

Abstract
Deregulated signaling via the phosphatidylinositol 3‐kinase (PI3K) pathway is common in many types of cancer, but its clinicopathological significance in endometrial cancer remains unclear. In the present study, we examined the status of the PI3K signaling pathway, especially in relation to PTEN and PIK3CA status, in endometrioid‐type endometrial cancer. The immunohistochemical analysis revealed a high level of phosphorylated (p)‐AKT expression, which is a hallmark of activated PI3K signaling, in approximately 60% of endometrial cancers. There was no correlation between p‐AKT expression and clinicopathological characteristics, such as International Federation of Gynecology and Obstetrics stage, tumor grade, and myometrial invasion. Unexpectedly, a high level of p‐AKT expression occurred independently of the presence of PTEN or PIK3CA mutations. Furthermore, p‐AKT expression did not correlate with the expression of potential downstream targets, including p‐mTOR and p‐FOXO1/3a. In turn, p‐AKT expression was strongly associated with extracellular‐regulated kinase 1/2 expression (P = 0.0031), which is representative of the activated RAS–MAP kinase pathway. Kaplan–Meier analysis suggested that low p‐AKT expression was associated with low rates of relapse‐free survival, although the difference was not statistically significant, indicating that AKT activation does not confer worse prognosis. The present study demonstrates the presence of complex signaling pathways that might mask the conventional tumorigenic PTEN–PI3K–AKT–mTOR pathway, and strongly suggests a close association between the extracellular‐regulated kinase and PI3K pathways in this tumor type. (Cancer Sci 2007; 98: 1881–1888)
Endometrial cancer is the second most common gynecological malignancy in Japan, and its incidence in Japan has increased dramatically over the last decade. Although premalignant lesions of endometrial cancer have been well characterized, the molecular pathways of endometrial carcinogenesis remain unclear. Inactivation of the PTEN gene, mainly via gene mutation, is the most common genetic abnormality detected in endometrioid‐type endometrial cancer.( 1 , 2 , 3 ) PTEN is a tumor‐suppressor gene encoding a lipid phosphatase that dephosphorylates phosphatidyl inositol 3,4,5‐triphosphate (PI[3,4,5]P3), thus opposing the action of phosphoinositide 3‐kinase (PI3K).( 4 , 5 ) PI(3,4,5)P3 is converted to PI(3,4)P2, which activates the proto‐oncogene AKT (also called PKB). Activation of AKT, which is caused by its phosphorylation, antagonizes apoptotic pathways via activation of mTOR or inactivation of the forkhead family, whereas dephosphorylation of PI(3,4,5)P3 by PTEN induces activation of the apoptotic pathway.( 6 , 7 , 8 ) Thus, the fundamental in vivo role of PTEN is to inhibit the PI3K–AKT pathway. Mutation of PTEN can disable this inhibitory function, thus inducing the antiapoptotic pathway, which is believed to be the key to endometrial carcinogenesis. Other critical factors involved in regulation of the PI3K–AKT pathway are mutations in genes that encode components of PI3K. Recent studies have identified non‐random somatic mutations in the PIK3CA gene, which encodes the p110α catalytic subunit of PI3K, in various cancers.( 9 ) Reported mutations have been of the missense type and clustered within two regions of the helical (exon 9) and kinase (exon 20) domains, and the mutant proteins have been shown to display enhanced lipid‐kinase activity. PIK3CA mutation has recently been identified in 20–40% of endometrial cancers.( 10 , 11 , 12 )
Other prevalent genetic abnormalities in endometrial carcinogenesis are mutations in the KRAS gene.( 13 , 14 ) The extracellular‐regulated kinase (ERK)–mitogen‐activated protein kinase (MAPK) pathway is activated by mitogenic stimuli mediated by receptor‐type tyrosine kinases and G protein‐coupled receptors, leading to sequential phosphorylation of RAS, RAF, MEK, and ERK1/2. Phosphorylated (p)‐ERK1/2 translocates to the nucleus and regulates a range of substrates that promote cell proliferation, motility, differentiation, and survival.( 15 , 16 , 17 ) A KRAS mutation can lead to continuous stimulation of its downstream targets, resulting in ERK1/2 activation in the absence of mitogenic stimuli.
Thus, most of these prevalent genetic alterations observed in endometrial cancer stimulate the PI3K–AKT and MAPK–ERK pathways, and may play major roles in endometrial carcinogenesis as well as disease progression. In a previous study, we examined the status of the ERK–MAPK pathway in endometrial cancer, and found that ERK1/2 activation was a significant prognostic factor.( 18 ) Unexpectedly, ERK1/2 activation frequently occurred irrespective of KRAS status, and was associated with a favorable prognosis. Thus, it appears that the widely recognized canonical RAS–RAF–MEK–ERK1/2 signaling pathway does not contribute to disease progression of endometrial carcinogenesis, at least in the late stage.
