anti‐Bid antibodies were purchased from MBL (Woburn, MA, USA). Anti‐Bcl‐XL and anticytochrome c antibodies were purchased from Santa Cruz (Santa Cruz, CA, USA). Monoclonal antibody to c‐FLIP (NF6) was purchased from ALEXIS (San Diego, CA, USA). Anti‐Bax antibody was purchased from Upstate (Lake Placid, NY, USA). Anti‐cytochrome oxidase subunit IV (COX IV) antibody was purchased (Abcam). Horseradish peroxidase linked antimouse IgG, antirabbit IgG and antigoat IgG secondary antibodies were purchased from Zhongshan Biotech (Beijing, China).
Cells and cell viability assay. Human lung cancer cell lines A549, H460, H1299 and Calu‐3 were sourced from the American Type Culture Collection (Rockville, MD, USA). Human small lung cancer cell line (SCLC) and Jurkat cell line SVT35 were obtained. These cells were cultured at 37°C in RPMI 1640, F12, MEM or DMEM (Gibco, Grand Island, NY, USA). Cell viability was quantified by 3‐(4,5‐dimethyl‐thiazole‐2‐yl)‐2,5‐biphenyl tetrazolium (MTT) assay (Sigma, St. Louis, MO, USA), as described previously.( 18 )
Apoptosis assay. Annexin V/PI staining was performed as described previously.( 9 )
Measurement of mitochondrial membrane potential. Changes in mitochondrial transmembrane potential (ΔΨM) were measured using 5,5′,6,6′‐tetrachloro‐1,1′,3,3′‐tetra‐ethylbenzimidazolylcarbocyanine iodide (JC‐1) as a probe. Cells were plated in six‐well plates at 1 × 105 cells/well, transfected with non‐silencing or c‐FLIPL siRNA, and then treated with or without AD5‐10 at 50 ng/mL for an indicated time. The treated cells were washed twice with PBS and re‐suspended in 1 mL PRMI‐1640 media containing JC‐1 (20 µg/mL), then incubated for 30 min at room temperature in the dark. The cells were washed twice with phosphate‐buffered saline (PBS), re‐suspended in 0.5 mL PBS, and immediately analyzed with a flow cytometer (FACScan, Becton Dickinson, Heidelberg, Germany).
Cellular fractionation and Western blot analysis. Cells in two 10‐cm culture dishes per sample were collected and washed twice in ice‐cold PBS, and re‐suspended in Fractionation Buffer Mix (20 mM HEPES–KOH pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM sodium EDTA, 1 mM sodium EGTA, 1 mM dithiothreitol and 0.1 mM PMSF) supplemented with protease inhibitors (5 mg/mL pepstatin A, 10 mg/mL leupeptin, 2 mg/mL aprotinin) and 250 mM sucrose, followed by incubation on ice overnight. The cells were homogenized, followed by centrifugation at 700g for 10 min at 4°C. The supernatants were collected and centrifuged again at 100 000g at 4°C for 30 min to collect a cytosolic fraction (supernatant) and a mitochondrial fraction (pellet). The pellets were suspended in Fractionation Buffer Mix. COX IV and β‐actin were used as loading controls for the mitochondrial and cytosolic fractions, respectively. Western blot analyses were performed as described previously.( 9 ) Equal loading was assessed using an anti‐β‐actin monoclonal antibody (Sigma).
siRNA. c‐FLIPL siRNAs (siFL1, siFL2 and siFL3) target the 5′‐AAGGAACAGCTTGGCGCTCAA‐3′, 5′‐CAGATTCTTGGCCA ATTTGCC‐3 and 5′‐GAGCTTCTTCGAGACACCTTC‐3′, respectively, as described previously,( 19 , 20 ) and control (non‐silencing) siRNA were synthesized by Genechem (Shanghai, China). One day prior to the transfection, cells were seeded without antibiotics at a density of 30 to 40%. c‐FLIPL siRNAs were transfected into cells using lipofectamineTM2000 (Invitrogen, Carlsbad, CA, USA).
Flow cytometric analysis for TRAIL receptor expression. The lung cancer cells were characterized for their surface expression of TRAIL receptors by flow cytometry. Briefly, 105 cells were incubated with the phycoerythrin‐conjugated anti‐DR4, anti‐DR5, anti‐DcR1, anti‐DcR2 antibody (R & D) or a respective isotype control for 45 min at 4°C. Cells were then washed with PBS and suspended in 300 µL of PBS for final flow cytometric analysis.
Immunohistochemical analysis. Primary paraffin‐embedded lung cancer tissues obtained from 33 patients and 27 cases of normal lung tissues during surgery were used for the immunohistochemical studies. These tissues were obtained from the Department of Thoracic Surgery, General Hospital of PLA, Beijing, China. The patients were fully informed and gave consent for collection of clinical samples. The study was in accordance with the principles in the Declaration of Helsinki and was approved by the ethics committee of the participating hospital. The characteristics of these patients are shown in Table 1. After deparaffinization and rehydration, slides were immersed in sodium citrate 10 mM, pH 6.0 and heated in a microwave oven. After blocking endogenous peroxidase activity, primary anti‐FLIPL antibody (Abcam, Cambridge, UK) was applied overnight at 4°C. The slides were then incubated in envision peroxidase, antirabbit IgG (DAKO, Carpinteria, CA, USA), visualized using 3,3′‐diaminobenzidine tetrahydrochloride (DAB, Carpinteria), lightly counterstained with hematoxylin for 60 s and mounted under a coverslip. All of the stained tumor samples were evaluated semiquantitatively for intensity and percent of staining: no staining (–), <10% of tumor cells showing weak positive staining (+), positive staining (++), or >60% of tumor cells showing strong positive staining (+++). For statistical analyses, the immunohistochemical stainings were considered as binary variables (positive versus negative). Moderate and strong staining in > 10% of tumor cells was considered positive, but tumors with negative or weakly positive staining were considered negative. Other clinical variables used as binary variables for statistical analysis were histology (squamous cell versus adenocarcinoma), stage (I/II versus III) and lymph node metastasis (0 versus 1/2).
