

Abstract
The RUNX1/EVI1 chimeric transcription factor produced by t(3;21) causes leukemic transformation in hematopoietic stem cell tumors, possibly through a differentiation block of malignant myeloid progenitors. A dominant negative effect over wild‐type RUNX1 has been shown to constitute one of the underlying molecular mechanisms. We introduced RUNX1/EVI1 cDNA into LG‐3 cells that differentiate along the myeloid lineage upon exposure to granulocyte colony stimulating factor, and confirmed that RUNX1/EVI1 suppressed the differentiation. To further investigate the molecular mechanisms of RUNX1/EVI1‐mediated differentiation block, we analyzed RUNX1/EVI1's effect on the functions of CCAAT–enhancer binding protein α (C/EBPα), a key transcriptional regulator that induces granulocytic differentiation. RUNX1/EVI1 was found to associate with C/EBPα. By using a reporter assay with the CEBPA promoter, we observed a dominant negative effect of RUNX1/EVI1 over C/EBPα‐mediated transcriptional activation via the carboxyl terminal‐binding protein (CtBP)‐binding site in the EVI1 portion. In a gel‐shift assay, RUNX1/EVI1 downregulated the DNA‐binding activity of C/EBPα. Therefore, recruitment of histone deacetylase via CtBP and disruption of DNA binding could be likely scenarios for the RUNX1/EVI1‐induced dominant repression on C/EBPα. Importantly, coexpression of C/EBPα restored the differentiation ability of the RUNX1/EVI1‐expressing LG‐3 cells. All of these data argue that inhibition of C/EBPα function may be causatively related to the leukemogenic potential of RUNX1/EVI1. (Cancer Sci 2007; 98: 1752–1757)
The t(3;21)(q26;q22) translocation is a cytogenetic hallmark of chronic myelogenous leukemia in blastic crisis, myelodysplastic syndrome in leukemic transformation and de novo acute myelogenous leukemia (AML).( 1 , 2 , 3 , 4 ) This translocation‐associated leukemia is of either myeloid or megakaryocytic origin. In the joining region of t(3;21), the RUNX1 gene on 21q22 is fused with the EVI1 gene on 3q26.( 5 ) The resultant RUNX1/EVI1 fusion gene is translated in‐frame to an aberrant transcription factor in which the N‐terminus of RUNX1, including its DNA‐binding domain Runt, is connected to almost the entire sequence of EVI1. This chimeric transcription factor could be behind the leukemogenesis caused by t(3;21).
RUNX1 is a member of the Runt family of transcription factors that regulates a number of hematopoietic cell‐specific genes. Depending on the Runt domain, RUNX1 binds to a specific DNA consensus sequence named PEBP2 (ACCRCA) and forms a heterodimeric active transcription factor complex with the non‐DNA binding β subunit (CBFβ–PEBP2β). RUNX1 plays an essential role in establishing definitive hematopoiesis in the fetal liver,( 6 , 7 ) and maturating megakaryocytes in the adult bone marrow.( 8 ) However, EVI1 is a zinc‐finger protein that displays versatile functions such as inhibition of transforming growth factor (TGF)‐β signaling,( 9 ) repression of c‐Jun N‐terminal kinase (JNK) activity( 10 ) and stimulation of activating protein (AP)‐1 activity.( 11 ) The molecular characterization of RUNX1/EVI1 points to two major functions: one is a dominant suppressive function over wild‐type RUNX1 and the other is EVI1's own function. Recent gene‐engineered studies have provided us with significant information on the in vivo functions of RUNX1/EVI1. RUNX1/EVI1 knock‐in heterozygous mice show defective hematopoiesis in the fetal liver similar to Runx1 knockout mice, but possess dysplastic hematopoietic progenitors with high self‐renewal capacity.( 12 ) Notably, RUNX1/EVI1 knock‐in chimeric mice have been reported to develop acute megakaryoblastic leukemia.( 13 )
CCAAT/enhancer binding protein α (C/EBPα) is a leucine zipper transcription factor that regulates the expression of specific target genes containing C/EBP sites in their promoters, and thus plays distinct roles in the differentiation process of various cell types.( 14 , 15 ) In the hematopoietic system, such genes include CEBPA itself,( 16 ) CEBPE ( 16 , 17 ) and granulocyte colony‐stimulating factor (G‐CSF) receptor.( 16 , 18 , 19 ) Conditional expression of C/EBPα is sufficient to trigger terminal granulocytic differentiation( 20 , 21 , 22 , 23 ) and block the monocytic differentiation program.( 20 , 22 ) Further, Cebpa knockout mice show profound defects in their granulocytic differentiation, whereas all other hematopoietic cells are present in normal numbers,( 18 ) indicating its critical role in granulopoiesis. Several lines of evidence suggest that disturbance of C/EBPα signaling is one of the major molecular events in myeloid malignancies. Ten percent of patients with AML that belong to M1 or M2 (according to the French–American–British [FAB] classification) and do not have a frequent cytogenetic abnormality, such as t(8;21)(q22;q22), carry heterozygous CEBPA gene mutations resulting in production of the truncated protein with a dominant negative function.( 17 , 24 , 25 ) RUNX1/ETO, generated by the t(8;21) translocation in AML (FAB‐M2), represses the transcription of CEBPA mRNA by suppressing C/EBPα's autoregulatory loop,( 16 ) whereas PML/RARα, caused by t(15;17)(q21;q22) in acute promyelocytic leukemia (FAB‐M3), inhibits the function of C/EBPα.( 26 ) Because RUNX1/EVI1 and RUNX1/ETO show high structural similarity, it could be possible that RUNX1/EVI1 mediates its differentiation block effect on myeloid progenitors through inhibiting transcription of the CEBPA gene.
