

Abstract
The role of methionine‐enkephalin (MENK) as an immunomodulator in colorectal carcinomas (CRC) was examined. MENK was produced in CT26, IEC6A, Colo320, and HT29 CRC cell lines but not in IEC6 intestinal cells. MENK secretion was associated with tumorigenicity and metastasis of CRC cells in syngeneic rodent models. The MENK concentration in subcutaneous tumors of CT26 and IEC6A CRC cells exhibited an inverse correlation with the number of tumor‐infiltrating T lymphocytes. MENK inhibited the growth of MOLT‐4 T‐lymphoblastic cells in a dose‐dependent manner. Furthermore, it increased the phosphorylation level of c‐Jun N‐terminal kinase and induced apoptosis in MOLT‐4 cells. MENK‐induced apoptosis was abrogated by a c‐Jun N‐terminal kinase inhibitor. Immunohistochemical analysis revealed moderate to strong expression of MENK in 33 (54%) of 61 CRC. MENK expression was associated with Dukes’ staging, nodal metastasis, and liver metastasis. The MENK concentration in tumor tissues was higher in Dukes’ C cases than in Dukes’ B cases. MENK expression was associated with tumor‐infiltrating T lymphocytes, especially those belonging to the CD4+ subset. These findings suggest that MENK secreted by CRC cells caused escape of the host from the effects of immunity. (Cancer Sci 2009; 100: 497–502)
Abbreviations:
- AOM
azoxymethane
- CRC
colorectal cancer
- DOR
δ‐opioid receptor
- ELISA
enzyme‐linked immunosorbent assay
- ENK
enkephalin
- ERK
extracellular signal‐regulated kinase
- HBSS
Hank's balanced saline solution
- JNK
c‐jun N‐terminal kinase
- MAPK
mitogen‐activated protein kinase
- MENK
Met‐enkephalin
- siRNA
short interfering RNA
In CRC, the anticancer immunity of the host is suppressed due to the secretion of several cytokines. High‐mobility group box 1 protein induces apoptosis of macrophages( 1 ) to reduce tumor‐associated macrophages, which enhances metastasis of CRC.( 2 ) Programmed death‐1 is an inhibitory receptor expressed in T lymphocytes. The expression of the programmed death‐1 ligand by CRC cells induces apoptosis of tumor‐infiltrating T lymphocytes.( 3 ) Serum receptor‐binding cancer antigen expressed on SiSo cells, a membrane molecule expressed on human cancer cells, suppresses the tumor‐infiltrating T lymphocytes by inducing apoptotic cell death.( 4 , 5 ) These factors cause the escape of cancer cells from host immunity, thereby enhancing disease progression and metastasis. In the present study, we focused on MENK as an immune‐evasion factor in CRC.
Methionine‐enkephalin is a neuropeptide known to exhibit opium‐like effects on the central nervous system. Morphine affects the immune system and inhibits cellular immunity.( 6 ) Opioids alter the second messenger cyclic adenosine monophosphate, intracellular calcium, and kinases activated by second messengers in immune cells.( 7 , 8 ) Similar to morphine, MENK is an immunomodulator that modifies immune responses to extracellular stimuli such as mitogens and antigens.( 9 ) DOR, the specific receptor of MENK, is expressed in T lymphocytes.( 9 ) In some situations, opiates induce apoptosis of T lymphocytes through the JNK pathway.( 10 ) MENK is produced in high concentrations in colon cancer.( 11 ) We therefore hypothesized that MENK produced by CRC cells suppresses T lymphocytes so that the cells escape the effects of the host's anticancer immunity. In the present study, we examined the effect of MENK on T lymphocytes in vitro and in human CRC materials to test the hypothesis.
Materials and Methods
Cell culture and reagents. The HT29 human colon cancer cell line, IEC6 rat intestinal cell line, and MOLT‐4 T‐lymphoblastic leukemia cell line were purchased from Dainihon Pharmaceutical Co. (Tokyo, Japan). The CT26 mouse colon carcinoma cell line was kindly provided by Professor Isaiah J. Fidler (MD Anderson Cancer Center).( 12 ) Colo320 was kindly provided by Professor Wataru Yasui (Hiroshima University).( 13 ) The IEC6A intestinal cell line from IEC6 cells transformed by AOM treatment was established in our laboratory.( 14 ) All cell lines were maintained in Dulbecco's modified essential medium (Sigma Chemical Co., St Louis, MO, USA) containing 10% fetal bovine serum (Sigma Chemical Co.) under the conditions of 5% CO2 in air at 37°C. MENK (Sigma Chemical Co.), p38 MAPK inhibitor SB239063, and thiorphan ([dl‐3‐mercapto‐2‐benzylpropanoyl]‐glycine) were purchased from Sigma Chemical Co. ERK inhibitor (Calbiochem‐Novabiochem International, Darmstadt, Germany) and JNK inhibitor (SP600125; Biomol, Humberg, Germany) were also purchased.