Table 1.
Patient characteristics
| Characteristic | Number of patients |
|---|---|
| Total | 33 |
| Male/female | 23/10 |
| Histology | |
| Squamous cell | 12 |
| Adenocarcinoma | 15 |
| Large cell | 2 |
| BAC | 4 |
| Stage I | |
| I a | 6 |
| I b | 13 |
| Stage II | |
| II a | 2 |
| II b | 4 |
| Stage III | |
| III a | 6 |
| III b | 2 |
BAC, bronchioloalveolar carcinoma.
Statistical analysis. Results were expressed as mean ± SD, and a Student's t‐test was used for evaluating statistical significance. To analyze the dependence of the immunohistochemical variables and their association with clinical variables, the χ2‐test was used. In cases of very low cell frequencies (expected frequency <5), the Fisher's exact test was used. P ≤ 0.05 was considered to be statistically significant.
Results
Various sensitivities of lung cancer cell lines to AD5‐10‐induced apoptosis. To determine the sensitivity of lung cancer cells to AD5‐10‐induced apoptosis, five lung cancer cell lines (A549, SCLC, H460, H1299 and Calu‐3) were treated with equimolar anti‐DR5 monoclonal antibody (AD5‐10)( 9 ) and recombinant soluble TRAIL (rsTRAIL)( 17 ) for 24 h, and the percent of apoptotic cells was determined by Annexin V/PI assay. TRAIL was used for comparison, and Jurkat leukemia cell line SVT35 was used as a positive control. As shown in Figure 1(a), A549 and SCLC were moderately sensitive to AD5‐10, whereas H460, H1299 and Calu‐3 showed a minimal response or no response to the antibody. Various sensitivities were observed in these cells treated with rsTRAIL: A549 and H460 were highly sensitive, SCLC moderately sensitive, and Calu‐3 and H1299 resistant to TRAIL cytotoxic activity. These data clearly demonstrate that three of five lung cancer cell lines tested possess either a minimal response or resistance to AD5‐10‐induced cell death; in particular, H460 is resistant to AD5‐10, even though it is highly sensitive to TRAIL. To explore the molecular mechanisms by which lung cancer cells are resistant to AD5‐10‐induced cell death, expression of the death receptors on the cell surface of H460, A549, SCLC, H1299 and Calu‐3 was determined by phycoerythrin (PE)‐labeled specific antibodies against DR4, DR5, DcR1 and DcR2, and followed by flow cytometry. As shown in Figure 1(b), DR5 was expressed on more than 89% of A549, H460 and Calu‐3, and about 48% of SCLC, while DR4 was expressed only on SCLC and H460 at a relatively low level. DcR1 and DcR2 were expressed only on SCLC and H460 cells, but were not detectable in the other lung cancer cells. These data indicate that DR5 is normally expressed on H460 as well as other lung cancer cells tested, and the levels of DR5 expression on these cell lines might not result in the various sensitivities to AD5‐10.
Figure 1.
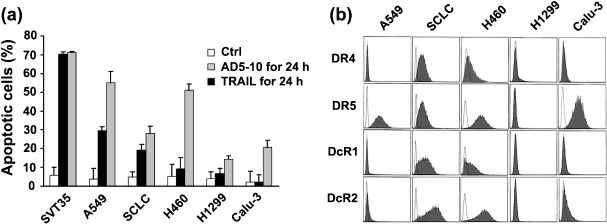
Sensitivity of lung cancer cells to AD5‐10‐induced apoptosis. (a) Cytotoxicities of equimolar AD5‐10 and TRAIL on A549, SCLC, H460, H1299, and Calu‐3 cell lines. Percentages represent the sum of Annexin V‐positive and Annexin V/PI double‐positive cells. SVT35 Jurkat T cell line was used as positive control. Columns, mean of three independent experiments; bars, SD. (b) Flow cytometric analysis of DR4, DR5, DcR1 and DcR2 expression on the cell surface of lung cancer cells. Shaded peaks represent phycoerythrin‐labeled cells, which shift to the right and indicate death receptor expression on the cell surfaces. Unshaded peaks represent isotype control antibody staining cells.