In the present study, we investigated whether RUNX1/EVI1 affects the expression and function of C/EBPα. We first confirmed that RUNX1/EVI1 blocked granulocytic differentiation in LG‐3 cells upon granulocyte colony‐stimulating factor (G‐CSF) exposure. Physical interaction between RUNX1/EVI1 and C/EBPα was detected in the immunoprecipitation assay. RUNX1/EVI1 significantly inhibited transcriptional activation of the CEBPA promoter induced by C/EBPα itself, depending on one of the two CtBP‐binding sites in EVI1. Further, RUNX1/EVI1 repressed the DNA‐binding activity of C/EBPα. These data indicate that RUNX1/EVI1 inhibits molecular functions of C/EBPα, possibly through recruiting the co‐repressor CtBP/histone deacetylase complex to the C/EBPα‐targeting promoter and suppressing DNA binding of C/EBPα. Importantly, the observation that coexpression of C/EBPα restored the RUNX1/EVI1‐induced differentiation block in LG‐3 cells suggests the influence of RUNX1/EVI1 on C/EBPα's biological function.
Materials and Methods
Plasmid construction. The pcDNA3‐C/EBPα and ptk81‐luc‐C/EBPα promoters were described previously.( 16 ) The FLAG‐tag (DYKDDDDK) was created upstream of the translation initiation site of wild‐type C/EBPα cDNA by the method of polymerase chain reaction (PCR) amplification. The resultant cDNA was inserted into the EcoRI site of pME18S in the sense orientation to give pME18S‐FLAG‐C/EBPα. pME18S‐RUNX1/EVI1 was described previously.( 27 ) pME18S‐FLAG‐RUNX1/EVI1 was also created in the same way. FLAG‐RUNX1/EVI1 and FLAG‐C/EBPα cDNA were cloned into the EcoRI sites of the pCXN2 and pCAGIPuro expression vectors, respectively. For construction of RUNX1/EVI1 deletion mutants, new restriction enzyme sites, NheI (140), NheI (536), EcoRV (1821), PvuII (3511), NheI (3664) and NheI (3844) (numbers in parentheses indicate nucleotide numbers from the start site of translation to the cutting site of the enzyme), were created in the pME18S‐RUNX1/EVI1 expression vector by site‐directed mutagenesis. Deletion mutants ΔRunt, ΔZF1, ΔZF2 and ΔAD were constructed by deleting internal fragments from mutagenic NheI (140) to mutagenic NheI (536), EcoRV (1102) to mutagenic EcoRV (1821), Eco473 (3227) to mutagenic PvuII (3511) and mutagenic NheI (3664) to mutagenic NheI (3844), respectively. For construction of the mCtBP mutant, adenine (2816), cytosine (2818) and thymine (2819) were substituted with cytosine, thymine and cytosine, respectively, by site‐directed mutagenesis. pME18S‐FLAG‐ΔRunt and pME18S‐FLAG‐mCtBP were constructed as described above.
Cell culture. LG‐3 cells( 28 ) were cultured in RPMI‐1640 medium supplemented with 10% fetal calf serum (FCS), 10 ng/mL mouse interleukin (IL)‐3 and 50 µM 2‐mercaptoethanol. COS‐7 and CV‐1 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FCS.
Establishment of stable transfectants and granulocytic differentiation assay. To establish stable transfectants of FLAG‐RUNX1/EVI1, 1 × 107 LG‐3 cells were electroporated with 20 µg of pCXN2‐FLAG‐RUNX1/EVI1 plasmid at 380 V and 975 µF using a Gene Pulser (Bio‐Rad Laboratories, Hercules, CA, USA). Electroporated cells were selected with 0.8 µg/mL G418 (Sigma‐Aldrich, St Louis, MO, USA) and cloned by limiting dilution. Surviving clones were screened for expression of RUNX1/EVI1 by western blot analysis using anti‐FLAG M2 antibody (Sigma‐Aldrich). To further obtain double transfectants of RUNX1/EVI1 and C/EBPα, 1 × 107 LG‐3 cells stably expressing FLAG‐RUNX1/EVI1 were electroporated with 20 µg of pCAGIPuro‐FLAG‐C/EBPα plasmid at 380 V and 975 µF using a Gene Pulser. Electroporated cells were selected with 0.75 µg/mL puromycin (Sigma‐Aldrich) and cloned by limiting dilution. Surviving clones were screened for concomitant expression of RUNX1/EVI1 and C/EBPα by western blot analysis using anti‐FLAG M2 antibody.
For the induction of granulocytic differentiation, LG‐3 cells were washed once with phosphate‐buffered saline and placed in RPMI‐1640 medium supplemented with 10% FCS, 50 µM 2‐mercaptoethanol and 2 ng/mL G‐CSF instead of IL‐3. After 7 days, morphological studies were carried out on cytospin preparations with Wright–Giemsa and myeloperoxidase stainings.
Western blotting and immunoprecipitation. COS‐7 cells were transfected with full‐length RUNX1/EVI1 or its mutant expression plasmids alone or in combination with the FLAG‐tagged C/EBPα expression plasmid using the DEAE‐dextran method as described previously.( 29 ) Western blot analyses were carried out as described previously( 29 ) using anti‐RUNX1 antiserum (Cell Signaling Technology, Beverly, MA, USA) and anti‐FLAG M2 antibody. Immunoprecipitation was carried out using anti‐FLAG M2 antibody conjugated with protein G‐Sepharose (Amersham Pharmacia Biotech, Piscataway, NJ, USA), and immunoprecipitates were analyzed by sodium dodecylsulfate–polyacrylamide gel electrophoresis.
