

Abstract
Breast cancer resistance protein (BCRP) is a half‐molecule ATP‐binding cassette transporter that forms a functional homodimer and pumps out various anticancer agents, such as 7‐ethyl‐10‐hydroxycamptothecin, topotecan, mitoxantrone and flavopiridol, from cells. Estrogens, such as estrone and 17β‐estradiol, have been found to restore drug sensitivity levels in BCRP‐transduced cells by increasing the cellular accumulation of such agents. Furthermore, synthetic estrogens, tamoxifen derivatives and phytoestrogens/flavonoids have now been identified that can effectively circumvent BCRP‐mediated drug resistance. Transcellular transport experiments have shown that BCRP transports sulfated estrogens and various sulfated steroidal compounds, but not free estrogens. The kinase inhibitor gefitinib inhibited the transporter function of BCRP and reversed BCRP‐mediated drug resistance both in vitro and in vivo. BCRP‐transduced human epidermoid carcinoma A431 (A431/BCRP) and BCRP‐transduced human non‐small cell lung cancer PC‐9 (PC‐9/BCRP) cells showed gefitinib resistance. Physiological concentrations of estrogens (10–100 pM) reduced BCRP protein expression without affecting its mRNA levels. Two functional polymorphisms of the BCRP gene have been identified. The C376T (Q126Stop) polymorphism has a dramatic phenotype as active BCRP protein cannot be expressed from a C376T allele. The C421A (Q141K) polymorphism is also significant as Q141K‐BCRP‐transfected cells show markedly low protein expression levels and low‐level drug resistance. Hence, individuals with C376T or C421A polymorphisms may express low levels of BCRP or none at all, resulting in hypersensitivity of normal cells to BCRP‐substrate anticancer agents. In summary, both modulators of BCRP and functional single nucleotide polymorphisms within the BCRP gene affect the transporter function of the protein and thus can modulate drug sensitivity and substrate pharmacokinetics and pharmacodynamics in affected cells and individuals. (Cancer Sci 2005; 96: 457–465)
In cancer chemotherapy, there are two major problems to be overcome. One is the innate or acquired resistance of cancer cells to anticancer drugs. The other is the toxic effects of chemotherapeutic drugs on some normal tissues, such as bone marrow and the digestive organs. The study of the mechanisms of drug resistance in cancer cells has led to the identification of some of the genes and gene products that confer drug resistance. For example, a family of ATP‐binding cassette (ABC) transporters, including the MDR1 gene product P‐glycoprotein (ABCB1)( 1 , 2 ) and MRP1 (ABCC1),( 3 ) have previously been shown to be responsible for multidrug resistance. Significantly, both P‐glycoprotein and the MRP proteins have internally duplicated structures with two membrane‐spanning domains and two ATP‐binding domains.( 1 , 2 , 3 )
Breast cancer resistance protein (BCRP), also called ABCG2, ABCP and MXR, is a half‐molecule ABC transporter with an N‐terminal ATP‐binding domain and a C‐terminal transmembrane domain (TM) (Fig. 1).( 4 , 5 , 6 , 7 , 8 , 9 ) We have shown that BCRP functions as a homodimer.( 10 ) BCRP mediates resistance to several anticancer drugs, such as 7‐ethyl‐10‐hydroxycamptothecin (SN‐38, an active metabolite of irinotecan), mitoxantrone, topotecan and flavopiridol.( 4 , 5 , 6 , 7 , 8 , 9 ) BCRP‐transduced human myelogenous leukemia K562 (K562/BCRP) cells showed 25‐fold higher resistance to SN‐38, 10‐fold higher resistance to mitoxantrone and 10‐fold higher resistance to topotecan.( 11 ) Hence, overexpression of BCRP in certain types of malignant cells would limit the effectiveness of some anticancer agents.
Figure 1.
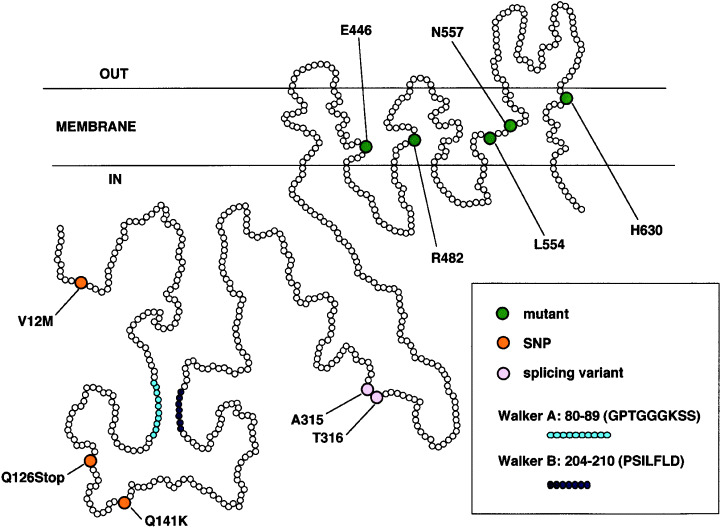
Schematic structure of the breast cancer resistance protein, representing the positions of amino acid changes in mutants, single nucleotide polymorphisms and a splicing variant.
Breast cancer resistance protein is usually expressed in a variety of normal tissues, such as placenta, intestine, kidney, liver, mammalian gland, ovary, testis, endothelium and in hematopoietic stem cells.( 12 , 13 , 14 ) BCRP is therefore assumed to play a role in the protective functions of the maternal–placental barrier, digestive tract and blood–testis barrier against toxic substances and metabolites. BCRP expression has also been reported in relapsed or refractory hematological malignancies.( 15 , 16 ) It has been shown that BCRP expression may be associated with poor responses to chemotherapy.( 16 , 17 ) It is thus possible that BCRP expression is responsible, at least in part, for many instances of clinical drug resistance. If this proves to be the case, overcoming BCRP‐mediated drug resistance would contribute greatly to improving the efficacy of many cancer chemotherapy treatments.