c‐FLIPL is associated with the resistance of H460 cells to AD5‐10‐induced apoptosis. As demonstrated above, H460 cells are resistant to AD5‐10 stimulation despite a high level of cell‐surface DR5 expression. We assumed that this might result from some intracellular molecules that inhibited AD5‐10‐induced cell death. It was reported that the antiapoptotic molecule c‐FLIP could interfere with the binding of caspase‐8 to FADD, and therefore, suppress apoptosis induced by death ligands.( 21 ) To confirm the hypothesis, we investigated expression of caspase‐8 and c‐FLIP in H460 cells treated with AD5‐10 or TRAIL by Western blot analysis. As illustrated in Figure 2(a), persistent expression of c‐FLIPL (55 kDa) was observed in H460 cells treated with AD5‐10, but the appearance of cleaved c‐FLIPL (44 kDa) and a decreased amount of c‐FLIPL (55 kDa) were observed in H460 treated with TRAIL. There was no obvious change in the expression of c‐FLIPS(25 kDa) when treated with AD5‐10 or TRAIL. The activation of caspase‐8 was evidenced by the appearance of the intermediate p43/41 form and the catalytically active p20/18 subunit in H460 cells treated with TRAIL, but not in the cells treated with AD5‐10. This result indicated that c‐FLIPL cleavage in H460 cells was well‐correlated to the activation of caspase‐8, which might be associated with the resistance of H460 cells to AD5‐10.
Figure 2.
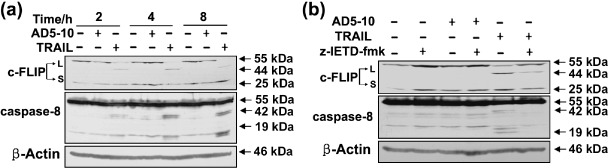
c‐FLIPL is associated with the resistance of H460 to AD5‐10‐induced apoptosis. (a) Western blot analysis of c‐FLIPL and caspase‐8 in absence (–) or presence (+) of AD5‐10 (50 ng/mL) or TRAIL (7 ng/mL) in H460 cell lines. Cells were either left untreated or stimulated with AD5‐10 (50 ng/mL) or TRAIL (7 ng/mL) for 2, 4 or 8 h, harvested and lyzed. β‐actin was used as the loading control. (b) Caspase‐8 inhibitor z‐IETD‐fmk prevents AD5‐10 or TRAIL induced c‐FLIPL cleavage. H460 cells were cultured for 4 h with or without AD5‐10 or TRAIL in the presence or absence of 50 mM z‐IETD‐fmk, respectively.
To further confirm the involvement of caspase‐8 in c‐FLIPL cleavage in H460 cells, the cells were treated with AD5‐10 or TRAIL in the presence of irreversible caspase‐8 inhibitor, z‐IETD‐fmk, then the caspase‐8 activation and cleavage of c‐FLIPL were detected by Western blot and individual specific antibodies. As shown in Figure 2(b), no matter the presence or absence of z‐IETD‐fmk, caspase‐8 was not activated and c‐FLIPL was not decreased or cleaved in H460 cells treated with AD5‐10; however, z‐IETD‐fmk partially blocked caspase‐8 activation and prevented the cleavage of c‐FLIPL in H460 cells treated with TRAIL. In summary, these data demonstrate that c‐FLIPL expression and caspase‐8 dependent c‐FLIPL cleavage are associated with the resistance of H460 lung cancer cells to AD5‐10‐induced apoptosis.
Down‐regulation of c‐FLIPL expression by siRNA sensitized lung cancer cells to AD5‐10‐induced cell death. As presented above, c‐FLIPL expression but not c‐FLIPS expression is associated with the resistance of H460 lung cancer cells to AD5‐10‐induced apoptosis. To further demonstrate the role of c‐FLIPL in the resistance of H460 lung cancer cells, the cells were treated with non‐silencing siRNA or specific c‐FLIPL siRNA, followed by treatment with AD5‐10 (50 ng/mL). Three oligonucleotide siRNAs (siFL1, siFL2, siFL3) designed from c‐FLIPL cDNA were used. Cell viability of H460 transfected with the different c‐FLIPL siRNAs and treated with AD5‐10 was evaluated by MTT. Figure 3(a) shows viabilities of H460 cells transfected with siFL1 or siFL3 siRNAs and treated with AD5‐10 decreased significantly. In contrast, siFL2 transfection has no obvious effect on the cell sensitivity to AD5‐10. As shown in Fig 3(b), c‐FLIPL expression was suppressed significantly by siFL1 in all the lung cancer cell lines tested, and the viabilities of A549, H460, SCLC and Calu‐3 cells transfected with siFL1 and treated with AD5‐10 were decreased to 60.38, 55.53, 73.24 and 68.09%, respectively. To further analyze whether caspase activation was involved, cleavage of pro‐caspases and the downstream substrates in the lysate of H460 treated with c‐FLIPL siRNA plus AD5‐10 were analyzed by Western blot. As shown in Figure 3(c), with the treatment of c‐FLIPL siRNA plus AD5‐10, expression levels of pro‐caspase‐10, ‐8, ‐9 and ‐3 significantly decreased, activation‐resulted cleaved fragments of 43/41 and 20/18 kDa for caspase‐8 and 37 kDa for caspase‐9 appeared, and the caspase substrate PARP was cut into smaller fragments. These results indicate that down‐regulation of c‐FLIPL expression by siRNA sensitizes H460 cells to AD5‐10‐induced cell death by activation of caspases.
Figure 3.