Luciferase assay. CV‐1 cells were transfected with 200 ng of ptk81‐luc‐C/EBPα reporter plasmid alone or along with 100 ng of expression plasmids using Lipofectamine2000 (Invitrogen, Rockville, MD, USA). Luciferase assays were carried out using a Dual‐luciferase Reporter Assay System (Promega, Madison, WI, USA). The phRL/CMV plasmid (10 ng; Promega) was cotransfected as an internal control of transfection efficacy and the data were normalized to Rennilla luciferase activity. All transfections were carried out at least three times and similar results were obtained.
Electrophoretic mobility shift assay. Preparation of FLAG‐C/EBPα or FLAG‐RUNX1/EVI1‐expressing COS‐7 lysates and binding reactions were carried out as described previously.( 16 ) The G‐CSF receptor promoter oligonucleotide had a sequence of 5′‐GGAAGGTGTTGCAATCCCCAGC‐3′, in which the C/EBP binding site is underlined. In competition studies, a 300‐fold molar excess of unlabeled specific or non‐specific oligonucleotide was added with the probe. The non‐specific oligonucleotide had a sequence of 5′‐GGAAGGTGTTGGATACCCCAGC‐3′, in which the C/EBP binding site was substituted with the GATA binding site. For supershift experiments, 5 µL of anti‐C/EBPα polyclonal antibody 14AA (Santa Cruz Biotechnology, Santa Cruz, CA) was added. Reactions were electrophoresed at 165 V on 10% Tris‐borate‐EDTA (TBE) gels in 0.25 × TBE at 25°C.
Results
RUNX1/EVI1 suppresses granulocytic differentiation in LG‐3 cells upon G‐CSF treatment. RUNX1/EVI1 has been reported to inhibit granulocytic differentiation in 32D cells upon G‐CSF stimulation.( 27 ) We confirmed the same effect of RUNX1/EVI1 using another murine IL‐3‐dependent myeloid progenitor cell line, LG‐3, which differentiates into mature granulocyte in response to G‐CSF. By transfecting the FLAG‐tagged RUNX1/EVI1 expression plasmid (pCXN2‐FLAG‐RUNX1/EVI1) into LG‐3 cells, we established several stable cell lines overexpressing RUNX1/EVI1. Western blot analysis with anti‐FLAG antibody verified that clones R/E11 and R/E14 expressed high levels of the 210‐kDa RUNX1/EVI1 protein (Fig. 1a). Two clones transfected with the empty plasmid were used as mock‐transfected controls (M5 and M20). R/E11 and R/E14 showed more rapid proliferation than M5 and M20 in the presence of IL‐3, although only to a slight degree (data not shown). However, the RUNX1/EVI1‐overexpressing cells did not become growth factor independent as IL‐3 was required for their continued growth. To test the effect of overexpressed RUNX1/EVI1 on granulocytic differentiation, LG‐3 cells were induced into terminal granulocytic differentiation by treatment with G‐CSF. As expected, Wright–Giemsa staining of the mock cells before and after 7 days of treatment with G‐CSF demonstrated dramatic morphological changes with myeloid blasts seen at day 0 and polymorphonuclear cells appearing at day 7 (data not shown). In contrast, the RUNX1/EVI1‐overexpressing cells hardly differentiated into mature granulocytes even after 7 days of the treatment. Differential counts of these cells at day 7 of culture are shown in Fig. 1b. The RUNX1/EVI1‐expressing clones reproducibly displayed lower percentages of mature granulocyte (percentages of the stab and segmented forms; 37% in M5 and 50% in M20 versus 10% in R/E11 and 12% in R/E14) and higher percentages of blast (6% in M5 and 10% in M20 versus 43% in R/E11 and 48% in R/E14) than the controls. Myeloperoxidase positivity, which indicates mature granulocytes, was also significantly lower in the RUNX1/EVI1‐expressing cells compared to the mock cells. These data demonstrate that RUNX1/EVI1‐positive LG‐3 cells arrest at the myeloblast stage even after induction with G‐CSF.
Figure 1.
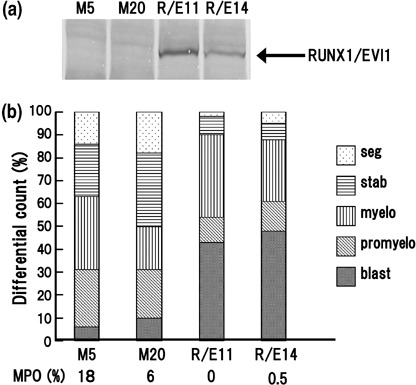
RUNX1/EVI1 represses granulocyte colony‐stimulating factor (G‐CSF)‐induced granulocytic differentiation in LG‐3 cells. (a) Western blot analysis using anti‐FLAG M2 antibody for RUNX/EVI1 proteins from whole cell lysates of RUNX1/EVI1‐expressing LG‐3 clones, R/E11 and R/E14, as well as mock clones, M5 and M20. (b) Granulocytic differential counts of the mock and RUNX1/EVI1‐expressing clones after 7 days of treatment with 2 ng/mL of G‐CSF are shown. Percentages of myeloperoxidase‐positive cells in each clone are shown below the graph. This experiment was carried out four times independently and similar results were obtained. Representative data are shown.