Various organic compounds have been identified as BCRP inhibitors. Some of these inhibitors include estrogens,( 18 ) anti‐estrogens,( 19 ) flavonoids,( 20 ) gefitinib( 21 , 22 , 23 , 24 , 25 ) and imatinib.( 26 , 27 , 28 ) Although a number of them are actively transported by BCRP, many act only as inhibitors of its function. In addition to anticancer agents, BCRP exports various dietary compounds, such as chlorophyll‐derived dietary phototoxin and protoporphyria, and dietary carcinogen 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine.( 29 , 30 , 31 ) As BCRP mediates the efflux of such agents from cells and tissues, suppression of its function by competitive inhibitors would be predicted to modulate the pharmacokinetics and pharmacodynamics of these drugs. This would potentially result in an increase in both the blood and tissue concentrations of the chemotherapeutics and cause more potent effects. However, there could also be increases in the undesirable side effects of these anticancer agents as a result of BCRP inhibition.
In this review we summarize the various factors that affect the expression and function of BCRP. These include substrates, inhibitors, regulators of protein expression and functional single nucleotide polymorphisms (SNP). We discuss the possible implications of the use of these factors in cancer chemotherapy.
Dimer formation of breast cancer resistance protein
We have demonstrated the homodimerization of BCRP using coexpression of Myc‐tagged and hemagglutinin (HA)‐tagged proteins (MycBCRP and HABCRP).( 10 ) Exogenous BCRP migrated as a 70‐kDa protein under reducing conditions in sodium dodecyl‐sulfate–polyacrylamide gel electrophoresis, but migrated as a 140‐kDa complex in the absence of reducing agents. The 140‐kDa BCRP complex was found to be heat stable but to dissociate into 70‐kDa species upon the addition of 2‐mercaptoethanol. We immunoprecipitated the 140‐kDa BCRP complex from lysates of PA317 cells doubly transfected with MycBCRP and HABCRP, using an anti‐Myc antibody. The 140‐kDa complex also reacted with anti‐HA and anti‐BCRP antibodies. In addition, following the addition of reducing agents, the 70‐kDa species also reacted with these antibodies. These results clearly indicate that BCRP forms a homodimer, bridged by disulfide bonds.
To test for possible dominant‐negative inhibition of the BCRP drug efflux pump, various mutant BCRP cDNAs were generated by polymerase chain reaction mutagenesis. These mutants were then expressed in parental PA317 cells and tested for drug‐resistance properties. HA‐tagged inactive BCRP cDNAs were subsequently transfected into MycBCRP‐expressing cells and tested for their ability to lower drug resistance. Among the eight inactive mutant cDNAs, L554P‐BCRP, with an amino acid change in TM5, was found to partially reverse drug resistance in MycBCRP‐transfected cells (Fig. 1). This result suggests that homodimer formation may be essential for the transporter function of BCRP, and that dominant‐negative inhibition of these complexes is a potential new strategy for circumventing drug resistance.( 10 )
Mutation analysis of breast cancer resistance protein
In earlier studies, BCRP cDNAs isolated from doxorubicin‐selected and mitoxantrone‐selected cells had mutations in R482.( 5 , 6 , 7 ) To test for possible alterations in substrate specificity and in the drug resistance patterns of different mutant BCRP, we generated 32 such mutants with amino acid substitutions in the TM (seven E446 mutants in TM2, 15 R482 mutants in TM3, four N557 mutants in TM5 and six H630 mutants in TM6) and examined the resulting effects of these substitutions on cellular drug resistance (Fig. 1).( 32 ) PA317 cells transfected with any one of the seven E446‐mutant BCRP cDNA did not show drug resistance. In contrast, cells transfected with any of the 13 R482X‐BCRP cDNA (X = N, C, M, S, T, V, A, G, E, W, D, Q and H, but not Y and K) showed a higher resistance to mitoxantrone and doxorubicin than wild‐type BCRP‐transfected cells. Cells transfected with N557D‐BCRP cDNA showed a similar level of resistance to mitoxantrone, but lower resistance to SN‐38 than wild‐type BCRP‐transfected cells. Cells transfected with N557E‐BCRP, H630E‐BCRP or H630L‐BCRP cDNA showed similar degrees of resistance to mitoxantrone and SN‐38. Cells transfected with R482G‐BCRP or R482S‐BCRP cDNA showed less intracellular accumulation of 3H‐mitoxantrone than wild‐type BCRP‐transfected cells. These results suggest that residues E446 in TM2, R482 in TM3, N557 in TM5 and H630 in TM6 play important roles in the drug recognition of BCRP.( 32 ) R482‐mutant BCRP was also shown to have a more effective pumping function for doxorubicin and mitoxantrone than the wild‐type protein, but high levels of doxorubicin transport may not be associated with the physiological role of BCRP, as wild‐type BCRP is less effective.( 33 , 34 ) In fact, we and others have previously reported that wild‐type BCRP effectively transports methotrexate and methotrexate polyglutamates. This suggests that BCRP may play a crucial role in the transport of folate derivatives.( 33 , 34 , 35 ) In addition, the R482 mutation was found only in in vitro drug‐selected cancer cells, and has not been found in any clinical specimens.
Estrogens and anti‐estrogens inhibit breast cancer resistance protein
Breast cancer resistance protein is normally expressed in a variety of tissues, such as placenta, intestine, kidney, liver, mammalian gland, ovary, testis, endothelium and in hematopoietic stem cells.( 12 , 13 , 14 ) Among these normal tissues, the highest level of BCRP expression has been seen in the placenta, and BCRP is therefore presumed to function in protecting the fetus against toxic compounds.( 12 ) In addition, BCRP may mediate the transport of placenta‐specific compounds across the blood–placenta barrier, as immunohistochemical analysis has shown that BCRP is highly expressed in the syncytiotrophoblast of the placenta, which produces female steroid hormones.( 12 ) We therefore examined for possible interactions of female steroid hormones with BCRP.( 18 ) The effects of steroid hormones and other related compounds on BCRP‐mediated drug resistance were evaluated by a cell growth inhibition assay using K562/BCRP cells. Among the compounds tested, estrone and estradiol (17β‐estradiol) were found to potentiate the cytotoxicity of SN‐38, mitoxantrone and topotecan in K562/BCRP cells.( 18 ) In contrast, these estrogens showed little effect on drug sensitivity in parental K562 cells.