Preparation of conditioned medium. For immunoblot analysis, cells were cultured in Dulbecco's modified essential medium containing 10% fetal bovine serum for 2 days. The conditioned medium was filtered through a 0.2‐µm filter (Becton‐Dickinson Labware, Bedford, MA, USA) and precipitated with acetone. The pellet was dissolved in lysis buffer (50 mM Tris‐HCl pH 7.4, 150 mM NaCl, 5 mM ethylenediaminetetraacetic acid, 50 µg/mL phenylmethylsulfonyl fluoride, 1 µg/mL aprotinin, and 0.5% Nonidet P‐40) to a concentration that was 50‐fold the original medium.
Immunoblot analysis. Whole‐cell lysates were prepared as described previously.( 15 ) Fifty‐microgram lysates were subjected to immunoblot analysis in 12.5% sodium dodecylsulfate–polyacrylamide gels followed by electrotransfer to nitrocellulose filters. The filters were incubated with primary antibodies, and then with peroxidase‐conjugated IgG antibody (Medical and Biological Laboratories, Nagoya, Japan). The primary antibodies used were anti‐CD10 antibody (Novocastra Laboratories, Newcastle upon Tyne, UK), anti‐ERK1/2 (Chemicon International, Temecula, CA, USA), anti‐phosphorylated ERK1/2 (Santa‐Cruz Biotechnology, Santa Cruz, CA, USA), anti‐JNK (JNK1, p46; Santa‐Cruz Biotechnology), anti‐phosphorylated JNK (Santa‐Cruz Biotechnology), anti‐p38 (clone A‐12; Santa‐Cruz Biotechnology), anti‐phosphorylated p38 (clone D‐8; Santa‐Cruz Biotechnology), anti‐MENK (Progen Biotechnic, Heidelberg, Germany), and anti‐DOR (Chemicon International). An α‐tubulin antibody was used to assess the levels of protein loaded per lane (Oncogene Research Products, Cambridge, MA, USA). The immune complex was visualized with an enhanced chemiluminescence (ECL) western blot detection system (Amersham, Aylesbury, UK).
Cell growth and apoptosis. MOLT‐4 cells were seeded at a density of 20 000 cells per well in 96‐well tissue culture plates. After cells were exposed to (3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide (Sigma Chemical Co.) for 1 h before harvest, and chromogenic granules from the cells were dissolved in dimethyl sulfoxide and subjected to examination at 540 nm. The experiments were carried out three times. Apoptosis was assessed by staining with Hoechst 33258 fluorescent dye (Wako Pure Chemical Industries, Osaka, Japan). The number of apoptotic cells was assessed by observing 1000 cells.
Animal model. BALB/c male mice and Fisher 344 male rats were purchased from Japan SLC (Shizuoka, Japan). The animals were maintained according to the institutional guidelines approved by the Committee for Animal Experimentation of Nara Medical University, in accordance with the current regulations and standards of the Ministry of Health, Labour, and Welfare. The animals were used according to the institutional guidelines when they were 5 weeks old. Their cells were briefly trypsinized and washed three times with HBSS. The cells were suspended in HBSS. For examination of tumorigenicity, CT26 cells (1 × 107 cells/100 µΛ) were inoculated subcutaneously into 10 BALB/c mice, and IEC6 (1 × 107 cells/100 µΛ) and IEC6A (1 × 107 cells/100 µΛ) cells were each inoculated subcutaneously into 10 Fisher 344 rats. Anti‐MENK antibody (Progen Biotechnic; 20 µg/mouse) or MENK (Progen Biotechnic, 12 µg/mouse) was injected into the tumors every 7 days. The last injection was carried out 24 h before death. For examination of metastasis, CT26 (1 × 106 cells/100 µΛ) cells were inoculated into the spleen of 10 BALB/c mice, and IEC6 (1 × 106 cells/100 µΛ) and IEC6A (1 × 106 cells/100 µΛ) cells were each inoculated into the spleen of 10 Fisher 344 rats.
Surgical specimens. Formalin‐fixed, paraffin‐embedded archival surgical specimens from 61 patients with primary colon adenocarcinomas that had invaded the subserosal layer were selected randomly from the JR Hiroshima General Hospital of West Japan Railway Company. Out of 61 cases, 25 were Dukes’ stage B (tumor invades beyond the muscularis propria without lymph node metastasis; all cases invaded into the subserosal layer), 21 were stage C (any cases with lymph node metastasis; all cases invaded into subserosal layer), and 15 were stage D (any case with or without lymph node metastases but with distant metastases; all cases metastasized to the liver). Because written informed consent was not obtained, identifying information for all samples was removed before analysis for strict privacy protection; the procedure was in accordance with the Ethical Guidelines for Human Genome/Gene Research enacted by the Japanese Government.