Given these unexpected features of the ERK1/2 pathway, in the present study, we assessed the status of the PI3K–AKT pathway in endometrial cancer, especially in relation to PTEN and PI3KCA mutation. We also examined whether the PI3K–AKT pathway affects tumor behavior or is a prognostic factor, attempted to identify downstream targets of AKT, and assessed the relationship between the ERK–MAPK and PI3K–AKT signaling pathways. We carried out these analyses using clinical samples of endometrial cancer and found unexpected results, raising questions about the roles of PTEN and PIK3CA mutation in activating the PI3K–AKT pathway or the significance of such signaling in the oncogenic potential of endometrial cancers.
Materials and Methods
Patients and tissue samples. A total of 63 patients with endometrioid‐type endometrial cancer (mean age, 57.5 years; range, 32–78 years) treated at the Department of Obstetrics and Gynecology, Kanazawa University Hospital, from January 1995 to December 2002, were enrolled in the study. All patients underwent a total abdominal or radical hysterectomy plus bilateral salpingo‐oophorectomy. Systemic retroperitoneal lymphadenectomy was carried out in approximately 70% of the patients. Staging was carried out for all patients using the International Federation of Gynecology and Obstetrics (FIGO) surgical staging system: 46 tumors were classified as stage I (substages: Ia, 10 tumors; Ib, 28 tumors; and Ic, eight tumors); six tumors were classified as stage II (substages: IIa, three tumors; and IIb, three tumors); nine tumors were classified as stage III (substages: IIIa, three tumors; and IIIc, six tumors); and two tumors were classified as stage IV. Based on histological examination, 34 tumors were classified as grade (G)1, 14 tumors were classified as G2, and 15 tumors were classified as G3. Patients with deep myometrial invasion, cervical involvement, and special histology underwent external radiotherapy, whereas those with positive peritoneal cytology or retroperitoneal lymph‐node metastasis were treated with four to six cycles of CAP chemotherapy (90 mg/m2 cisplatin; 50 mg/m2 doxorubicin; and 500 mg/m2 cyclophosphamide) or TJ chemotherapy (180 mg/m2 paclitaxel; and carboplatin, according to Chatelut's formula [area under the curve (AUC) = 5 mg/mL/min]) as postoperative adjuvant therapy. Patient treatment was followed up with a gynecological examination, recording of laboratory data, transvaginal and abdominopelvic ultrasonography, and a radiological examination. Data from regular follow‐up visits to the outpatient department were stored in a database designed specifically for endometrial carcinoma patients. A telephone survey to update the status of all surviving patients was made in August 2004. The exact date of disease recurrence was defined as the date when the apparent tumors were detected by ultrasonographic or radiological examinations.
Tumor samples were collected at the time of surgery, with written informed consent and approval of the Ethics Committee of Kanazawa University. Half of each tissue sample was examined histologically by pathologists, and the remaining portion of each sample was frozen at –80°C until DNA extraction for mutation analysis.
PTEN and PIK3CA mutation analysis. Of the 63 endometrial cancers, 45 were available for DNA sequencing, in which 38 cancers had been evaluated previously for mutations in PTEN. ( 3 , 19 ) All exons of the PTEN gene were amplified by polymerase chain reaction (PCR) using primer sets described previously.( 3 ) Exons 9 and 20 of the PIK3CA gene, which were known to be hot spots of gene mutations, were also amplified by PCR using primer sets described previously.( 9 ) PCR products were purified using the Qiagen PCR purification kit (Qiagen), and direct sequencing was carried out. PCR products with suspected mutations were reamplified and subsequently cloned into the pGEM‐T Easy vector (Promega), and sequencing was carried out using at least three clones.
Analysis of microsatellite instability. DNA samples from endometrial cancers and normal areas of the endometrium were analyzed using a panel of five microsatellite markers for the dinucleotide repeat sequences D2S123, D2S147, D10S197, D13S175, and D18S58, as described elsewhere.( 20 ) Myometrial samples from each patient served as controls. The PCR products of the microsatellite markers were analyzed using ABI GeneScan (Applied Biosystems, Foster City, CA, USA). The samples were classified as high‐frequency microsatellite instability (MSI) (PCR bands had shifted compared with those of the myometrial control samples for two or more of the five loci), low‐frequency MSI (one locus), or microsatellite stable (no loci).( 21 )
Immunohistochemistry. Immunohistochemical analysis was carried out using formalin‐fixed, paraffin‐embedded specimens from 63 endometrioid‐type endometrial cancer tissues. Sections were stained with rabbit monoclonal antibody to p‐AKT (#3787) or with rabbit polyclonal antibodies to p‐mTOR (#2974) and p‐FOXO1(Thr24)/FOXO3a(Thr32) (#9464) (Cell Signaling Technology, Beverly, MA, USA).