The reversal activities (measured as the ratios of the IC50 values in the absence or presence of the steroid) of 10 µM estrone were 3.6‐fold for SN‐38, 7.5‐fold for mitoxantrone and 4.1‐fold for topotecan. Similarly, estradiol enhanced the cytotoxicity of these antitumor agents in K562/BCRP cells, but not in parental K562 cells. Drug resistance levels were also slightly abrogated by estriol in a dose‐dependent manner but neither pregnenolone nor progesterone had any effect on the drug sensitivity of K562/BCRP cells. In order to determine whether this reversal might be associated with increased drug transport, effect of steroid hormones on the cellular accumulation of topotecan was evaluated by flow cytometric analysis. The intracellular accumulation of topotecan increased in the presence of estrone in a dose‐dependent manner in K562/BCRP cells, whereas levels were not altered in parental cells. In addition, increased cellular accumulation of topotecan was also observed in the presence of both estradiol and estriol. In contrast, pregnenolone and progesterone showed only a marginal effect on topotecan uptake. These results suggest that estrogens reverse BCRP‐mediated drug resistance by inhibiting its drug efflux function.( 18 )
We further examined the effects of other non‐steroidal estrogens and anti‐estrogens on BCRP‐mediated drug resistance.( 19 ) Initially, these compounds were tested for their effects on the cellular accumulation of topotecan in K562/BCRP cells. These compounds were then examined for their ability to reverse SN‐38 and mitoxantrone resistance in K562/BCRP cells. Among the commercially available estrogen antagonists and agonists that were tested, synthetic estrogen diethylstilbestrol showed the strongest BCRP‐reversing activity. Diethylstilbestrol was found to increase the cellular accumulation of topotecan and reverse BCRP‐mediated drug resistance in K562/BCRP cells, but showed only marginal or no effect in parental K562 cells. The reversal activities of estrone and diethylstilbestrol were more prominent for mitoxantrone than for SN‐38. Anti‐estrogens, tamoxifen and toremifene were also found to enhance topotecan uptake in K562/BCRP cells. Various tamoxifen derivatives were subsequently screened for anti‐BCRP activity and among the initial 14 compounds that were tested, TAG‐11 showed the strongest effects. In a second screening of 25 TAG‐11‐related compounds, TAG‐139 was found to show the strongest effect. Reversal of SN‐38 and mitoxantrone resistance in K562/BCRP cells by TAG‐139 was then found to be five‐fold greater than the results with estrone. The dose‐dependent characteristics of drug resistance reversal by estrone and TAG‐139 treatment were very similar, suggesting that derivatives of these compounds interact with the same binding site of BCRP. Next, the possible effects of TAG‐139 on P‐glycoprotein‐mediated and MRP1‐mediated drug resistance were evaluated. TAG‐139 strongly potentiated the cytotoxicity of doxorubicin and vincristine on K562/MDR cells. The reversal activity of TAG‐139 was more prominent for doxorubicin than for vincristine. TAG‐139 showed no effects on MRP1‐mediated doxorubicin resistance or VP‐16 resistance.( 19 )
To examine whether the BCRP‐reversing activities of these compounds are associated with anti‐estrogen activity, the effects of these agents on the binding of estradiol to estrogen receptors (ER) were evaluated.( 19 ) Tamoxifen and 4‐OH‐tamoxifen strongly inhibited the binding of estradiol to ERα. TAG‐11 showed weak inhibition of estradiol binding to ERα but TAG‐72 and TAG‐126, which both showed modest BCRP‐reversing activity, strongly inhibited this binding. TAG‐139, the most potent TAG‐compound BCRP inhibitor, showed a weak interaction with ERα. Similar results were obtained with ERβ. These results show that BCRP‐reversing activity and anti‐estrogen activity can be disassociated. Therefore, it should be possible to develop BCRP‐reversing agents that exhibit no other biological effects, including anti‐estrogen activity. Such compounds would have great potential to be used clinically in overcoming BCRP‐mediated drug resistance.( 19 )
Breast cancer resistance protein exports sulfated estrogens
Estrone and estradiol were both shown to reverse BCRP‐mediated multidrug resistance. However, this did not necessarily indicate that BCRP exports these estrogens. To clarify this point, we generated BCRP‐transduced porcine kidney LLC‐PK1 (LLC/BCRP) cells, in which exogenous BCRP is expressed in the apical membrane of the cell monolayer, and investigated the transcellular transport of 3H‐labeled compounds using cells plated on microporous filter membranes.( 36 ) The basal‐to‐apical transport (excretion) of 3H‐mitoxantrone, 3H‐estrone and 3H‐estradiol was greater in LLC/BCRP cells than in LLC‐PK1 cells. However, thin layer chromatography of transported steroids revealed that the transport of estrone and estradiol was independent of BCRP expression. In contrast, increased excretion of estrone sulfate and estradiol sulfate was observed in LLC/BCRP cells, which was shown to be completely abrogated by BCRP inhibitors. In addition, the conversion of estrogens into their sulfated conjugates occurred at a similar rate between LLC/BCRP and LLC‐PK1 cells, suggesting that the increased excretion of estrogen sulfates was attributable to BCRP‐mediated transport.( 36 )
The uptake of 3H‐labeled compounds in membrane vesicles from K562/BCRP cells was also investigated.( 36 ) 3H‐Labeled estrone sulfate, but not 3H‐labeled estrone or estradiol, was taken up by membrane vesicles from K562/BCRP cells, and this was ATP dependent. BCRP inhibitors suppressed the transport of estrone sulfate in membrane vesicles from K562/BCRP cells. Furthermore, sulfated steroidal compounds such as dehydroepiandrosterone sulfate, taurolithocholate and taurolithocholate sulfate, strongly inhibited the BCRP‐mediated transport of estrone sulfate across K562/BCRP membrane vesicles, suggesting that BCRP has a high affinity for sulfated steroids. These results clearly demonstrate that BCRP does not transport either free estrone or estradiol but exports sulfate conjugates of these estrogens, which were the first identified physiological substrates of BCRP.( 36 , 37 )