RUNX1/EVI1 associates with C/EBPαin vivo. To clarify the molecular effect of RUNX1/EVI1 on C/EBPα, we first tested whether RUNX1/EVI1 and C/EBPα physically interact in vivo. Mock or FLAG‐tagged C/EBPα expression plasmid (pME18S‐FLAG‐C/EBPα) was cotransfected with RUNX1/EVI1 expression plasmid (pME18S‐RUNX1/EVI1) in COS‐7 cells, and immunoprecipitation was carried out using an antibody against FLAG. Expression of RUNX1/EVI1 (Fig. 2, upper panel) and C/EBPα (middle panel) was confirmed by western blot analysis with anti‐RUNX1 and anti‐FLAG antibodies, respectively. RUNX1/EVI1 was immunoprecipitated with anti‐FLAG antibody only when FLAG‐tagged C/EBPα was co‐expressed (lower panel). This indicates that RUNX1/EVI1 binds to C/EBPαin vivo. To determine the C/EBPα‐binding region in RUNX1/EVI1, we then transfected a set of RUNX1/EVI1 plasmids expressing its deletion mutants ΔRunt, ΔZF1, ΔZF2 and ΔAD. ΔRunt is a mutant lacking the Runt domain of RUNX1, whereas ΔZF1, ΔZF2 and ΔAD are mutants lacking the first and second zinc finger, and the acidic domains of EVI1, respectively. The plasmid designated pME18S‐mCtBP, which produces the RUNX1/EVI1 point mutant harboring normal N‐terminal (PFDLT) but substituted C‐terminal (PLDLS to PLASS) CtBP‐binding motifs in the EVI1 portion, was also included in this study. This amino acid mutation has been reported to eliminate EVI1's binding to CtBP.( 30 ) All of these mutants were shown to be expressed at a comparable level by western blot analysis (Fig. 2, upper panel). Surprisingly, all of these mutants were again immunoprecipitated with anti‐FLAG antibody in the presence of FLAG‐tagged C/EBPα (lower panel). These data suggest that RUNX1/EVI1 associates with C/EBPα via regions other than the DNA‐binding domain in RUNX1 and the functional domains in EVI1, and that destruction of the critical CtBP‐binding motif does not modify C/EBPα‐binding activity of RUNX1/EVI1.
Figure 2.
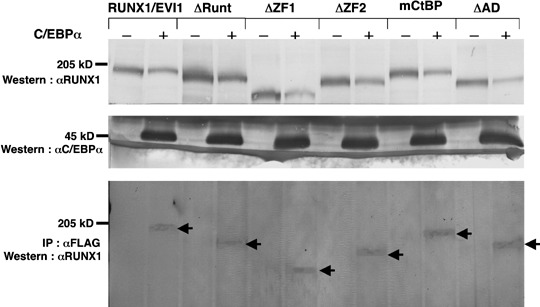
RUNX1/EVI1 binds to CCAAT/enhancer binding protein α (C/EBPα) in vivo. COS‐7 cells were transfected with 5 µg pME18S‐RUNX1/EVI1, pME18S‐ΔRunt, pME18S‐ΔZF1, pME18S‐ΔZF2, pME18S‐mCtBP or pME18S‐ΔAD with or without 5 µg pME18S‐FLAG‐C/EBPα and cultured in Dulbecco's modified Eagle's medium containing 10% fetal calf serum for 48 h before harvesting. Western blot analyses were carried out with anti‐RUNX1 antiserum to detect RUNX1/EVI1 or its mutant proteins (upper panel) or with anti‐FLAG M2 antibody to detect C/EBPα (middle panel) expressed in COS‐7 cells. RUNX1/EVI1 or its mutant proteins immunoprecipitated with anti‐FLAG M2 antibody were detected using anti‐RUNX1 antiserum (lower panel). Arrows indicate immunoprecipitated RUNX1/EVI1 and its mutant proteins.
RUNX1/EVI1 inhibits C/EBPα‐mediated transcriptional activity. Because a physical association between RUNX1/EVI1 and C/EBPα was demonstrated, we sought the effect of RUNX1/EVI1 on C/EBPα‐dependent transcription. C/EBPα has been shown to stimulate transcription of the reporter gene containing the human CEBPA promoter and so far is the only factor known to activate the promoter in synergy with the ubiquitous upstream stimulatory factor. We thus investigated whether RUNX1/EVI1 alters C/EBPα‐mediated transcriptional activity by transient transfection assay with a luciferase construct driven by a 562‐bp fragment of the human CEBPA promoter, ptk81‐luc‐CEBPA.( 16 ) To confirm that C/EBPα autoregulates its own promoter, we transfected the CEBPA reporter along with mock or C/EBPα expression plasmid into African green monkey kidney cell line CV‐1, in which C/EBPα is shown to activate its own promoter,( 16 ) and evaluated luciferase activities. Consistent with a previous report, cotransfection of the C/EBPα expression plasmid resulted in a 1.5‐fold increase in luciferase activity compared with that obtained with the mock plasmid (Fig. 3). RUNX1/EVI1 alone had no effect on the CEBPA promoter. Importantly, coexpression of RUNX1/EVI1 almost completely abolished the C/EBPα‐dependent activation of the promoter. These data suggest that RUNX1/EVI1 interferes with the autoregulatory loop of C/EBPα.
Figure 3.
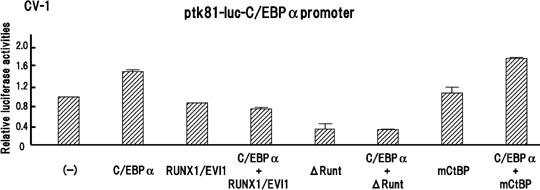
RUNX1/EVI1 represses CCAAT/enhancer binding protein α (C/EBPα)‐mediated transcriptional activity. CV‐1 cells were transfected with 200 ng ptk81‐luc‐C/EBPα reporter plasmid alone or along with 50 ng indicated expression plasmid (C/EBPα, pME18S‐C/EBPα; RUNX1/EVI1, pME18S‐RUNX1/EVI1; ΔRunt, pME18S‐ΔRunt; mCtBP, pME18S‐mCtBP) and cultured in Dulbecco's modified Eagle's medium containing 10% fetal calf serum for 48 h before harvesting. Bars show relative luciferase activities to the level when a control plasmid pME18S was cotransfected and present average results of duplicate experiments.