Immunohistochemistry. Consecutive 4‐µM sections were stained immunohistochemically using the immunoperoxidase technique described previously.( 12 ) Anti‐MENK, anti‐CD3, anti‐CD4, and anti‐CD8 (Dako) were used at a concentration of 0.5 µg/mL. Secondary antibodies (Medical and Biological Laboratories) were used at a concentration of 0.2 µg/mL. The specimens were color‐developed with diamine benzidine hydrochloride (Dako). Mayer's hematoxylin (Sigma Chemical Co.) was used for counterstaining. Immunoreactivity was classified according to Allred's score.( 16 ) We divided the immunoreactivity into 4 grades by Allred's score: grade 0, Allred's score 0; grade 1, Allred's score 1–4; grade 2, Allred's score 5–6; and grade 3, Allred's score 7–8.
To confirm the specificity of the immunoreactivity for each antibody, antibody absorbed with each antigen was used for immunostaining (absorption test).( 17 ) MENK was used for absorbing antigens. Antibody (1 nmol) was mixed with an excess amount of each antigen (10 nmol), and incubated for 2 h at 37°C. The solution was spun at 1000 g for 10 min and the supernatant was used for immunostaining.
Enzyme‐linked immunosorbent assay. The concentration of MENK was detected using a MENK ELISA kit (Calbiochem‐Novabiochem International) according to the provider's instructions.
Statistical analysis. Statistical analyses of experimental data were done by Mann–Whitney U‐test, anova test, χ2‐test, and Fisher's exact test. Statistical significance was defined as a two‐sided P‐value of less than 0.05.
Results
Expression of MENK in CRC cell lines. MENK protein expression was examined in intestinal and CRC cells (Fig. 1a). MENK expression was detected in CT26 mouse CRC cells and Colo320 and HT29 human CRC cells. In IEC6 rat intestinal cells, very low levels of MENK protein were observed, whereas increased MENK expression was observed in IEC6A and IEC6 cells that were subjected to AOM‐induced transformation.( 14 ) In order to examine the relationship between metastatic potential and MENK expression, the relationship between MENK concentrations in the culture medium of IEC6, IEC6A, and CT26 cells and the frequency of tumorigenesis and liver metastasis of the abovementioned cells was assessed in syngeneic animals (Fig. 1b). The MENK concentration in non‐metastatic IEC6A cells was higher than that in non‐tumorigenic IEC6 cells (P < 0.0001). Moreover, the MENK concentration in metastatic CT26 cells was higher than that in non‐metastatic IEC6A cells (P < 0.0001). Thus, MENK expression was associated with tumor progression of CRC cells.
Figure 1.
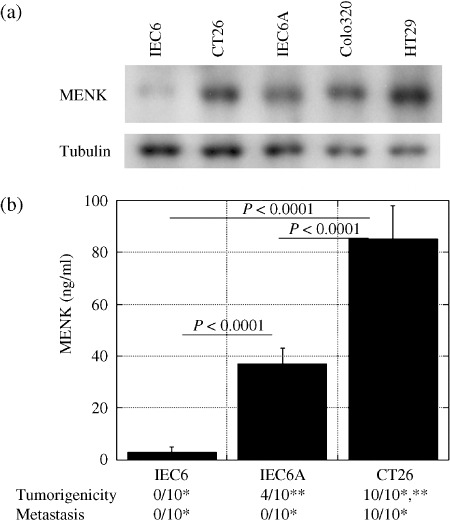
Expression of Met‐enkephalin (MENK) and tumorigenicity or metastasis in colon carcinoma cells. (a) MENK levels in IEC6 rat intestinal cells, IEC6A azoxymethane (AOM)‐induced transformed IEC6 cells, CT26 mouse colon cancer cells, and Colo320 and HT29 human colorectal cancer cells. Tubulin served as a loading control. (b) MENK concentration in cultured medium was compared with tumorigenicity and metastasis of IEC6, IEC6A, and CT26 cells in syngeneic rodent models. *P < 0.0001, **P = 0.0108. Error bar, SD.
Effect of MENK on the intratumoral infiltration of T lymphocytes. MENK is known to be an immunomodulator, especially of T lymphocytes, which express the MENK receptor DOR.( 9 , 18 ) We evaluated the relationship between the tissue concentration of MENK and intratumoral infiltration of CD3‐positive T lymphocytes (Table 1). Fewer T lymphocytes were observed in CT26 cells producing a higher level of MENK, whereas more T lymphocytes (P = 0.0001) infiltrated into IEC6 cells producing a lower level of MENK (P = 0.0005). When the MENK in tumors was neutralized by the anti‐MENK antibody, the number of infiltrating T lymphocytes increased in both CT26 and IEC6A tumors (P < 0.0001 and P = 0.0388, respectively). The growth of CT26 and IEC6A tumors was significantly suppressed by anti‐MENK antibody treatment (Fig. 2). In contrast, MENK treatment enhanced the tumor growth of IEC6A cells. The dosage of anti‐MENK antibody or MENK did not affect the lymphocyte number in the peripheral blood.
Table 1.