Flavonoids inhibit breast cancer resistance protein
We carried out additional screens of estrogenic compounds for anti‐BCRP activity and found that phytoestrogens and flavonoids, such as genistein, naringenin, acacetin and kaempferol, strongly potentiate the cytotoxicity of SN‐38 and mitoxantrone in K562/BCRP cells.( 20 ) Genistein and naringenin increased the cellular accumulation of topotecan in K562/BCRP cells. In addition, some glycosylated flavonoids, such as naringenin‐7‐glucoside, also effectively inhibited BCRP. These flavonoids showed marginal effects on drug sensitivity in K562 cells. Furthermore, neither genistein nor naringenin could reverse either P‐glycoprotein‐mediated vincristine resistance or MRP1‐mediated VP‐16 resistance. K562/BCRP cells accumulated less 3H‐genistein than parental K562 cells. Using a transcellular transport system we showed that 3H‐genistein transport in the basal‐to‐apical direction was greater in LLC/BCRP cells, which express exogenous BCRP in the apical membrane, than in parental LLC‐PK1 cells. BCRP inhibitors abolished this increased transport of 3H‐genistein in LLC/BCRP cells.( 38 ) Analysis by thin layer chromatography revealed that genistein was transported in its native but not in its metabolized form. These results suggest that genistein is in fact among the natural substrates of BCRP and competitively inhibits BCRP‐mediated drug efflux. BCRP therefore seems to function as an efflux pump for genistein and other plant‐derived flavonoids. These findings have two important clinical implications: (i) flavonoids and glycosylated flavonoids may be useful compounds for overcoming BCRP‐mediated drug resistance in cancer cells; and (ii) the intake of flavonoids, mostly from food or drink, together with the administration of BCRP‐substrate antitumor agents may alter the pharmacokinetics and consequently increase the toxicity of antitumor agents in cancer patients.( 20 )
Breast cancer resistance protein‐transduced cells show gefitinib resistance
We evaluated the possible interaction of gefitinib, a selective epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, with BCRP.( 21 ) BCRP‐transduced human epidermoid carcinoma A431 (A431/BCRP) and BCRP‐transduced human non‐small cell lung cancer PC‐9 (PC‐9/BCRP) cells have acquired cellular resistance to gefitinib, suggesting that BCRP is one of the determinants of gefitinib sensitivity in specific cell types (Fig. 2). However, BCRP expression did not alter gefitinib sensitivity in cells that were intrinsically insensitive to this drug, such as K562/BCRP cells. Gefitinib reversed SN‐38 resistance in both K562/BCRP cells and BCRP‐transduced murine lymphocytic leukemia P388 (P388/BCRP) cells, but not in the corresponding parental cells. In addition, gefitinib sensitized human colon cancer HT‐29 cells, which endogenously express BCRP, to SN‐38. Gefitinib was also demonstrated to increase the intracellular accumulation of topotecan in K562/BCRP cells and suppressed ATP‐dependent transport of estrone sulfate in membrane vesicles from K562/BCRP cells. These results suggest that gefitinib may overcome BCRP‐mediated drug resistance by inhibiting the pump function of BCRP. Furthermore, P388/BCRP‐transplanted mice that had been treated with a combination of irinotecan and gefitinib were observed to have significantly longer survival times than the P388/BCRP‐transplanted mice treated with either irinotecan or gefitinib alone. In conclusion, gefitinib interacts with BCRP, the expression of which in gefitinib‐responsive cells is likely to be one of the principal determinants of gefitinib sensitivity. Gefitinib also inhibits the transporter function of BCRP and reverses BCRP‐mediated drug resistance both in vitro and in vivo.( 21 )
Figure 2.
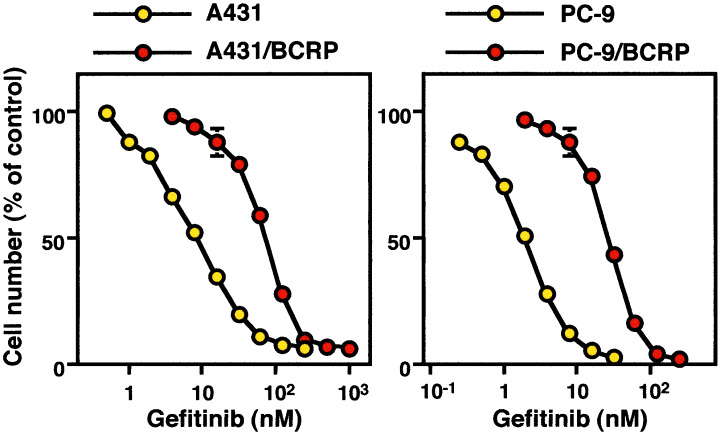
Resistance to gefitinib of breast cancer resistance protein (BCRP)‐transduced human epidermoid carcinoma A431 (A431/BCRP) and BCRP‐transduced human non‐small cell lung cancer PC‐9 (PC‐9/BCRP) cells. Cells were cultured for 5 days with increasing concentrations of gefitinib. Cell numbers were counted with a Coulter counter. Data are represented as mean ± SD from triplicate determinations.
Other breast cancer resistance protein inhibitors
Imatinib, an inhibitor of BCR‐ABL tyrosine kinase, has been reported to reverse BCRP‐mediated drug resistance.( 26 , 27 ) Imatinib was shown to significantly increase the accumulation of topotecan in the human osteosarcoma cell line Saos2, which expresses BCRP.( 26 ) Another report showed that imatinib is a substrate of BCRP by demonstrating that its accumulation is low in a BCRP‐overexpressing sub‐line, MCF‐7/MR.( 27 ) Moreover, Ko‐143, a specific inhibitor of BCRP, increased the accumulation of imatinib in MCF‐7/MR cells.( 27 ) In a separate study, imatinib was efficiently transported by mouse bcrp1 in bcrp1‐transfected Madin–Darby canine kidney strain II (MDCKII) monolayers.( 28 ) Furthermore, the clearance of intravenously injected imatinib was significantly decreased by 1.6‐fold in bcrp1‐knockout mice, compared with wild‐type mice.( 28 ) Taken together, BCRP seems to be a determinant for imatinib sensitivity. Another potent tyrosine kinase inhibitor, CI1033, has also been shown to enhance the uptake and cytotoxicity of SN‐38 and topotecan in BCRP‐transfected cells.( 39 ) CI1033 accumulation was diminished in BCRP‐expressing cells, suggesting that it may be transported by BCRP.( 39 ) It has been reported that BCRP exports dietary toxins and carcinogens such as chlorophyll‐derived dietary phototoxin and protoporphyria, and dietary carcinogen 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine.( 29 ) This study suggests that humans or animals with low or absent BCRP activity may be at an increased risk of developing diet‐dependent phototoxicity. BCRP thus seems to play a protective role against such toxins from normal food constituents.