To identify the critical portion of the RUNX1/EVI1 protein that contributes to the repression of C/EBPα transcriptional activity, we analyzed the functions of the RUNX1/EVI1 mutants ΔRunt and mCtBP. The ΔRunt mutant suppressed the basal CEBPA promoter activity, and still suppressed it when C/EBPα was co‐expressed. Notably, the mCtBP mutant lost the ability to repress C/EBPα‐mediated transcription, whereas it did not affect the promoter activity in the absence of C/EBPα. Therefore, we speculate that CtBP binding followed by histone deacetylase recruitment is required for RUNX1/EVI1 to suppress the molecular function of C/EBPα.
RUNX1/EVI1 decreases the DNA‐binding affinity of C/EBPα. Because RUNX1/EVI1 associated with C/EBPα and disturbed its transcriptional activity, we analyzed whether RUNX1/EVI1 influenced the DNA binding of C/EBPα. For this purpose, cell lysates prepared from COS‐7 cells expressing C/EBPα were at first subjected to electrophoretic mobility shift assay (EMSA) using a radioactive C/EBP‐site oligonucleotide derived from the G‐CSF receptor promoter (–57 to –38 bp).( 16 , 19 ) The expression of C/EBPα was demonstrated by western blot analysis with anti‐FLAG antibody (data not shown). In EMSA, C/EBPα generated a specific DNA–protein complex that was not seen in the mock lysate and was supershifted with anti‐C/EBPα antibody (Fig. 4). This band represented the specific binding of C/EBPα to the probe as the binding was reduced by the addition of the unlabeled wild‐type C/EBP site oligonucleotide but not the oligonucleotide mutated in the C/EBP site. We then carried out the EMSA assay in the same manner with the lysates expressing RUNX1/EVI1 added. The presence of RUNX1/EVI1 decreased the intensity of the specific band derived from the DNA–C/EBPα complex in a dose‐dependent manner. Thus, we conclude that RUNX1/EVI1 interferes with DNA binding of C/EBPα.
Figure 4.
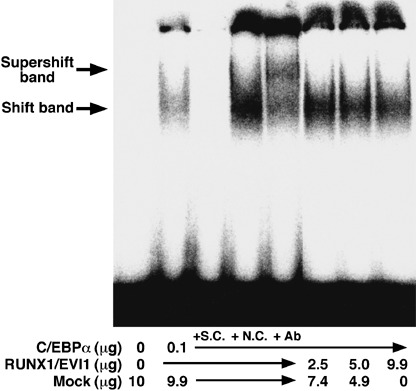
RUNX1/EVI1 reduces the DNA‐binding affinity of CCAAT/enhancer binding protein α (C/EBPα). Electrophoretic mobility shift assay was carried out using a [32P]‐labeled probe and lysates from COS‐7 cells transfected with pME18S, pME18S‐FLAG‐C/EBPα or pME18S‐FLAG‐RUNX1/EVI1. A 300‐fold molar excess of cold specific competitor (SC) or non‐specific competitor (NC) was added to the reaction. Anti‐C/EBPα antiserum (14 AA) was also added to the reaction.
Coexpression of C/EBPα restores the granulocytic differentiation suppressed by RUNX1/EVI1 in LG‐3 cells. Because it is conceivable that RUNX1/EVI1 blocks granulocytic differentiation at least partly by repressing the functions of endogenous C/EBPα in LG‐3 cells, we studied whether coexpression of C/EBPα was sufficient to induce granulocytic differentiation in RUNX1/EVI1‐expressing cells. To this end, we transfected the C/EBPα expression plasmid pCAGIPuro‐C/EBPα into R/E11 and successfully obtained several clones stably expressing both RUNX1/EVI1 and C/EBPα. The expression of RUNX1/EVI1 and C/EBPα in the representative clones R/E11‐C7 and R/E11‐C8 is shown in Fig. 5a. M15, R/E11 and the two R/E11‐derived clones were treated with G‐CSF and the degree of granulocytic differentiation was compared among them (Fig. 5b). Notably, R/E11‐C7 and R/E11‐C8 morphologically restored granulocytic differentiation suppressed by RUNX1/EVI1. The percentages of mature granulocytes were 44% in R/E11‐C7 and 31% in R/E11‐C8 versus 24% in R/E11. We speculated that restoration of C/EBPα function partly overcomes the block in differentiation mediated by RUNX1/EVI1 and progresses the granulocytic differentiation.
Figure 5.
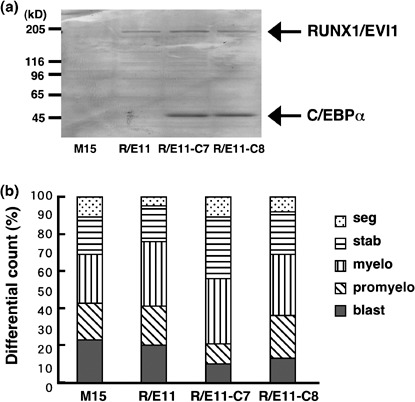
Co‐expression of CCAAT/enhancer binding protein α (C/EBPα) restores granulocytic differentiation in LG‐3 cells expressing RUNX1/EVI1. (a) Expression of RUNX1/EVI1 and C/EBPα proteins in the mock (M15), RUNX1/EVI1‐expressing (R/E11) or both RUNX1/EVI1‐ and C/EBPα‐expressing (R/E11‐C7 and R/E11‐C8) clones. (b) Granulocytic differential counts of these LG‐3 clones after 7 days of treatment with 2 ng/mL granulocyte colony‐stimulating factor (G‐CSF) are shown. This experiment was carried out four times independently and similar results were obtained. Representative data are shown.