Relationship between the Met‐enkephalin (MENK) levels and infiltration of T lymphocytes in subcutaneous tumors
| MENK (ng/g) | T lymphocytes (per mm2) | ||
|---|---|---|---|
| CT26 | |||
| None | 95 ± 16 | 18 ± 6 | |
| MENK antibody † | 17 ± 10 | 96 ± 23 | P < 0.0001 |
| IEC6A | |||
| None | 36 ± 9 | 78 ± 20 | |
| MENK anitbody † | 12 ± 6 | 104 ± 29 | P = 0.0388 |
| MENK injection † | 74 ± 15 | 24 ± 10 | P = 0.0002 |
Anti‐MENK antibody (20 µg/mouse) or MENK (12 µg/mouse) were injected into the tumors at 24 h before death.
Figure 2.
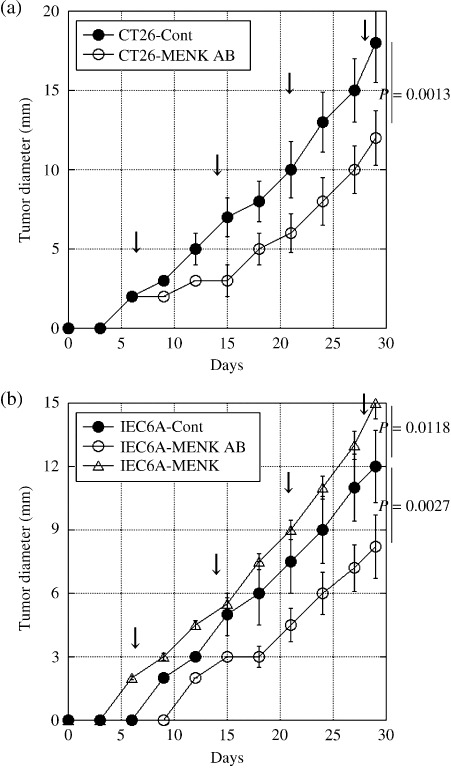
Effects of Met‐enkephalin (MENK) on the tumor growth of colon carcinoma cells. Subcutaneous tumor growth of (a) CT26 and (b) IEC6A cells was examined in syngeneic rodent models. Anti‐MENK antibody (MENK AB) or MENK was administrated intratumorally every 7 days (arrow). Error bar, SD. Cont, PBS.
Intracellular signaling of MENK in T lymphocytes. In order to examine the effect of MENK on the immune system, MOLT‐4 T‐lymphoblastic cells were examined. MOLT‐4 cells expressed DOR but not MENK or CD10 (Fig. 3a). DOR expression was decreased by treatment with DOR siRNA. As shown in Figure 2(b), MENK treatment decreased the number of MOLT‐4 cells exposed to the control siRNA in a dose‐dependent manner, whereas DOR siRNA treatment of MOLT‐4 cells recovered the cells from the MENK‐induced decrease in cell number (P = 0.0005). The effects of MENK on MAPK were examined in MOLT‐4 cells (Fig. 3c). MENK treatment decreased ERK1/2 and p38 phosphorylation but increased JNK phosphorylation in MOLT‐4 cells. Treatment with a JNK inhibitor partially recovered the MENK‐induced decrease in cell number, whereas treatment with ERK1/2 and p38 inhibitors did not show any significant effects on cell number (Fig. 3b). The increase in the number of apoptotic cells in MENK‐treated MOLT‐4 cells was more significantly reduced with JNK‐inhibitor treatment than with p38‐inhibitor and ERK‐inhibitor treatments (Fig. 3d).
Figure 3.
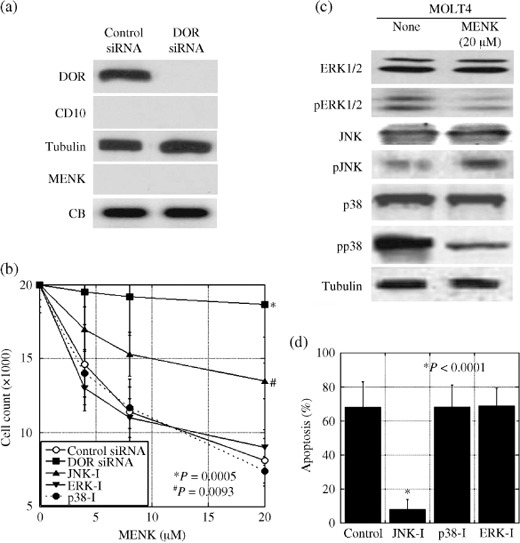
Effects of Met‐enkephalin (MENK) on cell growth and phosphorylation of mitogen‐activated protein kinase (MAPK) family members in MOLT‐4 cells. (a) δ‐Opioid receptor (DOR) and CD10 expression and MENK secretion were detected by immunoblotting in MOLT‐4 cells. (b) Effect of MENK and MAPK inhibitors on cell growth in MOLT‐4 cells. (c) Effect of MENK on the phosphorylation of MAPK in MOLT‐4 T lymphocytes. (d) Apoptotic cells were counted in MENK (20 µM)‐treated MOLT‐4 cells by Hoechst staining. Tubulin and Coomassie blue served as loading controls. ERK‐I, ERK1/2 inhibitor; JNK‐I, c‐jun N‐terminal kinase inhibitor; p38‐I, p38 inhibitor; pp38, phosphorylated p38; siRNA, short interfering RNA. Error bars, SD from three independent assays.