Suppression of breast cancer resistance protein expression by estrogens
Because estrogens induce the expression of various genes via ER‐mediated pathways, we examined the possible effect of estrogens on BCRP expression.( 40 ) We found that estrogens, such as estradiol at physiological concentrations (10–100 pM), markedly decrease endogenous BCRP expression in estrogen‐responsive and ERα‐positive human breast cancer MCF‐7 cells. These effects did not occur, however, in estrogen‐non‐responsive human lung cancer A549 cells. To examine the effects of estrogens on BCRP expression in various ERα‐positive and ERα‐negative cells, the human breast cancer cell lines MCF‐7, T‐47D and MDA‐MB‐231, the ovarian cancer cell line SKOV‐3 and the lung cancer cell line A549 were transduced with BCRP retrovirus. Among these cell lines, MCF‐7 and T‐47D were both estrogen responsive and ERα positive. Estradiol significantly reduced exogenous BCRP expression, driven by a retroviral constitutive promoter, in estrogen‐responsive MCF‐7/BCRP and T‐47D/BCRP cells, but not in estrogen‐non‐responsive MDA‐MB‐231/BCRP and SKOV‐3/BCRP cells. Estradiol also significantly potentiated the cytotoxicity of SN‐38, but not vincristine, in MCF‐7/BCRP cells, and increased cellular topotecan uptake in MCF‐7/BCRP cells. The anti‐estrogen compound tamoxifen was shown to reverse estradiol‐mediated BCRP downregulation in MCF‐7 and MCF‐7/BCRP cells. Treatment of MCF‐7/BCRP cells with an ERα small interfering RNA abolished estradiol‐mediated BCRP downregulation, suggesting that interaction of estradiol and ERα is necessary for this suppression. Estradiol did not alter endogenous BCRP mRNA levels in MCF‐7 cells or exogenous BCRP mRNA levels in MCF‐7/BCRP cells. Pulse‐chase labeling experiments using MCF‐7/BCRP cells suggested that decreased protein biosynthesis and maturation, but not alterations in protein turnover, might underlie estradiol‐mediated BCRP downregulation. These data indicate that estrogens downregulate BCRP expression by novel post‐transcriptional mechanisms (Fig. 3). Significantly, this was the first demonstration that small molecules can control cellular BCRP protein expression.( 40 ) Analysis of the regulation of BCRP expression by estrogens would assist in the development of a more rational anticancer treatment protocol, particularly against malignancies in women.( 40 , 41 )
Figure 3.
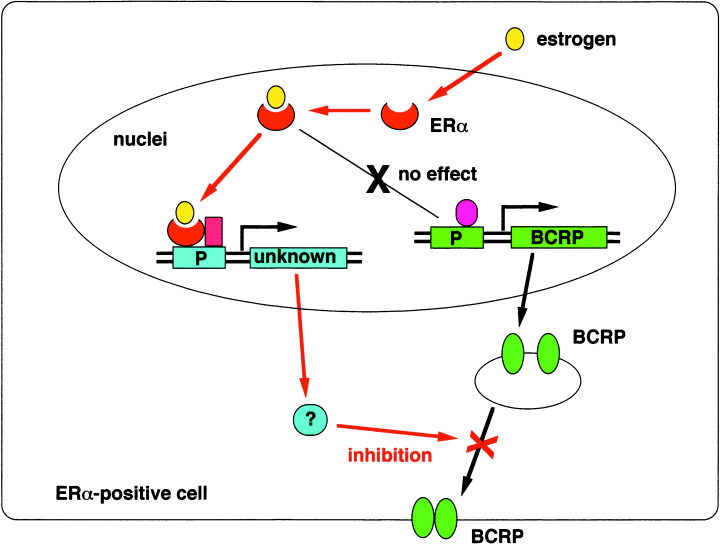
Putative mechanism of the suppression of breast cancer resistance protein (BCRP) expression by estrogens. Estrogens, such as estradiol at physiological concentrations (10–100 pM), markedly decrease BCRP expression in estrogen‐responsive and estrogen receptor (ER)α‐positive cells. Estrogen does not affect the BCRP mRNA expression in ERα‐positive cells. The BCRP downregulation seems to be associated with alterations in protein maturation, but not protein turnover.
Functional single nucleotide polymorphisms in the BCRP gene
Single nucleotide polymorphisms in the BCRP gene and BCRP cDNA variants were screened in genomic DNA samples from healthy Japanese volunteers and from 11 BCRP‐expressing human cancer cell lines, respectively. From these analyses, we identified three BCRP coding SNP, G34A (V12M), C376T (Q126Stop) and C421A (Q141K), and a splicing variant, Δ315‐6, that lacked nucleotides 944–949 (deletion of A315 and T316) (Fig. 1).( 42 ) It was expected that the C376T polymorphism in exon 4, which substituted Gln126 with a stop codon, would have the highest impact of the BCRP SNP, as an active BCRP protein cannot be expressed from the C376T allele. Of the 124 healthy Japanese volunteers that we sampled in our study, three were heterozygous for the C376T allele. In addition, Q141K‐BCRP‐transfected PA317 cells showed markedly lower levels of both BCRP protein expression and drug resistance than wild‐type BCRP‐transfected cells (Fig. 4a). It was noteworthy in this case that Q141K‐BCRP‐transfectants and wild‐type BCRP‐transfectants expressed similar levels of BCRP transcripts (Fig. 4a). V12M‐BCRP‐transfected and Δ315‐6‐BCRP‐transfected PA317 cells showed similar and somewhat lower BCRP protein expression and drug resistance levels compared with wild‐type BCRP‐transfected cells. Among the normal subjects in our analysis, 67 were wild type, 48 were heterozygous and nine were homozygous for the C421A allele. It has been reported that the C421A SNP is prevalent in 50–60% of Asians and 20–30% of Caucasians.( 42 ) Low expression levels of the Q141K BCRP protein have been confirmed using various experimental systems.( 43 , 44 , 45 ) These results suggest that some individuals harbor a C421A polymorphic BCRP gene and express low amounts of Q141K BCRP (Fig. 4b).