After the specimens were deparaffinized in xylene and graded alcohols, epitope retrieval was carried out. The sections were heated in a microwave oven at 700 W for 10 min in 1× antigen retrieval solution (Biogenex, San Ramon, CA, USA). Then, endogenous peroxidase was blocked by immersing the sections in 0.3% H2O2–methanol for 30 min. After blocking with horse serum, the (above‐described) appropriate primary antibodies at 1 : 50 (for p‐AKT and p‐FOXO1/3a) or at 1 : 100 (for p‐mTOR) were diluted and applied for 16 h at 4°C. The reaction was visualized using the EnVision Detection Kit (Dako Cytomation, Glostrup, Denmark) using diaminobenzidine tetrahydrochloride as the enzyme substrate. All sections were counterstained with GM hematoxylin stain solution (Muto Pure Chemicals, Tokyo, Japan). For negative controls, the primary antibody was omitted. For the absorption test, primary antibody against p‐AKT was incubated with two volumes of p‐Akt blocking peptide (#1140; Cell Signaling Technology) for 30 min prior to adding the entire volume to the slide. The expression of p‐Akt was scored into four levels: no, weak, moderate, and strong. Stained sections were evaluated blindly without prior knowledge of the clinicopathological parameters by two observers, who scored the percentage of positive nuclei and cytoplasms within tumors. The average staining score was registered, and there was no statistical significance in the scoring between observers. For the survival analysis, patients with no or weak expression were assigned to the low‐expression group, and those with moderate or strong expression were assigned to the high‐expression group.
Western blot analysis. Whole‐cell lysates were prepared by resuspending frozen cancer tissues in buffer containing 50 mM Tris‐HCl (pH 7.4), 1% Nonidet P40 (Fluka), and 1 mM ethylenediaminetetraacetic acid for 10 min at room temperature. The homogenate was centrifuged at 12 000 g for 5 min. The supernatant was recovered, and its protein concentration was measured by Bradford assay. Then, 30 µg of extract was electrophoresed on a sodium dodecylsulfate–polyacrylamide gel with a gradient of 10–20% (Readygels J, 161‐J351; Bio‐Rad, Hercules, CA, USA), and was then transferred to a polyvinylidene difluoride membrane. Membranes were blocked with TBS‐T (150 mM NaCl, 20 mM Tris‐HCl [pH 7.5], and 0.1% Tween 20) containing 5% non‐fat dried milk, and were then incubated with specific primary antibody against p‐AKT (#4058; Cell Signaling Technology), followed by incubation with horseradish peroxidase‐linked antirabbit IgG. Immunoreactive bands were visualized using the ECL detection system (Amersham), according to the manufacturer's instructions. As an internal control for equal protein loading, actin expression was examined simultaneously using a specific antibody (sc‐1615; Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Statistical analysis. Statistical analysis was carried out using the statistical package StatView version 5.0 (Abacus Concepts, Berkeley, CA, USA). We used the χ2‐test for 2 × 2 tables to compare the categorical data. Life tables were computed using the Kaplan–Meier method, whereas the log‐rank test was used to assess statistical significance. Cox's proportional hazards regression model with a stepwise manner was used to analyze the independent prognostic factors. A P‐value < 0.05 was considered to indicate statistical significance.
Results
Mutational analysis of PTEN and PI3KCA in endometrioid‐type endometrial cancer. We first identified PTEN mutations in endometrioid‐type endometrial cancer. Of the 45 cancers available for DNA sequencing, 18 cancers (40%) had mutations in the PTEN gene: point mutations in eight patients, and frameshift mutations in 10 patients. Some of these data have been reported previously.( 3 , 19 ) The clinicopathological and molecular features of patients with PTEN mutation are summarized in Table 1. PTEN mutation was observed in 13 of the 25 (52%) grade 1 cancers, 3 of the 11 (27.2%) grade 2 cancers, and two of the nine (22.2%) grade 3 cancers; thus, lower‐grade cancers were more likely to have a PTEN mutation, but the differences were not statistically significant. There were no significant correlations between PTEN mutation and other clinicopathological features (data not shown). As we reported previously,( 3 ) PTEN mutations, especially the frameshift mutations, were tightly associated with MSI (Table 1), indicating that some of the PTEN mutations are caused by mismatch repair deficiency.
Table 1.