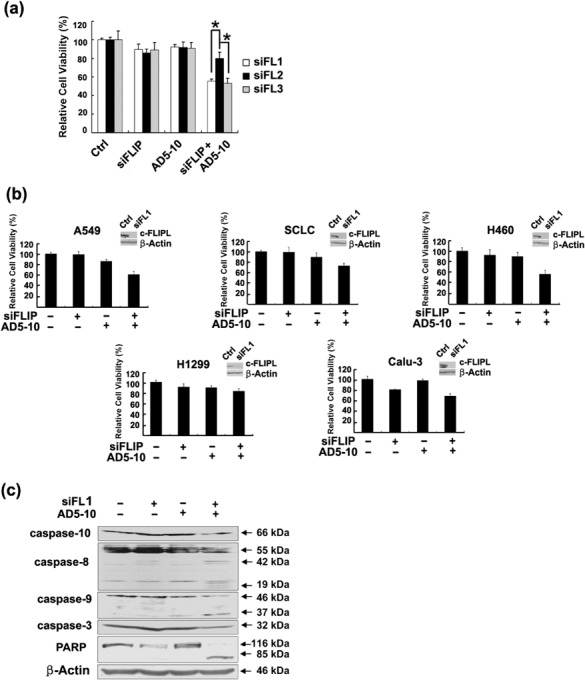
c‐FLIPL siRNA sensitized lung cancer cell lines to AD5‐10‐induced cell death. (a) H460 cells were transfected with c‐FLIPL siRNAs (siFL1, siFL2, siFL3) or non‐silencing siRNA for 24 h and then treated with or without AD5‐10 (50 ng/mL) for 24 h and cell viability was assayed. Columns, mean of three triplicate samples, and experiments were repeated three times; bars, SD; *, P < 0.05. (b) c‐FLIPL siRNA enhanced lung cancer cell viability reduction induced by AD5‐10. Indicated lung cancer cell line was transfected with non‐silencing or c‐FLIPL siRNA (siFL1) and then treated with or without AD5‐10 (50 ng/mL) for 24 h and cell viability was assayed. Columns, mean of three triplicate samples, and experiments were repeated three times; bars, SD. Insets show levels of c‐FLIPL protein in cells transfected with non‐silencing or c‐FLIPL siRNA (siFL1). Representative results of at least three different experiments shown. (c) Western blot analysis for the activation of caspase‐10, caspase‐8, caspase‐9, caspase‐3 and cleavage of Poly ADP‐ribose polymerase (PARP) in H460 cells.
c‐FLIPL siRNA‐enhanced AD5‐10‐induced cell death in lung cancer cells was via intrinsic death signaling pathway. To further investigate the signaling pathway in H460 cells treated with c‐FLIPL siRNA plus AD5‐10, the expression of Bcl‐XL, Bcl‐2, Bid and Bax in the cells were analyzed by Western blot assay. As shown in Figure 4(a), the expression levels of Bid and Bcl‐XL decreased significantly in H460 cells treated with c‐FLIPL siRNA plus AD5‐10 compared with the control, but there was no change in the expression of Bcl‐2 and Bax. This result suggests that proteins Bid and Bcl‐XL in the Bcl‐2 family were involved in the signaling events in c‐FLIPL siRNA‐enhanced AD5‐10‐induced cell death of H460 cells.
Figure 4.
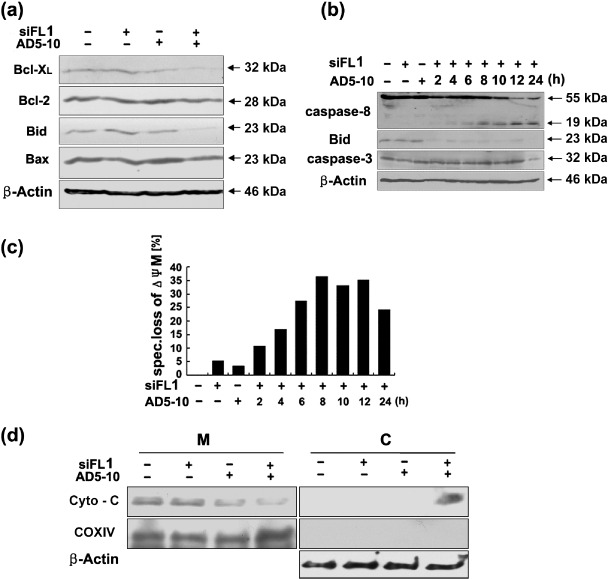
c‐FLIPL siRNA‐enhanced AD5‐10‐induced cell death via intrinsic death signaling pathway. (a) Expression of Bcl‐2 family proteins, Bcl‐XL, Bcl‐2, Bid and Bax in H460 cells. (b) Kinetics of caspase‐8, Bid and caspase‐3 cleavage in H460 cells. c‐FLIPL siRNA (siFL1) transfected cells treated with AD5‐10 at indicated time were harvested and lyzed with lysis buffer. The cell lysates were subjected to SDS‐PAGE and Western blot with specific antibodies against caspase‐8, Bid, and caspase‐3, respectively. (c) Changes of mitochondrial membrane potential (ΔΨM) in H460 cells. The cells were transfected with non‐silencing or c‐FLIPL siRNA (siFL1) and then incubated with or without 50 ng/mL AD5‐10 for indicated time. Subsequently, the cells were stained with the mitochondrial membrane potential sensitive dye JC‐1 and followed by flow cytometry. Specific ΔΨM was calculated as follows: (% experimental ΔΨM – % spontaneous ΔΨM)/(100 – % spontaneous ΔΨM) × 100. Mean of three experiments is shown. (d) Western blot and subcellular fractionation analysis of cytochrome c release from mitochondria. Mitochondrial (M) and cytosolic (C) fractions were prepared and subjected to Western blot analysis to detect cytochrome c release. COX IV and β‐actin were used as controls for the mitochondrial and cytosolic fractions, respectively.