Figure 4.

Effect of C376T (Q126Stop) and C421A (Q141K) single nucleotide polymorphisms in the breast cancer resistance protein (BCRP) gene on protein expression. (a) Upper panel: western blotting of BCRP in PA317 cells and BCRP transfectants. PA317 cells transfected with wild‐type, G34A, C421A and Δ944‐949 BCRP cDNAs were termed PA/WT, PA/V12M, PA/Q141K and PA/Δ315‐6, respectively. Western blot analysis processed under non‐reducing conditions. The BCRP dimer was detected as a band at approximately 140 kDa (ref. 42). Lower panel: northern blot analysis of PA317 cells and BCRP transfectants. The blot was hybridized with an internal BCRP cDNA probe (ref. 42). (b) Putative BCRP expression levels in the C376T and/or C421A allele carriers. Left, putative BCRP expression levels relative to that of homozygous wild‐type allele carriers. Right, putative frequencies of each genotype with respect to nucleotides 376 and 421 of the BCRP gene.
The possible significance of the C421A BCRP SNP was evaluated recently in a phase I study of diflomotecan, a new camptothecin derivative anticancer agent.( 46 ) In this study, five patients who were heterozygous for the C421A allele showed three‐fold higher plasma levels of diflomotecan than 15 patients who were wild type. Following intravenous administration of this drug, the area‐under‐curve (AUC) analysis of patients with the C421A allele and in patients who were homozygous wild type was 138 ng·h/mL·mg and 46.1 ng·h/mL·mg, respectively (P = 0.015).( 46 ) A similar trend was noted after oral administration of this agent, although the differences were found not to be statistically significant. This study suggests that the BCRP genotype of a patient may impact strongly on the resulting pharmacokinetics of diflomotecan administration. Although the C376T allele is rare, a combination of the C376T and C421A SNP or a homozygous C421A genotype would be expected to occur frequently in Japanese individuals, considering the high incidence of this allele in Japan. In summary, individuals with C376T or C421A polymorphisms may express low amounts of BCRP, resulting in hypersensitivity of normal cells to anticancer drugs such as irinotecan, topotecan and diflomotecan.( 42 , 43 , 44 , 45 , 47 )
Conclusions
Breast cancer resistance protein, an ATP‐binding cassette transporter, confers resistance to a series of anticancer agents, such as mitoxantrone, SN‐38 (an active metabolite of irinotecan), topotecan and flavopiridol. BCRP expression is one of the key determinants of the sensitivity of cells to these drugs. We found that estrone and estradiol reverse drug resistance in K562/BCRP cells. Estrone and estradiol also increase the cellular accumulation of topotecan in K562/BCRP cells, but not in the parental K562 cells. BCRP‐dependent and ATP‐dependent uptake of estrone sulfate, but not estrone or estradiol themselves, was also observed in K562/BCRP vesicles. BCRP‐dependent excretion of estrone sulfate was observed in porcine kidney LLC/BCRP cells. Taken together, BCRP exports estrone sulfate, and sulfated estrogens seem to be physiological substrates of BCRP. Based on these findings, we have identified various BCRP inhibitors among the different estrogens, anti‐estrogens, phytoestrogens, flavonoids and kinase inhibitors. Some compounds, such as the flavonoid derivatives and the EGFR kinase inhibitor gefitinib, were effective against P388/BCRP in vivo.
Many clinical studies of P‐glycoprotein inhibitors have shown that the inhibition of drug efflux pumps not only increases the sensitivity of malignant cells to anticancer agents but also modulates the pharmacokinetics and pharmacodynamics of these drugs, and increases their concentrations in blood and tissues.( 48 ) In the case of BCRP inhibitors, it is possible that these factors could also alter the bioavailability and pharmacokinetics of the drugs that are targeted by BCRP.( 49 , 50 ) An example of this is seen with GF120918, a dual inhibitor of P‐glycoprotein and BCRP that increases the oral bioavailability of topotecan through the inhibition of BCRP function.( 50 ) The camptothecins are good BCRP substrates and are being used increasingly in cancer chemotherapy. Modulation of BCRP activity by inhibitors should alter the pharmocokinetics of such chemotherapeutic drugs in a number of clinical contexts. These effects might be used advantageously in improving several aspects of chemotherapy, such as a reduction of the variability in exposure to orally administered topotecan and potentiation of the cytotoxic activity of irinotecan. In addition, however, unintentional side effects may be caused by modulations to drug bioavailability by the inhibition of transporters. These must be considered in any new strategies that combine chemotherapeutic treatments with BCRP inhibitors.
We have identified two important functioning BCRP SNP, C376T (Q126Stop) and C421A (Q141K), that greatly diminish the expression of this protein. No active BCRP can be expressed from the C376T allele. Furthermore, cells transfected with Q141K‐BCRP cDNA express low amounts of BCRP protein and show only low levels of drug resistance. Polymorphisms within the BCRP genes of individuals that cause low transporter expression are likely to be associated with the hypersensitivity of their normal cells to substrate anticancer agents. Because BCRP may play crucial roles in the absorption and excretion of anticancer drugs such as DNA topoisomerase I inhibitors, the use of BCRP SNP should be considered carefully during the clinical development of novel anticancer agents and BCRP‐reversing drugs.