Clinicopathological and molecular features of 21 endometrial cancers with mutations in the PTEN or PIK3CA genes
| Patient no. | Age (years) | FIGO stage | Grade | MSI | PTEN mutation | PIK3CA mutation | p‐AKT expression † |
|---|---|---|---|---|---|---|---|
| 1 | 70 | Ib | 1 | H | Ex5; G389T, R130L | Ex20; A3140T, H1047L | + |
| 2 | 58 | Ib | 3 | L | Ex20; A3140G, H1047R | − | |
| 3 | 71 | Ib | 1 | H | Ex8; G871A, E291K Ex8; 863 del A, fs (stop at 868) | Ex20; A3140T, H1047L | + |
| 4 | 52 | Ic | 2 | H | Ex8; 946 ins G, fs (stop at 970) Ex8; 987 del TAAA, fs (stop at 1024) | − | |
| 5 | 41 | Ic | 2 | H | Ex5; A446G, Q149R Ex8; 863 del A, fs (stop at 868) | ++ | |
| 6 | 44 | Ia | 1 | H | Ex8; A938G, K313R Ex8; 963 del A, fs (stop at 1027) | ++ | |
| 7 | 47 | Ib | 1 | H | Ex8; 963 del A, fs (stop at 1027) | ± | |
| 8 | 58 | Ib | 1 | H | Ex8; 923 ins A, fs (stop at 931) | − | |
| 9 | 62 | Ib | 2 | MSS | Ex20; A3140G, H1047R | − | |
| 10 | 54 | Ib | 1 | MSS | Ex5; C388G, R130G | + | |
| 11 | 64 | Ic | 2 | MSS | Ex1; A76C, T26P | − | |
| 12 | 43 | Ia | 1 | H | Ex8; A815G, H272R Ex8; 950 del TACT, fs (stop at 955) | + | |
| 13 | 67 | Ia | 1 | MSS | Ex5; G389A, R130Q | Ex9; C1636G, Q546E | ++ |
| 14 | 33 | Ia | 1 | H | Ex5; 336 del AAGTG, fs (stop at 340) | + | |
| 15 | 68 | Ia | 1 | H | Ex8; 956 del CTTT (stop at 1024) | + | |
| 16 | 49 | Ia | 1 | MSS | Ex5; C388T, R130stop | − | |
| 17 | 55 | Ic | 3 | L | Ex5; C462A, F154L | ++ | |
| 18 | 59 | Ib | 1 | H | Ex20; A3036G, E1002E | ++ | |
| 19 | 63 | Ib | 1 | MSS | Ex5; G389A, R130Q Ex5; C424T, R142W | Ex20; A3062G, Y1021C | ± |
| 20 | 55 | Ib | 1 | ND | Ex5; C388T, R130stop | ± | |
| 21 | 53 | Ic | 3 | ND | Ex5; 493 ins A, fs (stop at 499) | + |
– negative, +/– weak, + moderate, ++ strong. Some of the data have been reported previously.( 3 , 19 ) EX, exon; FIGO, International Federation of
Gynecology and Obstetrics; H, high‐frequency microsatellite instability; L, low‐frequency microsatellite instability; MSI, microsatellite instability; MSS, microsatellite stable; ND, not done; p‐AKT, phosphorylated AKT.
We next identified PIK3CA mutations. A total of seven cancers (15.6%) had mutations in exons 9 or 20 of the PI3KCA gene. All were missense mutations (Table 1). Five of them (71%) were observed in grade 1 cancers, whereas one was grade 2 and one was grade 3. There were no significant correlations between PIK3CA mutation and clinicopathological features (data not shown). Unlike the PTEN gene, PIK3CA mutations appeared to be independent of MSI status. Four cancers had both PTEN and PIK3CA mutations. PI3KCA mutations were more common in cancers with PTEN mutations (4 of 18, 22.2%) compared to those without PTEN mutations (3 of 27, 11.1%).
Activation of AKT frequently occurs in endometrial cancer, but is independent of PTEN or PIK3CA status. Immunohistochemical analysis was then carried out for the expression of p‐AKT in endometrial cancer. Expression of p‐AKT was basically detected in the cytoplasm but some samples had both cytoplasmic and nuclear staining, especially in samples with strong expression (Fig. 1). There were 13 patients with weak p‐AKT expression, 27 with moderate expression, and nine with strong expression. Thus, 36 of the 63 cancers (57.1%) had moderate or strong expression of p‐AKT. The expression of p‐AKT was further confirmed by western blot analysis in some patients whose protein extracts were available, and there was a tight correlation between the results of immunohistochemistry and western blot analysis (Fig. 2). We then assessed the correlation between p‐AKT expression and clinicopathological characteristics (Table 2). There was no correlation between p‐AKT expression and any of the following: patient age, FIGO stage, lymph‐node metastasis, depth of myometrial invasion, pathological grade, menopause, or body mass index (BMI). We also assessed the correlation between PTEN or PIK3CA status and p‐AKT expression (1, 3). Unexpectedly, there was no statistically significant correlation between PTEN or PIK3CA mutations and p‐AKT expression (Table 3). We paid special attention to cancers with the coexistence of PTEN and PIK3CA mutations, because this may induce p‐AKT expression more efficiently. However, of the four cancers that had both PTEN and PIK3CA mutations, one had strong, two had moderate, and one had weak expression of p‐AKT (Table 1), indicating that coexistence of the mutations did not necessarily lead to enhanced expression of p‐AKT in endometrial cancer. Finally, we assessed the involvement of KRAS mutation in p‐AKT expression, because it is known that RAS activates PI3K both directly and indirectly in some cancer types.( 20 ) We found a total of 10 cancers with KRAS mutations in codon 12 or codon 13 of exon 1,( 18 ) but no significant correlation was observed between KRAS mutation and p‐AKT expression (P = 0.15, data not shown).
Figure 1.
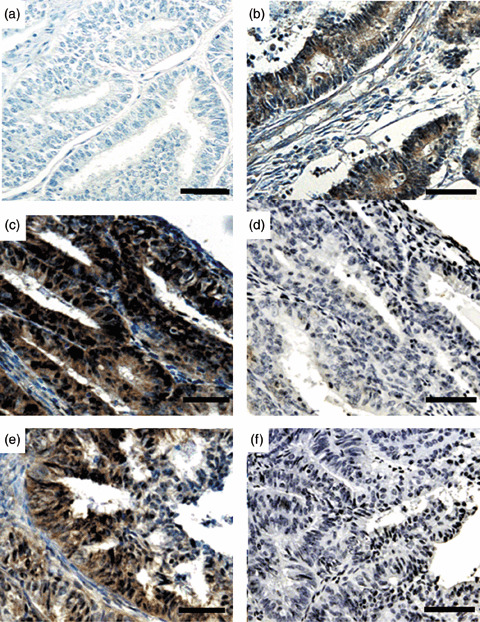
Representative results of phosphorylated (p)‐AKT immunohistochemistry in endometrial cancer specimens. (a) A sample with negative staining. (b) A sample with moderate expression. (c,e) Cases with strong expression, showing both nuclear and cytoplasmic expression. (d,f) Absorption test using p‐AKT blocking peptide in (d) case C and (f) case E. The staining completely disappeared with the addition of blocking peptide. (Original magnification ×200. Scale bars = 100 µm.)