Expression of MENK in human CRC. Next, we examined the expression of MENK in 61 CRC by immunohistochemistry (Fig. 4; Table 2). MENK immunoreactivity was detected in the cytoplasm of cancer cells (Fig. 4b,c) but not in normal epithelium (Fig. 4a). The immunohistochemical staining disappeared after treatment with antigen‐adsorbed antibodies; this showed the specificity of the staining technique (Fig. 4d). In the 61 CRC, all tumors showed MENK expression (Table 2), which was associated with Dukes’ stage (P < 0.0001), nodal metastasis (P = 0.0003), and liver metastasis (P < 0.0001), but not with histological differentiation. To confirm the secretion of MENK in 19 CRC, the MENK concentration in tumor tissues was examined (Fig. 4e). The MENK concentration in Dukes’ C cases (53 ± 24 ng/g) was higher than that in Dukes’ B cases (10 ± 10 ng/g) (P = 0.0032). Immunoreactivity and tissue concentration of MENK were not associated with serum levels of carcinoembryonic antigen and sialyl Lewis X antigen nor with Ki‐67 labeling in CRC cells (data not shown).
Figure 4.
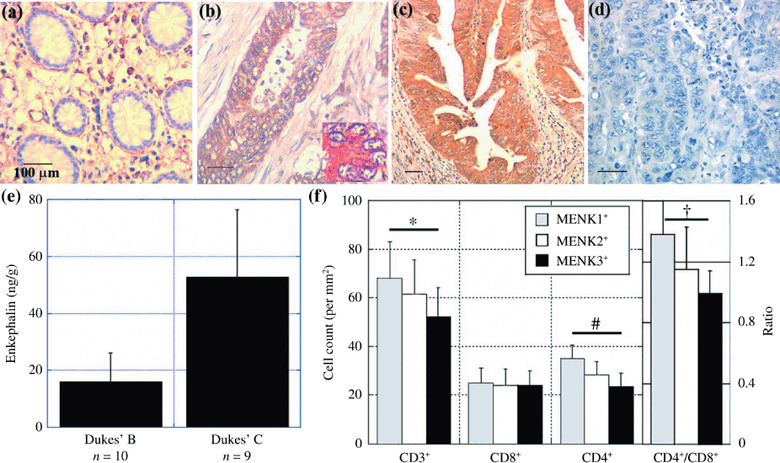
Expression of Met‐enkephalin (MENK) and infiltration of T lymphocytes in human colorectal cancer (CRC). The expression of MENK was examined by immunohistochemistry in (a) normal colonic mucosa, (b) Dukes’ B case, and (c) Dukes’ D case (with liver and nodal metastasis). The inset shows MENK immunostaining revealing cytoplasmic localization of MENK immunoreactivity. (d) The antibody adsorbed by the antigen was used to confirm the specificity of immunoreactivity. Scale bar = 100 µM. (e) Tissue concentration of MENK in 19 CRC cases. (f) Tumor‐infiltrating T lymphocytes were counted using cell markers CD3+, CD4+, and CD8+ and compared with MENK expression in CRC. Bar, SD. *P = 0.0085, # P < 0.0001, † P = 0.0005
Table 2.
Expression of Met‐enkephalin (MENK) in 61 human human colorectal cancers
| Pathological parameters | Immunohistochemical grade ‡ | P‐value | ||
|---|---|---|---|---|
| 1 | 2 | 3 | ||
| Differentiation | ||||
| Well | 13 | 15 | 6 | |
| Mod | 15 | 7 | 5 | NS § |
| Pathological stage † | ||||
| B | 21 | 4 | 0 | |
| C | 7 | 11 | 3 | |
| D | 0 | 7 | 8 | <0.0001 |
| Nodal metastasis | ||||
| Negative | 21 | 4 | 5 | |
| Positive | 7 | 18 | 6 | 0.0003 |
| Liver metastasis | ||||
| Negative | 28 | 15 | 3 | |
| Positive | 0 | 7 | 8 | <0.0001 |
Well, well‐differentiated adenocarcinoma; Mod, moderately differentiated adenocarcinoma.
Dukes’ stage B, tumor invades beyond the muscularis propria without lymph node metastasis; Dukes’ stage C, any case with lymph node metastasis; Dukes’ stage D, any case with or without lymph node metastasis but with distant metastasis.
We classified the immunoreactivity into four grades by Allred's score( 16 ): grade 0, Allred's score 0; grade 1, Allred's score between 1 and 4; grade 2, Allred's score between 5 and 6; grade 3, Allred's score between 7 and 8.