References
- 1. Gottesman MM, Hrycyna CA, Schoenlein PV, Germann UA, Pastan I. Genetic analysis of the multidrug transporter. Annu Rev Genet 1995; 29: 607–49. [DOI] [PubMed] [Google Scholar]
- 2. Chen CJ, Chin JE, Ueda K et al. Internal duplication and homology with bacterial transport proteins in the mdr1 (P‐glycoprotein) gene from multidrug‐resistant human cells. Cell 1986; 47: 381–9. [DOI] [PubMed] [Google Scholar]
- 3. Cole SP, Bhardwaj G, Gerlach JH et al. Overexpression of a transporter gene in a multidrug‐resistant human lung cancer cell line. Science 1992; 258: 1650–4. [DOI] [PubMed] [Google Scholar]
- 4. Allikmets R, Schriml L, Hutchinson A, Romano‐Spica V, Dean M. A human placenta‐specific ATP‐binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res 1998; 58: 5337–9. [PubMed] [Google Scholar]
- 5. Doyle LA, Yang W, Abruzzo LV et al. A multidrug resistance transporter from human MCF‐7 breast cancer cells. Proc Natl Acad Sci USA 1998; 95: 15 665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miyake K, Mickley L, Litman T et al. Molecular cloning of cDNA which is highly overexpressed in mitoxantrone‐resistant cells: demonstration of homology to ABC transport genes. Cancer Res 1999; 59: 8–13. [PubMed] [Google Scholar]
- 7. Maliepaard M, Van Gastelen MA, De Jong LA et al. Overexpression of the BCRP/MXR/ABCP gene in a topotecan‐selected ovarian tumor cell line. Cancer Res 1999; 59: 4559–63. [PubMed] [Google Scholar]
- 8. Kawabata S, Oka M, Shiozawa K et al. Breast cancer resistance protein directly confers SN‐38 resistance of lung cancer cells. Biochem Biophys Res Commun 2001; 280: 1216–23. [DOI] [PubMed] [Google Scholar]
- 9. Robey RW, Medina‐Perez WY, Nishiyama K et al. Overexpression of the ATP‐binding cassette half‐transporter, ABCG2 (Mxr/BCrp/ABCP1), in flavopiridol‐resistant human breast cancer cells. Clin Cancer Res 2001; 7: 145–52. [PubMed] [Google Scholar]
- 10. Kage K, Tsukahara S, Sugiyama T et al. Dominant‐negative inhibition of breast cancer resistance protein as drug efflux pump through the inhibition of S‐S dependent homodimerization. Int J Cancer 2002; 97: 626–30. [DOI] [PubMed] [Google Scholar]
- 11. Imai Y, Tsukahara S, Ishikawa E, Tsuruo T, Sugimoto Y. Estrone and 17β‐estradiol reverse breast cancer resistance protein‐mediated multidrug resistance. Jpn J Cancer Res 2002; 93: 231–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maliepaard M, Scheffer GL, Faneyte IF et al. Subcellular localization and distribution of the breast cancer resistant protein transporter in normal human tissue. Cancer Res 2001; 61: 3458–64. [PubMed] [Google Scholar]
- 13. Eisenblatter T, Galla HJ. A new multidrug resistance protein at the blood–brain barrier. Biochem Biophys Res Commun 2002; 293: 1273–8. [DOI] [PubMed] [Google Scholar]
- 14. Zhou S, Schuetz JD, Bunting KD et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side‐population phenotype. Nat Med 2001; 7: 1028–34. [DOI] [PubMed] [Google Scholar]
- 15. Van Der Kolk DM, Vellenga E, Scheffer GL et al. Expression and activity of breast cancer resistance protein (BCRP) in de novo and relapsed acute myeloid leukemia. Blood 2002; 99: 3763–70. [DOI] [PubMed] [Google Scholar]
- 16. Van Den Heuvel‐Eibrink MM, Wiemer EA, Prins A et al. Increased expression of the breast cancer resistance protein (BCRP) in relapsed or refractory acute myeloid leukemia (AML). Leukemia 2002; 16: 833–9. [DOI] [PubMed] [Google Scholar]
- 17. Steinbach D, Sell W, Voigt A, Hermann J, Zintl F, Sauerbrey A. BCRP gene expression is associated with a poor response to remission induction therapy in childhood acute myeloid leukemia. Leukemia 2002; 16: 1443–7. [DOI] [PubMed] [Google Scholar]
- 18. Imai Y, Tsukahara S, Ishikawa E, Tsuruo T, Sugimoto Y. Estrone and 17β‐estradiol reverse breast cancer resistance protein‐mediated multidrug resistance. Jpn J Cancer Res 2002; 93: 231–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sugimoto Y, Tsukahara S, Imai Y, Sugimoto Y, Ueda K, Tsuruo T. Reversal of breast cancer resistance protein‐mediated drug resistance by estrogen antagonists and agonists. Mol Cancer Ther 2003; 2: 105–12. [PubMed] [Google Scholar]
- 20. Imai Y, Tsukahara S, Asada S, Sugimoto Y. Phytoestrogens/flavonoids reverse breast cancer resistance protein/ABCG2‐mediated multidrug resistance. Cancer Res 2004; 64: 4346–52. [DOI] [PubMed] [Google Scholar]
- 21. Yanase K, Tsukahara S, Asada S, Ishikawa E, Imai Y, Sugimoto Y. Gefitinib reverses breast cancer resistance protein‐mediated drug resistance. Mol Cancer Ther 2004; 3: 1119–25. [PubMed] [Google Scholar]
- 22. Ozvegy‐Laczka C, Hegedus T, Varady G et al. High‐affinity interaction of tyrosine kinase inhibitors with the ABCG2 multidrug transporter. Mol Pharmacol 2004; 65: 1485–95. [DOI] [PubMed] [Google Scholar]
- 23. Stewart CF, Leggas M, Schuetz JD et al. Gefitinib enhances the antitumor activity and oral bioavailability of irinotecan in mice. Cancer Res 2004; 64: 7491–9. [DOI] [PubMed] [Google Scholar]
- 24. Nakamura Y, Oka M, Soda H et al. Gefitinib (‘Iressa’, ZD1839), an epidermal growth factor receptor tyrosine kinase inhibitor, reverses breast cancer resistance protein/ABCG2‐mediated drug resistance. Cancer Res 2005; 65: 1541–6. [DOI] [PubMed] [Google Scholar]
- 25. Elkind NB, Szentpetery Z, Apati A et al. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, Gefitinib). Cancer Res 2005; 65: 1770–7. [DOI] [PubMed] [Google Scholar]