Figure 2.
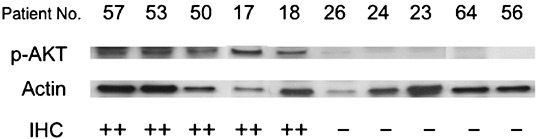
Representative results of western blot analysis in endometrial cancer specimens. Ten patients with strong (++) or no/weak (–) phosphorylated (p)‐AKT expression assessed by immunohistochemistry were analyzed for p‐AKT expression by western blot analysis. Actin expression was examined as an internal control to verify the amounts of protein loading.
Table 2.
Correlation between phosphorylated (p)‐AKT expression and clinicopathological factors in patients with endometrial cancer
| Variable | p‐AKT expression | P‐value (χ2‐test) | |
|---|---|---|---|
| High (n = 36) | Low (n = 27) | ||
| Age (years) | |||
| <60 (n = 39) | 24 | 15 | |
| ≥60 (n = 24) | 12 | 12 | 0.3688 |
| FIGO stage | |||
| I (n = 46) | 26 | 20 | |
| II, III, IV (n = 17) | 10 | 7 | 0.8698 |
| Lymph‐node metastasis | |||
| Negative (n = 58) | 33 | 25 | |
| Positive (n = 5) | 3 | 2 | 0.8930 |
| Depth (myometrial invasion) | |||
| a, b (n = 47) | 25 | 22 | |
| c (n = 16) | 11 | 5 | 0.2774 |
| Histopathological degree of differentiation | |||
| Grade 1 (n = 34) | 21 | 13 | 0.5601 |
| Grade 2, 3 (n = 29) | 15 | 14 | |
| Menopause | |||
| Peri, pre (n = 23) | 14 | 9 | 0.6504 |
| Post (n = 40) | 22 | 18 | |
| Body mass index | |||
| <25 (n = 38) | 23 | 15 | 0.5035 |
| ≥25 (n = 25) | 13 | 12 | |
| FIGO, International Federation of Gynecology and Obstetrics. | |||
Table 3.
Correlation between phosphorylated (p)‐AKT expression and PTEN or PIK3CA status in endometrial cancer
| p‐AKT expression | n | PTEN | PIK3CA | PTEN or PIK3CA | |||
|---|---|---|---|---|---|---|---|
| Mutation | Wild type | Mutation | Wild type | Mutation | Wild type | ||
| Negative | 10 | 4 | 6 | 2 | 8 | 6 | 4 |
| Weak | 8 | 3 | 5 | 1 | 7 | 3 | 5 |
| Moderate | 19 | 7 | 12 | 2 | 17 | 7 | 12 |
| Strong | 8 | 4 | 4 | 2 | 6 | 5 | 3 |
| 45 | 18 | 27 | 7 | 38 | 21 | 24 | |
| P‐value | 0.93 | 0.77 | 0.47 | ||||
Activation of AKT does not lead to phosphorylation of mTOR or members of the forkhead family, but is strongly associated with ERK1/2 activation in endometrial cancer. The known representative downstream targets of AKT are mTOR and the FOXO family, two members of which (FOXO1 and FOXO3a) play key roles in cell‐cycle regulation and apoptosis.( 22 ) Therefore, we immunohistochemically examined the expression of p‐mTOR and p‐FOXO1/3a (Fig. 3), and assessed their correlations with the expression of p‐AKT. We observed p‐mTOR expression in 58 of the 63 patients (92%): seven (11%) with weak expression, 39 (62%) with moderate expression, and 12 (19%) with strong expression. We observed p‐FOXO1/3a expression in 51 of the 63 patients (81%): 16 (25%) with weak expression, 27 (43%) with moderate expression, and eight (13%) with strong expression. There was no significant correlation between the expression of p‐AKT and the expression of p‐mTOR or p‐FOXO1/3a (4, 5), indicating that AKT activation does not always trigger the canonical downstream signaling pathway in endometrial cancer. We further examined the relationship between PTEN and PIK3A mutations and mTOR or p‐FOXO1/3a expression, but no significant correlation was observed (PTEN mutation vs p‐mTOR, P = 0.074; PTEN or PIK3CA mutation vs p‐mTOR, P = 0.098; PTEN mutation vs p‐FOXO1/3a, P = 0.77; PTEN or PIK3CA mutation vs p‐FOXO1/3a, P = 0.51).
Figure 3.