It is known that proteins in the Bcl‐2 family are functionally localized on the membrane of mitochondria. To investigate possible activation of mitochondrial pathway, H460 cells were transfected with c‐FLIPL siRNA and followed with AD5‐10 for 2–24 h. The kinetics of caspase‐8, Bid and caspase‐3 cleavage, and the changes of mitochondrial membrane potential (ΔΨM) were detected by Western blot analysis and JC‐1 staining assay, respectively. As shown in Figure 4(b), caspase‐8 was activated in the c‐FLIPL siRNA transfected cell from 2 h of AD5‐10 treatment. The proapoptotic protein Bid was cleaved from 2 h AD5‐10 treatment in the c‐FLIPL siRNA transfected cells. No obvious decrease of procaspase‐3 was detected in the c‐FLIPL siRNA transfected cells until 12 h, but decrease in procaspase‐3 was detected at 24 h of AD5‐10 treatment. As shown in Figure 4(c), the ΔΨM was reduced markedly in the c‐FLIPL siRNA transfected cells from 2 h of AD5‐10 treatment. The release of cytochrome c from the mitochondria into the cytosol was significantly increased (Fig. 4d). These results strongly suggest that c‐FLIPL siRNA‐enhanced AD5‐10‐induced H460 apoptosis is through the activation of caspase‐8, cleavage of Bid, alteration in mitochondrial membrane potential, and release of cytochrome c.
Expression of cFLIPL in human lung carcinoma. To understand the biological significance of c‐FLIPL siRNA‐enhanced AD5‐10‐induced cell death in lung cancer cells and its possibility of being applied to clinical lung cancer therapy, we further investigated c‐FLIPL expression in patients with various lung cancers. The paraffin‐embedded sections from 33 cases of lung carcinoma tissues and 27 cases of tumor‐surrounding tissues more than 3 cm away from the tumor edges as normal controls were stained with the specific antibody against c‐FLIPL by immunohistochemistry. The representative micrographs of the tissue sections in Figure 5(b,d,f) show c‐FLIPL was preferentially and highly expressed in the cytoplasm of malignant lung cancer cells (brown color), but little in the tumor‐surrounding tissues, bronchus or alveolus in most of the normal controls (Fig. 5a,c). Statistically using a 10% cutoff, c‐FLIPL was expressed in 29 of 33 (87.9%) lung cancer tissues, but only in 4 of 27 (14.8%) controls (Table 2, χ2‐test, P < 0.001). The representative micrographs of those four controls in Figure 5(e,g) show c‐FLIPL was moderately expressed in bronchial epithelium and type II alveolar cells, and highly expressed in the pulmonary macrophage. In the cohort of various lung cancer patients, no significant correlation was observed between c‐FLIPL expression and various stages of tumor types, but the c‐FLIPL expression in the tumor of lymph node metastasis was slightly more common than that in tumors without metastasis; however, there was no statistical significance (Fisher's exact test, P = 0.426). These results indicate that c‐FLIPL expression in human lung cancers is significantly higher than that in normal controls.
Figure 5.
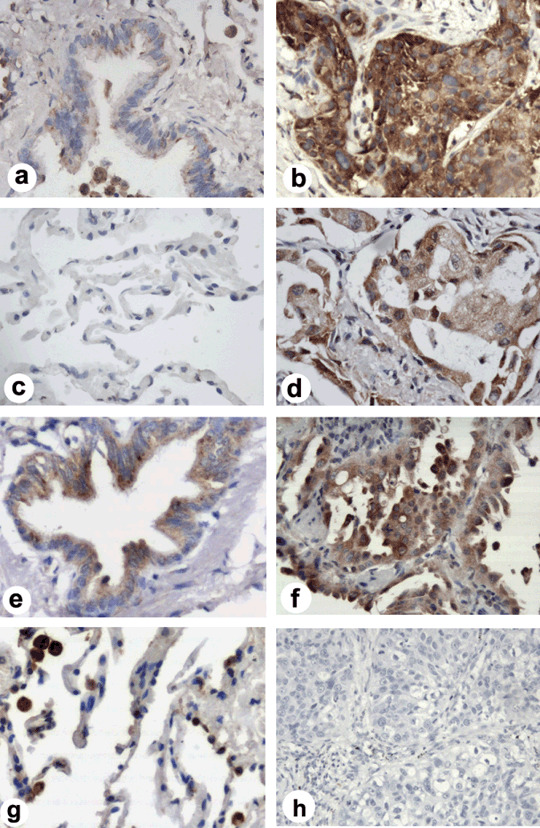
Immunohistochemical analysis of c‐FLIPL expression in lung cancer tissues. Little c‐FLIPL expression in (a) most normal lung bronchus and (c) alveolar tissues (400 × magnification). Representative (e) normal lung bronchus and (g) alveolar staining (brown color) in c‐FLIPL positive staining (++) normal controls (400 × magnification). Representative examples of c‐FLIPL high expression (brown color) in different lung cancer tissues from (b) patients with squamous cancer, (d) adenocarcinoma, and (f) bronchioloalveolar carcinoma (400 × magnification). (h) Negative control of immunostaining section from a squamous cancer tissue, in which the primary antibodies were substituted by PBS (200 × magnification).
Table 2.