Statistical difference was calculated by a χ2‐test. NS, not significant.
In the 61 CRC, tumors with stronger MENK immunoreactivity were associated with less infiltration of CD3+ T lymphocytes; the numbers of infiltrating CD3+ T lymphocytes in tumors with MENK immunoreactivity grades 1, 2, and 3 were 64 ± 19, 61 ± 16, and 52 ± 12, respectively (P = 0.0085) (Fig. 4f). In the T lymphocyte subsets, MENK immunoreactivity was associated with less infiltration of CD4+ T lymphocytes (P < 0.0001) and a lower CD4+/CD8+ ratio (P = 0.0005) (Fig. 4f).
Discussion
In the present study, we have reported that CRC cells express MENK. MENK suppresses T lymphocytes, especially CD4+ T lymphocytes, and is subsequently associated with disease progression and metastasis.
Morphine can decrease the effectiveness of several functions of both natural and adaptive immunity and significantly reduce cellular immunity.( 6 ) The differentiation function of immune cells is significantly affected by opioids.( 19 ) In animal studies, morphine is consistently associated with increased morbidity and mortality because it leads to the worsening of cancer.( 6 ) Chronic administration of opioids decreases the proliferative capacity of macrophage progenitor cells and lymphocytes. T‐lymphocyte responses are suppressed by morphine, as assessed by the inhibition of induction of delayed‐type hypersensitivity reactions and cytotoxic T‐lymphocyte activity, modulation of T‐lymphocyte antigen expression, and suppression of responses to T‐lymphocyte mitogens.( 20 ) Morphine mainly activates the µ‐opioid receptor, whereas MENK activates DOR specifically. DOR activation is associated with suppression of immunity. DOR is expressed in phytohemagglutinin‐stimulated CD4+ and CD8+ T lymphocytes, and modulates T‐lymphocyte proliferation, interleukin‐2 production, chemotaxis, and intracellular signaling.( 21 ) Exposure of a pure DOR antagonist results in dose‐dependent suppression of concanavalin A‐induced rat T‐lymphocyte proliferation.( 22 ) In contrast, some reports claim that in vivo administration of a relatively low dose (2.5 mg/kg, intraperitoneal) of MENK stimulates the proliferative ability of both T and B lymphocytes in mice.( 23 ) The discrepancy might be explained by differences in the intracellular signaling pathway. Our data suggest that in MENK‐induced MAPK activation, activation of JNK might induce apoptosis, whereas activation of ERK1/2 might induce proliferation. MENK concentration, concurrent effects of other cytokines, growth factors with MENK, and other factors might affect the differential activation of the MAPK family. We also examined the effect of MENK on macrophages; however, macrophages express CD10, a MENK‐degrading enzyme, which abrogates MENK‐induced cell suppression (data not shown).
To elucidate the inhibitory effects of MENK on T lymphocytes, we focused on the intracellular signaling pathway of MENK. DOR, a receptor with high affinity for MENK, is a member of the G‐protein‐related receptor family.( 24 ) DOR transmits intracellular signals through heterotrimeric G proteins to the MAPK family.( 25 ) In DOR‐transfected Jurkat cells, DOR agonists stimulate MAPK phosphorylation in a Ras‐independent and protein kinase C‐dependent manner.( 26 ) The diversity of the roles of DOR depends on the multifunctionality of MAPK, which is attributed to the activation profiles of the members of the MAPK family.( 27 ) DOR and κ‐opioid receptor are expressed in T lymphocytes in the same way as they are expressed in MOLT‐4 cells.( 28 , 29 ) We observed that in MOLT‐4 cells, MENK suppressed cell growth in a dose‐dependent manner and decreased the phosphorylation of ERK1/2 and p38 but increased the phosphorylation of JNK. These results are supported by the findings of Singhal et al., in which DOR‐induced JNK activation is associated with T‐lymphocyte apoptosis.( 10 ) Additionally, ERK phosphorylation also attenuates T‐lymphocyte activation through DOR, which is expressed by activated T lymphocytes.( 30 )
With the exception of MAPK activation, DOR affects the activation of T lymphocytes in all other intracellular events. DOR activation affects intracellular calcium concentration and DOR agonists increase intracellular free calcium concentrations, whereas µ‐opioid receptor is not associated with calcium concentration.( 31 ) In contrast, DOR is negatively coupled to adenylate cyclase for the production of cyclic AMP.( 7 ) DOR also plays a role in transcriptional regulation by activating activator protein‐1, activator protein‐2, c‐fos, Ikaros‐1, Ikaros‐2, nuclear factor‐κB, and activating transcription factor‐2 in immune cells, including T lymphocytes.( 8 , 32 ) Ikaros enhances DOR gene expression transcriptionally in phytohemagglutinin‐activated T lymphocytes.( 33 ) Thus, DOR might provide a negative feedback for T‐lymphocyte activation through several intracellular signaling mechanisms.