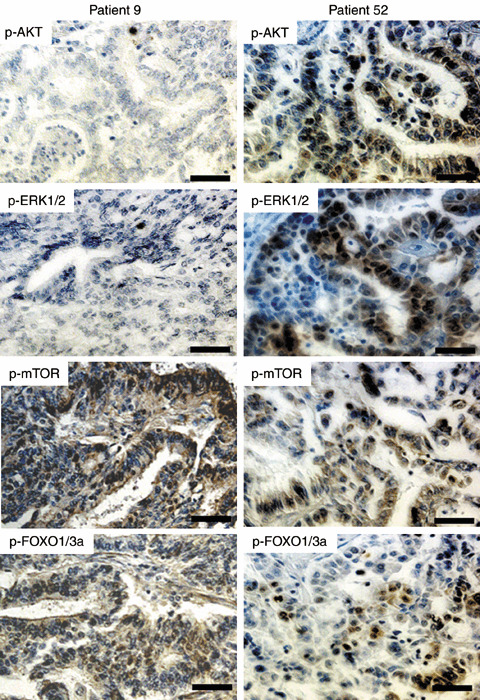
Concordance of phosphorylated (p)‐AKT and p‐ERK1/2 expression in endometrial cancer specimens. Patient 9 showed negative staining for p‐AKT and p‐ERK1/2, whereas both were positive in patient 52. Staining for p‐mTOR and p‐FOXO1/3a was positive in both patients. Note the concordance of p‐AKT and p‐ERK1/2 expression in each patient. (Original magnification ×200. Scale bars = 50 µm.)
Table 4.
Correlation between phosphorylated (p)‐AKT and p‐mTOR expression in endometrial cancer
| p‐AKT expression | n | p‐mTOR expression | |||
|---|---|---|---|---|---|
| Negative | Weak | Moderate | Strong | ||
| Negative | 14 | 3 | 0 | 9 | 2 |
| Weak | 13 | 1 | 2 | 7 | 3 |
| Moderate | 27 | 1 | 3 | 18 | 5 |
| Strong | 9 | 0 | 2 | 5 | 2 |
| Total | 63 | 5 | 7 | 39 | 12 |
P = 0.54.
Table 5.
Correlation between phosphorylated (p)‐AKT and p‐FOXO1/03a expression in endometrial cancer
| p‐AKT expression | n | p‐FOXO1/3a expression | |||
|---|---|---|---|---|---|
| Negative | Weak | Moderate | Strong | ||
| Negative | 14 | 5 | 5 | 3 | 1 |
| Weak | 13 | 2 | 4 | 6 | 1 |
| Moderate | 27 | 4 | 5 | 13 | 5 |
| Strong | 9 | 1 | 2 | 5 | 1 |
| Total | 63 | 12 | 16 | 27 | 8 |
P = 0.60.
In a previous study, we observed the expression of p‐ERK1/2 in 41 out of 63 endometrial cancers (65.1%).( 18 ) Consequently, in the present study, we assessed the correlation between p‐AKT and p‐ERK1/2 expression in endometrial cancer, and found a strong association between them (P = 0.0031) (Table 6; Fig. 3). Categorization of expression levels can significantly affect the results of association assessments. Therefore, we also tested another categorization of p‐AKT and p‐ERK1/2 expression into two groups: low expression (no expression or weak expression), and high expression (moderate or strong expression). Even using that two‐group categorization of expression, we observed a significant association between p‐AKT and p‐ERK1/2 expression (P = 0.018).
Table 6.
Correlation between phosphorylated (p)‐AKT and p‐ERK expression
| p‐AKT expression | n | p‐ERK1/2 expression | |||
|---|---|---|---|---|---|
| Negative | Weak | Moderate | Strong | ||
| Negative | 14 | 11 | 3 | 0 | 0 |
| Weak | 13 | 4 | 4 | 3 | 2 |
| Moderate | 27 | 4 | 11 | 7 | 5 |
| Strong | 9 | 3 | 0 | 5 | 1 |
| Total | 63 | 22 | 18 | 15 | 8 |
P = 0.0031.
Effect of p‐AKT expression on the survival of patients with endometrial cancer. We next examined the prognostic effect of p‐AKT expression. The mean follow‐up time for all patients was 4.08 years for relapse‐free survival (RFS; median, 3.51 years; range, 0.15–9.63 years) and 4.31 years for overall survival (OS; median, 3.75 years; range, 0.56–9.63 years). First, the effect of p‐AKT expression on RFS was analyzed. At the time of the last follow up, 11 of the 63 patients (15.9%) had suffered relapses of endometrial cancer. Figure 4 illustrates univariate outcomes based on p‐AKT expression. The RFS for the high‐ and low‐expression groups was 88.9 and 74.0%, respectively (P = 0.148), suggesting that low p‐AKT expression may be associated with lower RFS. Next, the effect of p‐AKT on OS was analyzed. At the time of the last follow up, 12 of the 63 patients (20.6%) had died (eight from endometrial cancer, and four from other causes [breast cancer, melanoma, liver cancer, and lung embolism]). There was little difference in OS between the high‐ and low‐p‐ERK1/2‐expression groups (83.3 and 77.8%, respectively; P = 0.478).
Figure 4.