- 26. Houghton PJ, Germain GS, Harwood FC et al. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN‐38 in vitro . Cancer Res 2004; 64: 2333–7. [DOI] [PubMed] [Google Scholar]
- 27. Burger H, Van Tol H, Boersma AW et al. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood 2004; 104: 2940–2. [DOI] [PubMed] [Google Scholar]
- 28. Breedveld P, Pluim D, Cipriani G et al. The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): implications for the use of breast cancer resistance protein and P‐glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer Res 2005; 65: 2577–82. [DOI] [PubMed] [Google Scholar]
- 29. Jonker JW, Buitelaar M, Wagenaar E et al. The breast cancer resistance protein protects against a major chlorophyll‐derived dietary phototoxin and protoporphyria. Proc Natl Acad Sci USA 2002; 99: 15 649–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van Herwaarden AE, Jonker JW, Wagenaar E et al. The breast cancer resistance protein (Bcrp1/Abcg2) restricts exposure to the dietary carcinogen 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine. Cancer Res 2003; 63: 6447–52. [PubMed] [Google Scholar]
- 31. Jonker JW, Merino G, Musters S et al. The breast cancer resistance protein BCRP (ABCG2) concentrates drugs and carcinogenic xenotoxins into milk. Nat Med 2005; 11: 127–9. [DOI] [PubMed] [Google Scholar]
- 32. Miwa M, Tsukahara S, Ishikawa E, Asada S, Imai Y, Sugimoto Y. Single amino acid substitutions in the transmembrane domains of breast cancer resistance protein (BCRP) alter cross resistance patterns in the transfectants. Int J Cancer 2003; 107: 757–67. [DOI] [PubMed] [Google Scholar]
- 33. Chen ZS, Robey RW, Belinsky MG et al. Transport of methotrexate, methotrexate polyglutamates, and 17β‐estradiol 17‐(β‐d‐glucuronide) by ABCG2: effects of acquired mutations at R482 on methotrexate transport. Cancer Res 2003; 63: 4048–54. [PubMed] [Google Scholar]
- 34. Volk EL, Schneider E. Wild‐type breast cancer resistance protein (BCRP/ABCG2) is a methotrexate polyglutamate transporter. Cancer Res 2003; 63: 5538–43. [PubMed] [Google Scholar]
- 35. Ifergan I, Shafran A, Jansen G, Hooijberg JH, Scheffer GL, Assaraf YG. Folate deprivation results in the loss of breast cancer resistance protein (BCRP/ABCG2) expression. A role for BCRP in cellular folate homeostasis. J Biol Chem 2004; 279: 25 527–34. [DOI] [PubMed] [Google Scholar]
- 36. Imai Y, Asada S, Tsukahara S, Ishikawa E, Tsuruo T, Sugimoto Y. Breast cancer resistance protein exports sulfated estrogens but not free estrogens. Mol Pharmacol 2003; 64: 610–18. [DOI] [PubMed] [Google Scholar]
- 37. Suzuki M, Suzuki H, Sugimoto Y, Sugiyama Y. ABCG2 transports sulfated conjugates of steroids and xenobiotics. J Biol Chem 2003; 278: 22 644–9. [DOI] [PubMed] [Google Scholar]
- 38. Rabindran SK, Ross DD, Doyle A, Yang W, Greenberger LM. Fumitremorgin C reverses multidrug resistance in cells transfected with the breast cancer resistance protein. Cancer Res 2000; 60: 47–50. [PubMed] [Google Scholar]
- 39. Erlichman C, Boerner SA, Hallgren CG et al. The HER tyrosine kinase inhibitor CI1033 enhances cytotoxicity of 7‐ethyl‐10‐hydroxycamptothecin and topotecan by inhibiting breast cancer resistance protein‐mediated drug efflux. Cancer Res 2001; 61: 739–48. [PubMed] [Google Scholar]
- 40. Imai Y, Ishikawa E, Asada S, Sugimoto Y. Estrogen‐mediated post‐transcriptional down‐regulation of breast cancer resistance protein/ABCG2. Cancer Res 2005; 65: 596–604. [PubMed] [Google Scholar]
- 41. Merino G, Van Herwaarden AE, Wagenaar E, Jonker JW, Schinkel AH. Sex‐dependent expression and activity of the ATP‐binding cassette transporter breast cancer resistance protein (BCRP/ABCG2) in liver. Mol Pharmacol 2005; 67: 1765–71. [DOI] [PubMed] [Google Scholar]
- 42. Imai Y, Nakane M, Kage K et al. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low‐level drug resistance. Mol Cancer Ther 2002; 1: 611–16. [PubMed] [Google Scholar]
- 43. Kondo C, Suzuki H, Itoda M et al. Functional analysis of SNPs variants of BCRP/ABCG2. Pharm Res 2004; 21: 1895–903. [DOI] [PubMed] [Google Scholar]
- 44. Kobayashi D, Ieiri I, Hirota T et al. Functional assessment of abcg2 (bcrp) gene polymorphisms to protein expression in human placenta. Drug Metab Dispos 2005; 33: 94–101. [DOI] [PubMed] [Google Scholar]
- 45. Zamber CP, Lamba JK, Yasuda K et al. Natural allelic variants of breast cancer resistance protein (BCRP) and their relationship to BCRP expression in human intestine. Pharmacogenetics 2003; 13: 19–28. [DOI] [PubMed] [Google Scholar]
- 46. Sparreboom A, Gelderblom H, Marsh S et al. Diflomotecan pharmacokinetics in relation to ABCG2 421C>A genotype. Clin Pharmacol Ther 2004; 76: 38–44. [DOI] [PubMed] [Google Scholar]
- 47. De Jong FA, Marsh S, Mathijssen RH et al. ABCG2 pharmacogenetics: ethnic differences in allele frequency and assessment of influence on irinotecan disposition. Clin Cancer Res 2004; 10: 5889–94. [DOI] [PubMed] [Google Scholar]
- 48. Oza AM. Clinical development of P‐glycoprotein modulators in oncology. Novartis Found Symp 2002; 243: 103–15. [PubMed] [Google Scholar]
- 49. Jonker JW, Smit JW, Brinkhuis RF et al. Role of breast cancer resistance protein in the bioavailability and fetal penetration of topotecan. J Natl Cancer Inst 2000; 92: 1651–6. [DOI] [PubMed] [Google Scholar]
- 50. Kruijtzer CM, Beijnen JH, Rosing H et al. Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P‐glycoprotein inhibitor GF120918. J Clin Oncol 2002; 20: 2943–50. [DOI] [PubMed] [Google Scholar]