Previous reports show that MENK is associated with the development and progression of CRC. High levels of MENK are detected in colon cancer.( 11 ) MENK enhances AOM‐induced colon carcinogenesis in Wistar rats.( 34 ) The inhibitory effect of MENK on T lymphocytes is thought to be associated with the pro‐tumoral effect of MENK. We assessed the relationship of MENK expression with T‐lymphocyte infiltration into the tumors in CRC; these parameters were found to be correlated. In particular, CD4+ T lymphocytes decreased according to MENK expression. Moreover, all metastatic tumors we examined showed a high grade of MENK expression (grade 3), and reduced CD3+ and CD4+ T‐lymphocyte infiltration (data not shown). These findings suggest that MENK expression by CRC cells inhibits anticancer immunity in the host. In our study, because MENK was detected at various concentrations in all cancer tissues in Dukes’ B and Dukes’ C cases, MENK expression is suggested to be a common event that enhances the progression and metastasis of CRC. In the present study, anti‐MENK antibody treatment showed significant inhibition of tumor growth. We also attempted to detect the MENK concentration of CRC tissues to evaluate their metastatic potential. Therefore, targeting MENK might be a reasonable approach for suppressing the progression and metastasis of CRC.
Acknowledgments
This work was supported in part by a Grant‐in‐Aid for Scientific Research from the Ministry of Health, Labour, and Welfare, Japan.
References
- 1. Kuniyasu H, Yano S, Sasaki T, Sasahira T, Sone S, Ohmori H. Colon cancer cell‐derived high mobility group 1/amphoterin induces growth inhibition and apoptosis in macrophages. Am J Pathol 2005; 166: 751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kuniyasu H, Sasaki T, Sasahira T, Ohmori H, Takahashi T. Depletion of tumor‐infiltrating macrophages is associated with amphoterin expression in colon cancer. Pathobiology 2004; 71: 129–36. [DOI] [PubMed] [Google Scholar]
- 3. Dong H, Strome SE, Salomao DR et al . Tumor‐associated B7–H1 promotes T‐cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002; 8: 793–800. [DOI] [PubMed] [Google Scholar]
- 4. Nakashima M, Sonoda K, Watanabe T. Inhibition of cell growth and induction of apoptotic cell death by the human tumor‐associated antigen RCAS1. Nat Med 1999; 5: 938–42. [DOI] [PubMed] [Google Scholar]
- 5. Oshikiri T, Miyamoto M, Morita T et al . Tumor‐associated antigen recognized by the 22‐1‐1 monoclonal antibody encourages colorectal cancer progression under the scanty CD8+ T cells. Clin Cancer Res 2006; 12: 411–16. [DOI] [PubMed] [Google Scholar]
- 6. Sacerdote P. Opioids and the immune system. Palliat Med 2006; 20: s9–15. [PubMed] [Google Scholar]
- 7. Sharp BM, Shahabi NA, Heagy W et al . Dual signal transduction through δ opioid receptors in a transfected human T‐cell line. Proc Natl Acad Sci USA 1996; 93: 8294–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martin‐Kleiner I, Balog T, Gabrilovac J. Signal transduction induced by opioids in immune cells: a review. Neuroimmunomodulation 2006; 13: 1–7. [DOI] [PubMed] [Google Scholar]
- 9. Sharp BM. Multiple opioid receptors on immune cells modulate intracellular signaling. Brain Behav Immun 2006; 20: 9–14. [DOI] [PubMed] [Google Scholar]
- 10. Singhal P, Kapasi A, Reddy K, Franki N. Opiates promote T cell apoptosis through JNK and caspase pathway. Adv Exp Med Biol 2001; 493: 127–35. [DOI] [PubMed] [Google Scholar]
- 11. Davis WG, Tormey WP, Delaney PV. Enkephalins in large bowel malignancy and in acute appendicitis. Gut 1979; 20: 865–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kuniyasu H, Yasui W, Shinohara H et al . Induction of angiogenesis by hyperplastic colonic mucosa adjacent to colon cancer. Am J Pathol 2000; 157: 1523–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuniyasu H, Yoshida K, Yokozaki H et al . Expression of cripto, a novel gene of the epidermal growth factor family, in human gastrointestinal carcinomas. Jpn J Cancer Res 1991; 82: 969–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sasaki T, Yoshida K, Shimura H et al . Inhibitory effect of linoleic acid on transformation of IEC6 intestinal cells by in vitro azoxymethane treatment. Int J Cancer 2006; 118: 593–9. [DOI] [PubMed] [Google Scholar]