Discussion
We demonstrated in this study that RUNX1/EVI1 disturbs the C/EBPα‐mediated transcriptional activity of the CEBPΑ promoter containing the C/EBP site. Because we could not identify the PEBP2 site in the promoter used in the assay and expression of RUNX1/EVI1 alone had no effect on it, RUNX1/EVI1 is thought to inhibit autoregulation of C/EBPα. We observed the association between RUNX1/EVI1 and C/EBPαin vivo. However, analysis with a set of RUNX1/EVI1 mutants failed to identify the C/EBPα‐binding region, because none of the mutants tested lost their ability to bind to C/EBPα. This indicates that regions outside of the functional domains deleted in this study, the Runt domain in RUNX1 and the zinc finger and acidic domains in EVI1, are required for interaction with C/EBPα, or that RUNX1/EVI1 associates with C/EBPα through multiple binding sites including the functional domains. It is interesting to remember that RUNX1/ETO generated by t(8;21) in AML (FAB‐M2) also associates with C/EBPα and inhibits its transcriptional activity.( 16 ) The association between RUNX1/ETO and C/EBPα occurs at the DNA‐binding domains of both proteins, namely the Runt domain in RUNX1/ETO and the basic‐region leucine zipper domain in C/EBPα. Despite the structural similarity between RUNX1/EVI1 and RUNX1/ETO, deletion of the Runt domain did not abolish the C/EBPα binding of RUNX1/EVI1, suggesting that the domain is the sole binding domain in RUNX1/EVI1.
Based on the observation of a physical association between these molecules, we propose two possible underpinning mechanisms in the suppressive effect of RUNX1/EVI1 on C/EBPα function. One is recruitment of histone deacetylase via CtBP bound to the EVI1 portion and the other is interference of C/EBPα's DNA‐binding activity. Considering that introduction of the point mutation in the C‐terminal CtBP‐binding motif in the EVI1 portion of RUNX1/EVI1 significantly repressed RUNX1/EVI1's negative effect on C/EBPα‐induced transcription, binding with the co‐repressor CtBP and subsequent recruitment of histone deacetylase could play a critical role in the suppression of C/EBPα function. There are two putative CtBP‐binding motifs, PFDLT (amino acid 553–557) and PLDLS (584–588) located between the two zinc finger domains of EVI1. Of the two motifs, the C‐terminal PLDLS motif has been shown to be responsible for the interaction between EVI1 and CtBP.( 30 ) Collectively with the previous report, the indirect association with histone deacetylase via the C‐terminal CtBP‐binding motif could be required for RUNX1/EVI1 to disturb the molecular function of C/EBPα. Notably, the ΔRunt mutant that retained both the C/EBPα‐ and CtBP‐binding abilities appeared to be able to repress C/EBPα function. However, RUNX1/EVI1 inhibits the DNA‐binding activity of C/EBPα in a dose‐dependent manner. Thus, dissociation of C/EBPα from DNA also contributes to the suppressive function of RUNX1/EVI1 on C/EBPα. However, if C/EBPα leaves the DNA, recruitment of histone deacetylase by RUNX1/EVI1 should not be effective to suppress C/EBPα function. Therefore, the former mechanism is not compatible with the latter.
Our data postulate the possibility that RUNX1/EVI1 could reduce the transcription of C/EBPα's target genes in vivo. By using real‐time reverse transcription–PCR assay, we compared the mRNA levels of its target genes including Cebpa, Cebpe and G‐CSF receptor between the mock and RUNX1/EVI1‐expressing LG‐3 cells. The levels of Cebpa and Cebpe mRNA were unchanged and that of G‐CSF receptor mRNA was rather increased in the presence of RUNX1/EVI1. Because the amount of Cebpa mRNA was extremely small in parental and mock‐transfected LG‐3 cells, it may have been difficult to detect a decrease in expression, if present. Notably, higher expression of G‐CSF receptor mRNA was also observed in the LG‐3 cells ectopically expressing RUNX1/ETO.( 31 ) We used western blot analysis to evaluate the levels of the C/EBPɛ and G‐CSF receptor proteins, but found no differences between the mock and RUNX1/EVI1‐expressing cells. Helbling et al. have reported that RUNX1/EVI1 reduces the level of C/EBPα protein but not its mRNA in U937 cells, and that a putative inhibitor of CEBPA translation (calreticulin) is upregulated by RUNX1/EVI1.( 32 ) Calreticulin is a ubiquitous protein with calcium storage and chaperone functions and is postulated to be involved in the development of leukemia.( 32 , 33 ) In an experiment with small interfering RNA for the calreticulin gene, they concluded that RUNX1/EVI1 inhibits C/EBPα expression through a post‐transcriptional mechanism of calreticulin. However, the level of calreticulin protein was unaltered in the RUNX1/EVI1‐expressing LG‐3 cells compared to the mock cells (data not shown), suggesting that the post‐transcriptional mechanism of calreticulin may not be activated by RUNX1/EVI1 in LG‐3 cells. RUNX1/EVI1 could modify C/EBPα expression at either the transcriptional or translational level in a context‐dependent manner.
We demonstrated that exogenous expression of RUNX1/EVI1 in LG‐3 cells resulted in the maturation block induced by G‐CSF, as reported in 32D cells.( 27 ) Co‐expression of C/EBPα in the RUNX1/EVI1‐expressing cells clearly restored their ability to differentiate along the myeloid lineage. These data support the concept of RUNX1/EVI1 as an inhibitor of C/EBPα‐mediated transcription required for myeloid differentiation. However, we could not identify which target genes of C/EBPα are transcriptionally repressed by RUNX1/EVI1 in LG‐3 cells, because the levels of the candidate mRNA tested were not decreased as described above. Other critical target genes may be regulated by C/EBPα in LG‐3 cells and downregulation of those genes could lead to the differentiation block in the RUNX1/EVI1‐expressing cells.