Immunohistochemical staining results in patients with lung carcinoma
| n | c‐FLIPL | |||||
|---|---|---|---|---|---|---|
| – | + | ++ | +++ | (%) † | ||
| Normal tissue | 27 | 15 | 8 | 4 | 0 | 14.8 |
| Patients | 33 | 2 | 2 | 8 | 21 | 87.9* |
| I a I b | 19 | 1 | 2 | 5 | 11 | 84.2 |
| II a II b | 6 | 0 | 0 | 1 | 5 | 100 |
| III a III b | 8 | 1 | 0 | 2 | 5 | 87.5 |
| Lymph node metastasis | ||||||
| N0 | 19 | 1 | 2 | 5 | 11 | 84.2 |
| N1‐3 | 14 | 1 | 0 | 3 | 10 | 92.9 |
Significant difference (P < 0.001) versus normal tissue.
Positively stained/total no. tissues × 100%.
c‐FLIPL, cellular FLICE inhibitory protein; n, number of patients.
Discussion
The agonistic antibodies to TRAIL receptors have been proved to have tumoricidal activity either alone or in combination with chemotherapy against human cancers without cytotoxicity to normal hepatocytes.( 22 , 23 , 24 ) Takeda et al.( 25 ) reported that the anti‐mouse DR5 agonistic antibody not only eliminated TRAIL‐sensitive tumor cells in vivo, but also induced tumor specific cytotoxic T cells leading to eradication of those TRAIL‐resistance tumors. A fully humanized agonistic antibody, Lexatumumab, is currently in phase I/II clinical trials in patients with advanced malignant cancer. However, some patients are resistant to Lexatumumab‐induced cytotoxicity, and the molecular mechanism remains to be clarified.( 5 ) In the present study, we have demonstrated that five lung cancer cell lines tested possess various sensitivities to AD5‐10 induced cell death. The H460 cells were resistant to AD5‐10 despite a high level of cell‐surface DR5 expression, which is a good model for exploring the resistance mechanism of anti‐DR5 monoclonal antibody‐induced apoptosis. We showed that the sensitivity of H460 lung cancer cells to AD5‐10 were not related to the expression level of DR5, but related with the expression and cleavage of c‐FLIPL. Knockdown of c‐FLIPL by specific siRNA enhanced AD5‐10 induced apoptosis in lung cancer cells. Moreover, c‐FLIPL was expressed in 87.9% of the tumor tissues from lung carcinoma patients, but little in the normal controls. These data provide evidence that knockdown of c‐FLIPL expression might be a potential strategy for lung cancer therapy, especially for those lung cancers resistant to the agonistic antibody against death receptors.
TRAIL receptors DR4 and DR5 are highly expressed in various human cancer tissues and cells including breast cancer, leukemia, colorectal cancer, pancreatic cancer and lung cancers.( 10 , 26 ) The expression of TRAIL receptors on colon cancer cells( 27 ) and lymphocytes( 28 ) plays a major role in the regulation of TRAIL‐induced apoptosis; however, reports indicate that there is no affirmative correlation between the expression of death receptors and cell sensitivity to TRAIL.( 29 ) Straughn et al.( 30 ) showed that, despite a high level of DR5 expression, not all cervical cancer cells were necessarily susceptible to anti‐DR5 antibody TRA‐8‐induced apoptosis. Consistent with this report, our data demonstrate that the sensitivity of lung cancer cells to AD5‐10‐induced apoptosis was not related to the surface expression of DR5; for example, H460 and Calu‐3 were relative resistant to AD5‐10 despite high‐level DR5 expression. Here, we have demonstrated that the susceptibility of the lung cancer cells to AD5‐10‐mediated apoptosis is not due to DR5 expression but due to the intracellular signaling mechanism conferring resistance to AD5‐10‐induced apoptosis in lung cancer cells.
More recently, Zhang et al.( 5 ) reported that DR5 was up‐regulated and c‐FLIPL was down‐regulated in Lexatumumab‐sensitive renal cancer cells. Whether persistent c‐FLIPL expression results in Lexatumumab resistance in renal cancer cells is unknown. In this study, we demonstrated c‐FLIPL knockdown by the specific siRNA significantly sensitized H460 lung cancer cells to AD5‐10‐induced cell death and strongly enhanced activation of caspase‐8 and ‐10 as well as cleavage of the substrate PARP. Treatment of c‐FLIPL siRNA plus AD5‐10 markedly decreased the viability of these lung cancer cells (Fig. 3b), but not in normal control cells such as MRC‐5, HFTF and PHH (data not shown), indicating that c‐FLIPL‐specific interfering RNA might be a potential agent in lung cancer therapy.
It has been reported that both caspase‐8 and caspase‐10 are the apical caspases in the TRAIL signaling pathway.( 31 ) In our study, we showed that both caspase‐8 and ‐10 were activated, Bid and Bcl‐XL expression decreased, mitochondrial membrane potential ΔΨM changed, and cytochrome c released from the mitochondria into the cytosol in c‐FLIPL siRNA transfected and AD5‐10 treated H460 cells. These observations suggest that the resistant of H460 to AD5‐10 is developed by a blockage of apical caspase activation and thereafter a mitochondrial signaling pathway.