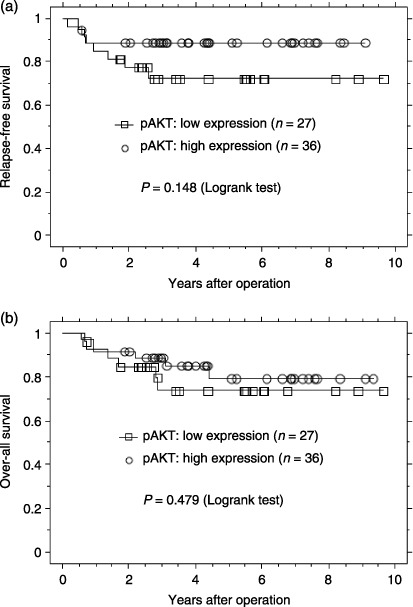
Kaplan–Meier plots of (a) relapse‐free survival and (b) overall survival of groups defined by phosphorylated (p)‐AKT immunohistochemistry. The tumors were divided into two groups (high and low expression of p‐AKT).
Discussion
In the present study, we found that AKT is frequently activated in endometrial cancer, based on the finding that approximately 60% of the patients expressed high levels of p‐AKT. The first notable finding in the present study is that PTEN mutation is not significantly associated with p‐AKT expression in endometrial cancer, indicating that PTEN mutation does not necessarily lead to activation of the PI3K–AKT pathway. This is in contrast to a previous study by Kanamori et al., which found that PTEN expression (evaluated by immunohistochemistry) was inversely correlated with p‐AKT expression in endometrial cancer.( 23 ) Ideally, inactivation of PTEN would be evaluated by examining protein expression using immunohistochemistry or western blot analysis, rather than by examining gene mutation. However, our attempts at PTEN immunohistochemistry have produced non‐specific staining and unstable results, possibly due to a lack of a reliable antibody (data not shown). In a recent study, Velasco et al. found no significant association between PTEN mutation and the expression of p‐AKT in endometrial cancer.( 11 ) Similar results have been reported for other tumor types. In a study of more than 600 breast cancer patients, Panigrahi et al. found that PTEN expression does not correlate with p‐AKT expression.( 24 ) Osman et al. obtained similar results in a study of 204 prostate cancer patients.( 25 ) The present results are also consistent with previous results of an in vitro analysis indicating that PTEN‐knockdown by short hairpin (sh)RNA in endometrial immortalized cell lines does not effectively induce p‐AKT expression.( 19 ) These findings support the theory that there are complex regulatory pathways upstream of AKT other than PTEN mutation. One candidate for such an upstream factor may be PIK3CA mutation, which has been observed in a variety of cancers, including colon,( 26 ) breast,( 27 ) stomach,( 28 ) and ovary.( 29 ) The PIK3CA gene, mapped to 3q26, encodes the p110α catalytic subunit of PI3K, and mutant p110α proteins have been shown to have enhanced lipid‐kinase activity compared to the wild‐type protein, thereby potentially activating the PI3K–AKT pathway and inducing oncogenic transformation. Recent studies have identified PIK3CA mutation in 20–40% of endometrial cancers.( 10 , 11 , 12 ) The present study identified PIK3CA mutations in 16% of endometrial cancers. However, our study found no significant association between PIK3CA mutation and p‐AKT expression. We found four cancers with both PTEN and PIK3CA mutations, but even those cancers did not show enhanced p‐AKT expression. This is consistent with the study by Velasco et al. that failed to find an association between PIK3CA mutation and p‐AKT expression.( 11 ) These findings suggest that there might be additional factors required for activating the PI3K–AKT pathway in endometrial cancer, or either PTEN or p110α has additional functions distinct from PI3K–AKT regulation.
Also in the present study, no correlation was observed between p‐AKT and downstream targets, including p‐mTOR and p‐FOXO expression, indicating that AKT phosphorylation does not necessarily lead to the activation of possible downstream targets, again suggesting that complex downstream signaling pathways are involved in endometrial carcinogenesis.
The second notable finding in the present study is the strong association between AKT and ERK1/2 activation in endometrial cancer. This association may be due to several different molecular events. First, there may be common factors upstream of AKT and ERK1/2, such as epidermal growth factor receptor (EGFR) and Her2. Obviously, a significant proportion of endometrial cancers receive extracellular growth stimuli that initiate intracellular signaling via receptor‐type tyrosine kinases.( 30 , 31 ) Eventually, these common upstream signals induce expression of both AKT and ERK. Simultaneous activation of AKT and ERK is therefore not surprising. No data of a direct association between AKT and ERK1/2 have been reported. Some relevant studies indicate that AKT phosphorylates RAF1 directly, which is an upstream factor of ERK, but this results in a decrease in RAF1 activity.( 32 ) In one study, ERK1/2 was coimmunoprecipitated with AKT in an assembly of ERK1/2, AKT, RSK, and PDK1 in kidney tubular cells, in which RSK acts as a bridge bringing ERK1/2 into proximity with PDK1‐associated AKT.( 33 ) However, this association also resulted in downregulation of AKT activity by ERK1/2. Thus, there has been no reported evidence of a direct biochemical association between AKT and ERK1/2 as coactivators. Thus, it appears that common upstream factors or crosstalk between signals is responsible for the simultaneous activation of AKT and ERK1/2. One candidate mediator for crosstalk might be KRAS mutation, which is known to activate PI3K directly or indirectly.( 22 ) However, we failed to find any correlation between KRAS mutation and p‐AKT expression, indicating that the story is not so simple and complex regulatory pathways may exist for the concomitant activation of AKT and ERK1/2 in endometrial cancer. A recent study indicates that the RAF–MEK–ERK and PI3K–PDK–AKT pathways cooperate to promote G0‐G1‐S‐phase cell‐cycle progression in normal and cancer cells.( 34 ) The present data suggest that the proliferation of endometrial cancer cells is maintained via such cooperative signaling pathways.