RUNX1/EVI1 causes various kinds of leukemia, including de novo or therapy‐related AML, myelodysplastic syndrome‐transformed leukemia and blastic crisis of chronic myelogenous leukemia, through the following mechanisms:( 34 ) dominant negative effect over wild‐type RUNX1,( 27 , 35 ) blockade of TGF‐β‐mediated signal,( 36 ) inhibition of JNK( 10 ) and stimulation of AP‐1 activity.( 37 ) Our study points to another function for RUNX1/EVI1, that is, suppression of C/EBPα, as the molecular mechanism leading to the block in maturation seen in myeloid leukemia characterized by the t(3;21) translocation. From these data, we argue that transfer of exogenous C/EBPα protein into leukemia cells could represent a specific therapeutic option for the treatment of this type of leukemia by recovering their differentiation ability. Further, considering that recruitment of histone deacetylase seems to be critical for RUNX1/EVI1 to block the autoregulatory loop and suppress the molecular function of C/EBPα, administration of histone deacetylase inhibitor could be another potential modality to restore the function of C/EBPα and thereby differentiate the leukemic cells expressing RUNX1/EVI1.
Acknowledgments
The pcDNA3‐C/EBPα and ptk81‐luc‐C/EBPα promoters were generous gifts from Dr Daniel G. Tenen (Beth Israel Deaconess Medical Center, Boston, MA, USA). LG‐3 cells were generous gifts from Dr T. Honjo (Kyoto University Graduate School of Medicine, Kyoto, Japan) and Dr I. Kitabayashi (National Cancer Center Research Institute, Tokyo, Japan). pCXN2 and pCAGIPuro were kindly provided by Dr J. Miyazaki (Osaka University Graduate School of Medicine, Osaka, Japan). Recombinant human G‐CSF and murine IL‐3 were provided by KIRIN Brewery. This work was financially supported in part by the Japan Health Sciences Foundation, Grants‐in‐Aid from the Ministries in Japan of Education, Culture, Sports, Science and Technology, and Health, Labour and Welfare, and from the Japanese Society for the Promotion of Science.
References
- 1. Rubin CM, Larson RA, Bitter MA et al . Association of a chromosomal 3;21 translocation with the blast phase of chronic myelogenous leukemia. Blood 1987; 70: 1338–42. [PubMed] [Google Scholar]
- 2. Coyle T, Najfeld V. Translocation (3;21) in Philadelphia chromosome‐positive chronic myelogenous leukemia prior to the onset of blast crisis. Am J Hematol 1988; 27: 56–9. [DOI] [PubMed] [Google Scholar]
- 3. Rubin CM, Larson RA, Anastasi J et al . t(3;21)(q26;q22): a recurring chromosomal abnormality in therapy‐related myelodysplastic syndrome and acute myeloid leukemia. Blood 1990; 76: 2594–8. [PubMed] [Google Scholar]
- 4. Schneider NR, Bowman WP, Frenkel EP. Translocation (3;21)(q26;q22) in secondary leukemia. Report of two cases and literature review. Ann Genet 1991; 34: 256–63. [PubMed] [Google Scholar]
- 5. Mitani K, Ogawa S, Tanaka T et al . Generation of the AML1‐EVI‐1 fusion gene in the t(3;21)(q26;q22) causes blastic crisis in chronic myelocytic leukemia. EMBO J 1994; 13: 504–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Okuda T, Van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996; 84: 321–30. [DOI] [PubMed] [Google Scholar]
- 7. Wang Q, Stacy T, Binder M, Marin‐Padilla M, Sharpe AH, Speck NA. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc Natl Acad Sci USA 1996; 93: 3444–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ichikawa M, Asai T, Saito T et al . AML‐1 is required for megakaryocytic maturation and lymphocytic differentiation, but not for maintenance of hematopoietic stem cells in adult hematopoiesis. Nat Med 2004; 10: 299–304. [DOI] [PubMed] [Google Scholar]
- 9. Kurokawa M, Mitani K, Irie K et al . The oncoprotein Evi‐1 represses TGF‐β signalling by inhibiting Smad3. Nature 1998; 394: 92–6. [DOI] [PubMed] [Google Scholar]
- 10. Kurokawa M, Mitani K, Yamagata T et al . The evi‐1 oncoprotein inhibits c‐Jun N‐terminal kinase and prevents stress‐induced cell death. EMBO J 2000; 19: 2958–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tanaka T, Nishida J, Mitani K, Ogawa S, Yazaki Y, Hirai H. Evi‐1 raises AP‐1 activity and stimulates c‐fos promoter transactivation with dependence on the second zinc finger domain. J Biol Chem 1994; 269: 24 020–6. [PubMed] [Google Scholar]
- 12. Maki K, Yamagata T, Asai T et al . Dysplastic definitive hematopoiesis in AML1/EVI1 knock‐in embryos. Blood 2005; 106: 2147–55. [DOI] [PubMed] [Google Scholar]
- 13. Maki K, Yamagata T, Yamazaki I, Oda H, Mitani K. Development of megakaryoblastic leukaemia in Runx1‐Evi1 knock‐in chimaeric mouse. Leukemia 2006; 20: 1458–60. [DOI] [PubMed] [Google Scholar]
- 14. Ramji DP, Foka P. CCAAT/enhancer‐binding proteins: structure, function and regulation. Biochem J 2002; 365: 561–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schuster MB, Porse BT. C/EBPα: a tumour suppressor in multiple tissues? Biochim Biophys Acta 2006; 1766: 88–103. [DOI] [PubMed] [Google Scholar]