Several studies in vivo reported that high levels of c‐FLIP might contribute to development of many carcinomas such as Epstein‐Barr virus infected Burkitt's lymphoma, Hodgkin's lymphoma, melanomas and colorectal and gastric tumors, and facilitate tumor immune escape.( 32 ) Horak et al.( 33 ) reported there was a strong tendency towards epithelial c‐FLIPL expression in early stage non‐serous tumors. Here we report that lung carcinoma is yet another tumor with frequent higher expression of c‐FLIPL in lung cancer tissues than that in normal controls. However, no statistically significant association was observed between c‐FLIPL expression, disease stage and different tumor types in our cohort of patients with lung cancer. Further investigation is required to determine whether c‐FLIPL contributes to the progression of lung carcinoma.
In conclusion, our data show that not all lung cancer cells were responsive to AD5‐10‐induced apoptosis despite high levels of cell‐surface DR5 expression. c‐FLIP, especially c‐FLIPL, is associated with AD5‐10 resistance in human lung carcinoma cells. The resistance could be reversed by knockdown c‐FLIPL expression and activation of a mitochondrial signaling pathway. Furthermore, c‐FLIPL was highly expressed in most lung cancer tissues from the patients, but not in the controls. These results raise the possibility that strategies aimed at lowering c‐FLIPL expression may improve the efficacy of AD5‐10 in lung cancer therapy.
Acknowledgments
The human small lung cancer cell line (SCLC) was kindly provided by Professor Xiaoguang Chen, Institute of Materia Medica, Chinese Academy of Medical Sciences, Beijing, China and Jurkat cell line SVT35 was kindly provided by Dr Brian Seed, Department of Molecular Biology, Massachusetts General Hospital, Boston. This work was partially supported by Natural Science Foundation of China Grant no. 30721063 and 30571687, and State Key Basic Research Program of China Grant no. 2007CB507404.
References
- 1. Greenlee RT, Hill‐Harmon MB, Murray T, Thun M. Cancer statistics. CA Cancer J Clin 2001; 51: 15–36. [DOI] [PubMed] [Google Scholar]
- 2. Ferreira CG, Huisman C, Giaccone G. Novel approaches to the treatment of non‐small cell lung cancer. Crit Rev Oncol Hematol 2002; 41: 57–77. [DOI] [PubMed] [Google Scholar]
- 3. Carlo‐Stella C, Lavazza C, Locatelli A, Vigano L, Gianni AM, Gianni L. Targeting TRAIL agonistic receptors for cancer therapy. Clin Cancer Res 2007; 13: 2313–7. [DOI] [PubMed] [Google Scholar]
- 4. Walczak H, Miller RE, Ariail K et al . Tumoricidal activity of tumor necrosis factor‐related apoptos‐inducing ligand in vivo . Nat Med 1999; 5: 157–63. [DOI] [PubMed] [Google Scholar]
- 5. Zhang L, Zhang X, Barrisford GW, Olumi AF. Lexatumumab (TRAIL‐receptor 2 mAb) induces expression of DR5 and promotes apoptosis in primary and metastatic renal cell carcinoma in a mouse orthotopic model. Cancer Lett 2007; 251: 146–57. [DOI] [PubMed] [Google Scholar]
- 6. Jo M, Kim TH, Seol DW et al . Apoptosis induced in normal human hepatocytes by tumor necrosis factor related apoptosis‐inducing ligand. Nat Med 2000; 6: 564–7. [DOI] [PubMed] [Google Scholar]
- 7. Lawrence D, Shahrokh Z, Marsters S et al . Differential hepatocyte toxicity of recombinant Apo2L/TRAIL versions. Nat Med 2001; 7: 383–5. [DOI] [PubMed] [Google Scholar]
- 8. LeBlanc HN, Ashkenazi A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ 2003; 10: 66–75. [DOI] [PubMed] [Google Scholar]
- 9. Guo Y, Chen C, Zheng Y et al . A novel anti‐human DR5 monoclonal antibody with tumoricidal activity induces caspase‐dependent and caspase‐independent cell death. J Biol Chem 2005; 280: 41 940–52. [DOI] [PubMed] [Google Scholar]
- 10. Shi J, Liu Y, Zheng Y et al . Therapeutic expression of an anti‐death receptor 5 single‐chain fixed‐variable region prevents tumor growth in mice. Cancer Res 2006; 66: 11 946–53. [DOI] [PubMed] [Google Scholar]
- 11. Hu S, Vincenz C, Ni J, Gentz R, Dixit VM. I‐FLICE, a novel inhibitor of tumor necrosis factor receptor 1 and CD95 induced apoptosis. J Biol Chem 1997; 272: 17 255–7. [DOI] [PubMed] [Google Scholar]
- 12. Golks A, Brenner D, Fritsch C, Krammer PH, Lavrik IN. c‐FLIPR, a new regulator of death receptor‐induced apoptosis. J Biol Chem 2005; 280: 14 507–13. [DOI] [PubMed] [Google Scholar]
- 13. Yeh WC, Itie A, Elia AJ et al . Requirement for casper (c‐FLIP) in regulation of death receptor‐induced apoptosis and embryonic development. Immunity 2000; 12: 633–42. [DOI] [PubMed] [Google Scholar]