- 15. Kuniyasu H, Yasui W, Kitahara K et al . Growth inhibitory effect of interferon‐β is associated with the induction of cyclin‐dependent kinase inhibitor p27Kip1 in a human gastric carcinoma cell line. Cell Growth Differ 1997; 8: 47–52. [PubMed] [Google Scholar]
- 16. Allred DC, Harvey JM, Berardo M, Clark GM. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol 1998; 11: 155–68. [PubMed] [Google Scholar]
- 17. Larsson LI. Peptide immunocytochemistry. Prog Histochem Cytochem 1981; 13: 1–85. [PubMed] [Google Scholar]
- 18. Cadet P, Rasmussen M, Zhu W, Tonnesen E, Mantione KJ, Stefano GB. Endogenous morphinergic signaling and tumor growth. Front Biosci 2004; 9: 3176–86. [DOI] [PubMed] [Google Scholar]
- 19. Roy S, Loh HH. Effects of opioids on the immune system. Neurochem Res 1996; 21: 1375–86. [DOI] [PubMed] [Google Scholar]
- 20. Eisenstein TK, Hilburger ME. Opioid modulation of immune responses: effects on phagocyte and lymphoid cell populations. J Neuroimmunol 1998; 83: 36–44. [DOI] [PubMed] [Google Scholar]
- 21. Sharp BM, McAllen K, Gekker GA, Shahabi NA, Peterson PK. Immunofluorescence detection of δ opioid receptors (DOR) on human peripheral blood CD4+ T cells and DOR‐dependent suppression of HIV‐1 expression. J Immunol 2001; 167: 1097–102. [DOI] [PubMed] [Google Scholar]
- 22. Spetea M, Harris HE, Berzetei‐Gurske IP, Klareskog L, Schmidhammer H. Binding, pharmacological and immunological profiles of the δ‐selective opioid receptor antagonist HS 378. Life Sci 2001; 69: 1775–82. [DOI] [PubMed] [Google Scholar]
- 23. Gabrilovac J, Marotti T. Gender‐related differences in murine T‐ and B‐lymphocyte proliferative ability in response to in vivo Met5 enkephalin administration. Eur J Pharmacol 2000; 392: 101–8. [DOI] [PubMed] [Google Scholar]
- 24. Mayer P, Tischmeyer H, Jayasinghe M et al . A delta opioid receptor lacking the third cytoplasmic loop is generated by atypical mRNA processing in human malignomas. FEBS Lett 2000; 480: 156–60. [DOI] [PubMed] [Google Scholar]
- 25. Fukuda K, Kato S, Morikawa H, Shoda T, Mori K. Functional coupling of the δ‐, µ‐, and κ‐opioid receptors to mitogen‐activated protein kinase and arachidonate release in Chinese hamster ovary cells. J Neurochem 1996; 67: 1309–16. [DOI] [PubMed] [Google Scholar]
- 26. Shahabi NA, Daaka Y, McAllen K, Sharp BM. δ Opioid receptors expressed by stably transfected jurkat cells signal through the map kinase pathway in a ras‐independent manner. J Neuroimmunol 1999; 94: 48–57. [DOI] [PubMed] [Google Scholar]
- 27. Gutkind JS. The pathways connecting G protein‐coupled receptors to the nucleus through divergent mitogen‐activated protein kinase cascades. J Biol Chem 1998; 273: 1839–42. [DOI] [PubMed] [Google Scholar]
- 28. Sharp BM. Opioid receptor expression and intracellular signaling by cells involved in host defense and immunity. Adv Exp Med Biol 2003; 521: 98–105. [PubMed] [Google Scholar]
- 29. Wick MJ, Minnerath SR, Roy S, Ramakrishnan S, Loh HH. Differential expression of opioid receptor genes in human lymphoid cell lines and peripheral blood lymphocytes. J Neuroimmunol 1996; 64: 29–36. [DOI] [PubMed] [Google Scholar]
- 30. Shahabi NA, McAllen K, Matta SG, Sharp BM. Expression of δ opioid receptors by splenocytes from SEB‐treated mice and effects on phosphorylation of MAP kinase. Cell Immunol 2000; 205: 84–93. [DOI] [PubMed] [Google Scholar]
- 31. Sharp BM, McKean DJ, McAllen K, Shahabi NA. Signaling through δ opioid receptors on murine splenic T cells and stably transfected Jurkat cells. Ann N Y Acad Sci 1998; 840: 420–4. [DOI] [PubMed] [Google Scholar]
- 32. Shahabi NA, McAllen K, Sharp BM. Phosphorylation of activating transcription factor in murine splenocytes through δ opioid receptors. Cell Immunol 2003; 221: 122–7. [DOI] [PubMed] [Google Scholar]
- 33. Sun PLH. Transcriptional regulation of mouse δ‐opioid receptor gene. Ikaros‐2 and upstream stimulatory factor synergize in trans‐activating mouse δ‐opioid receptor gene in T cells. J Biol Chem 2003; 278: 2304–8. [DOI] [PubMed] [Google Scholar]
- 34. Iishi H, Tatsuta M, Baba M, Okuda S, Taniguchi H. Enhancement by methionine enkephalin of colon carcinogenesis induced by azoxymethane. Cancer Res 1991; 51: 785–8. [PubMed] [Google Scholar]