- 16. Pabst T, Mueller BU, Harakawa N et al . AML1‐ETO downregulates the granulocytic differentiation factor C/EBα in t(8;21) myeloid leukemia. Nat Med 2001; 7: 444–51. [DOI] [PubMed] [Google Scholar]
- 17. Pabst T, Mueller BU, Zhang P et al . Dominant‐negative mutations of CEBPA, encoding CCAAT/enhancer binding protein‐α (C/EBPα), in acute myeloid leukemia. Nat Genet 2001; 27: 263–70. [DOI] [PubMed] [Google Scholar]
- 18. Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ, Tenen DG. Absence of granulocyte colony‐stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein α‐deficient mice. Proc Natl Acad Sci USA 1997; 94: 569–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smith LT, Hohaus S, Gonzalez DA, Dziennis SE, Tenen DG. PU.1 (Spi‐1) and C/EBPα regulate the granulocyte colony‐stimulating factor receptor promoter in myeloid cells. Blood 1996; 88: 1234–47. [PubMed] [Google Scholar]
- 20. Radomska HS, Huettner CS, Zhang P, Cheng T, Scadden DT, Tenen DG. CCAAT/enhancer binding protein α is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol Cell Biol 1998; 18: 4301–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang X, Scott E, Sawyers CL, Friedman AD. C/EBPα bypasses granulocyte colony‐stimulating factor signals to rapidly induce PU.1 gene expression, stimulate granulocytic differentiation, and limit proliferation in 32D cl3 myeloblasts. Blood 1999; 94: 560–71. [PubMed] [Google Scholar]
- 22. Miyamoto T, Iwasaki H, Reizis B et al . Myeloid or lymphoid promiscuity as a critical step in hematopoietic lineage commitment. Dev Cell 2002; 3: 137–47. [DOI] [PubMed] [Google Scholar]
- 23. Tavor S, Park DJ, Gery S, Vuong PT, Gombart AF, Koeffler HP. Restoration of C/EBPα expression in a BCR‐ABL+ cell line induces terminal granulocytic differentiation. J Biol Chem 2003; 278: 52 651–9. [DOI] [PubMed] [Google Scholar]
- 24. Gombart AF, Hofmann WK, Kawano S et al . Mutations in the gene encoding the transcription factor CCAAT/enhancer binding protein α in myelodysplastic syndromes and acute myeloid leukemias. Blood 2002; 99: 1332–40. [DOI] [PubMed] [Google Scholar]
- 25. Snaddon J, Smith ML, Neat M et al . Mutations of CEBPA in acute myeloid leukemia FAB types M1 and M2. Genes Chromosomes Cancer 2003; 37: 72–8. [DOI] [PubMed] [Google Scholar]
- 26. Mueller BU, Pabst T. C/EBPα and the pathophysiology of acute myeloid leukemia. Curr Opin Hematol 2006; 13: 7–14. [DOI] [PubMed] [Google Scholar]
- 27. Tanaka T, Mitani K, Kurokawa M et al . Dual functions of the AML1/Evi‐1 chimeric protein in the mechanism of leukemogenesis in t(3;21) leukemias. Mol Cell Biol 1995; 15: 2383–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kinashi T, Lee KH, Ogawa M et al . Premature expression of the macrophage colony‐stimulating factor receptor on a multipotential stem cell line does not alter differentiation lineages controlled by stromal cells used for coculture. J Exp Med 1991; 173: 1267–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Waga K, Nakamura Y, Maki K et al . Leukemia‐related transcription factor TEL accelerates differentiation of Friend erythroleukemia cells. Oncogene 2003; 22: 59–68. [DOI] [PubMed] [Google Scholar]
- 30. Izutsu K, Kurokawa M, Imai Y, Maki K, Mitani K, Hirai H. The corepressor CtBP interacts with Evi‐1 to repress transforming growth factor beta signaling. Blood 2001; 97: 2815–22. [DOI] [PubMed] [Google Scholar]
- 31. Shimizu K, Kitabayashi I, Kamada N et al . AML1‐MTG8 leukemic protein induces the expression of granulocyte colony‐stimulating factor (G‐CSF) receptor through the up‐regulation of CCAAT/enhancer binding protein epsilon. Blood 2000; 96: 288–96. [PubMed] [Google Scholar]
- 32. Helbling D, Mueller BU, Timchenko NA et al . The leukemic fusion gene AML1‐MDS1‐EVI1 suppresses CEBPA in acute myeloid leukemia by activation of Calreticulin. Proc Natl Acad Sci USA 2004; 101: 13 312–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Helbling D, Mueller BU, Timchenko NA et al . CBFB‐SMMHC is correlated with increased calreticulin expression and suppresses the granulocytic differentiation factor CEBPA in AML with inv(16). Blood 2005; 106: 1369–75. [DOI] [PubMed] [Google Scholar]
- 34. Mitani K. Molecular mechanisms of leukemogenesis by AML1/EVI‐1. Oncogene 2004; 23: 4263–9. [DOI] [PubMed] [Google Scholar]
- 35. Izutsu K, Kurokawa M, Imai Y et al . The t(3;21) fusion product, AML1/Evi‐1, blocks AML1‐induced transactivation by recruiting CtBP. Oncogene 2002; 21: 2695–703. [DOI] [PubMed] [Google Scholar]
- 36. Kurokawa M, Mitani K, Imai Y, Ogawa S, Yazaki Y, Hirai H. The t(3;21) fusion product, AML1/Evi‐1, interacts with Smad3 and blocks transforming growth factor‐β‐mediated growth inhibition of myeloid cells. Blood 1998; 92: 4003–12. [PubMed] [Google Scholar]
- 37. Kurokawa M, Ogawa S, Tanaka T et al . The AML1/Evi‐1 fusion protein in the t(3;21) translocation exhibits transforming activity on Rat1 fibroblasts with dependence on the Evi‐1 sequence. Oncogene 1995; 11: 833–40. [PubMed] [Google Scholar]