- 14. Chaudhary PM, Eby MT, Jasmin A, Kumar A, Liu L, Hood L. Activation of the NF‐κB pathway by caspase 8 and its homologs. Oncogene 2000; 19: 4451–60. [DOI] [PubMed] [Google Scholar]
- 15. Kataoka T, Budd RC, Holler N et al . The caspase‐8 inhibitor FLIP promotes activation of NF‐κB and Erk signaling pathways. Curr Biol 2000; 10: 640–8. [DOI] [PubMed] [Google Scholar]
- 16. Shu HB, Halpin DR, Goeddel DV. Casper is a FADD‐ and caspase‐related inducer of apoptosis. Immunity 1997; 6: 751–63. [DOI] [PubMed] [Google Scholar]
- 17. Shi J, Zhang J, Chang X, Liu S, Liu Y, Zheng D. Expression and biological activity of the soluble recombinant tumor necrosis factor related apoptosis inducing ligand. Chin J Bioengineering 2003; 23: 46–9. [Google Scholar]
- 18. Shi J, Zheng D, Liu Y et al . Overexpression of soluble TRAIL induces apoptosis in human lung adenocarcinoma and inhibits growth of tumor xenografts in nude mice. Cancer Res 2005; 65: 1687–92. [DOI] [PubMed] [Google Scholar]
- 19. Flahaut M, Mühlethaler‐Mottet A, Auderset K et al . Persistent inhibition of FLIP (L) expression by lentiviral small hairpin RNA delivery restores death‐receptor‐induced apoptosis in neuroblastoma cells. Apoptosis 2006; 11: 255–63. [DOI] [PubMed] [Google Scholar]
- 20. Longley DB, Wilson TR, McEwan M et al . c‐FLIP inhibits chemotherapy‐induced colorectal cancer cell death. Oncogene 2006; 25: 838–48. [DOI] [PubMed] [Google Scholar]
- 21. Irmler M, Thome M, Hahne M et al . Inhibition of death receptor signals by cellular FLIP. Nature 1997; 388: 190–5. [DOI] [PubMed] [Google Scholar]
- 22. Buchsbaum DJ, Zhou T, Grizzle WE et al . Antitumor efficacy of TRA‐8 anti‐DR5 monoclonal antibody alone or in combination with chemotherapy and/or radiation therapy in a human breast cancer model. Clin Cancer Res 2003; 9: 3731–41. [PubMed] [Google Scholar]
- 23. Chuntharapai A, Dodge K, Grimmer K et al . Isotype‐dependent inhibition of tumor growth in vivo by monoclonal antibodies to death receptor 4. J Immunol 2001; 166: 4891–8. [DOI] [PubMed] [Google Scholar]
- 24. Ichikawa K, Liu W, Zhao L et al . Tumoricidal activity of a novel anti‐human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nature Med 2001; 7: 954–60. [DOI] [PubMed] [Google Scholar]
- 25. Takeda K, Yamaguchi N, Akiba H, Kojima Y, Hayakawa Y, Tanner JE. Induction of tumor‐specific T cell immunity by anti‐DR5 antibody. Ther J Exp Med 2004; 199: 437–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koornstra JJ, Kleibeuker JH, van Geelen CM et al . Expression of TRAIL (TNF‐related apoptosis inducing ligand) and its receptors in normal colonic mucosa, adenomas, and carcinomas. J Pathol 2003; 200: 327–35. [DOI] [PubMed] [Google Scholar]
- 27. Lacour S, Hammann A, Wotawa A, Corcos L, Solary E, Dimanche‐Boitrel MT. Anticancer agents sensitize tumor cells to tumor necrosis factor‐related apoptosis‐inducing ligand mediated caspase‐8 activation and apoptosis. Cancer Res 2001; 6: 1645–51. [PubMed] [Google Scholar]
- 28. Mongkolsapaya J, Cowper AE, Xu XN et al . Lymphocyte inhibitor of TRAIL (TNF related apoptosis‐inducing ligand): a new receptor protecting lymphocytes from the death ligand TRAIL. J Immunol 1998; 160: 3–6. [PubMed] [Google Scholar]
- 29. Yamaguchi Y, Shiraki K, Fuke H, Inoue T, Miyashita K, Yamanaka Y. Adenovirus‐mediated transfection of caspase‐8 sensitizes hepatocellular carcinoma to TRAIL‐ and chemotherapeutic agent‐induced cell death. Biochim Biophys Acta 2006; 1763: 844–53. [DOI] [PubMed] [Google Scholar]
- 30. Straughn JM Jr, Oliver PG, Zhou T et al . Anti‐tumor activity of TRA‐8 anti‐death receptor 5 (DR5) monoclonal antibody in combination with chemotherapy and radiation therapy in a cervical cancer model. Gynecol Oncol 2006; 101: 46–54. [DOI] [PubMed] [Google Scholar]
- 31. Mitsiades N, Poulaki V, Tseleni‐Balafouta S, Koutras DA, Stamenkovic I. Thyroid carcinoma cells are resistant to FAS‐mediated apoptosis but sensitive to tumor necrosis factor‐related apoptosis‐inducing ligand. Cancer Res 2000; 60: 4122–9. [PubMed] [Google Scholar]
- 32. Zhou XD, Yu JP, Liu J, Luo HS, Chen HX, Yu HG. Overexpression of cellular FLICE‐inhibitory protein (FLIP) in gastric adenocarcinoma. Clin Sci (London) 2004; 106: 397–405. [DOI] [PubMed] [Google Scholar]
- 33. Horak P, Pils D, Kaider A et al . Perturbation of the tumor necrosis factor related apoptosis‐inducing ligand cascade in ovarian cancer. overexpression of FLIPL and deregulation of the functional receptors DR4 and DR5. Clin Cancer Res 2005; 11: 8585–91. [DOI] [PubMed] [Google Scholar]