Finally, in the present study, we did not find an effect of p‐AKT expression on the prognosis of endometrial cancer. In univariate analysis, higher p‐AKT expression appeared to be associated with better RFS, which is not consistent with the theory that AKT has an antiapoptotic function. Whereas some previous studies have observed a correlation between higher p‐AKT expression and poor clinical outcome in a variety of cancers,( 35 , 36 , 37 , 38 ) others have shown a correlation between lower p‐AKT expression and poor clinical outcome.( 23 , 39 ) Of particular interest is the finding that higher AKT expression correlates with better prognosis for tumors in estrogen‐responsive tissues, including breast, ovarian, and endometrial cancer tissue,( 21 , 39 ) as was found in the present study. In a recent study, we found that endometrial cancers with high ERK1/2 expression had better overall and RFS than those with low expression;( 18 ) the present results suggest a similar relationship for AKT. How can we explain such a relationship between favorable prognosis and high ERK1/2 and AKT expression? Although these physiologically important signaling pathways are essential for the proliferation of normal and cancer cells, disease progression in the late stage of endometrial carcinogenesis probably requires other signaling pathways. For example, during tumorigenesis in estrogen‐dependent organs, cancer cells are likely to lose expression of estrogen receptors and achieve hormone‐independent growth, leading to more aggressive phenotypes in which genetic rearrangements such as mutation, deletion, and translocation of genes make a greater contribution to the malignant potential of cells. This theory can be applied to p‐AKT expression in endometrial cancer.
In summary, in the present study, AKT expression was observed frequently in endometrial cancers in a PTEN‐independent manner, was strongly associated with ERK1/2 activation, and was associated with relatively favorable prognosis. The present study supports the presence of complex signaling pathways, including the novel signaling cascades that might mask the conventional tumorigenic PTEN–PI3K–AKT–mTOR pathway, and suggests a close association between the ERK and PI3K pathways in this tumor type.
Acknowledgments
We wish to thank the members of the Department of Pathology, Kanazawa University Hospital, for the preparation of tissue for immunohistochemistry. This study was supported by a Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science and the Megumi Medical Foundation of Kanazawa University.
References
- 1. Tashiro H, Blazes MS, Wu R et al . Mutations in PTEN are frequent in endometrial carcinoma but rare in other common gynecological malignancies. Cancer Res 1997; 57: 3935–40. [PubMed] [Google Scholar]
- 2. Mutter GL, Lin MC, Fitzgerald JT et al . Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J Natl Cancer Inst 2000; 92: 924–30. [DOI] [PubMed] [Google Scholar]
- 3. Kanaya T, Kyo S, Sakaguchi J et al . Association of mismatch repair deficiency with PTEN frameshift mutations in endometrial cancers and the precursors in a Japanese population. Am J Clin Pathol 2005; 124: 89–96. [DOI] [PubMed] [Google Scholar]
- 4. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3–5‐trisphosphate. J Biol Chem 1998; 273: 13 375–8. [DOI] [PubMed] [Google Scholar]
- 5. Myers MP, Pass I, Batty IH et al . The lipid phosphatase activity of PTEN is critical for its tumor suppressor function. Proc Natl Acad Sci USA 1998; 95: 13 513–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Franke TF, Kaplan DR, Cantley LC. PI3K: Downstream AKTion blocks apoptosis. Cell 1997; 88: 435–7. [DOI] [PubMed] [Google Scholar]
- 7. Downward J. Ras signalling and apoptosis. Curr Opin Genet Dev 1998; 8: 49–54. [DOI] [PubMed] [Google Scholar]
- 8. Brunet A, Bonni A, Zigmond MJ et al . Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999; 96: 857–68. [DOI] [PubMed] [Google Scholar]
- 9. Samuels Y, Wang Z, Bardelli A et al . High frequency of mutations of the PIK3CA gene in human cancers. Science 2004; 304: 554. [DOI] [PubMed] [Google Scholar]
- 10. Oda K, Stokoe D, Taketani Y, McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res 2005; 65: 10 669–73. [DOI] [PubMed] [Google Scholar]
- 11. Velasco A, Bussaglia E, Pallares J et al . PIK3CA gene mutations in endometrial carcinoma: correlation with PTEN and K‐RAS alterations. Hum Pathol 2006; 37: 1465–72. [DOI] [PubMed] [Google Scholar]
- 12. Hayes MP, Wang H, Espinal‐Witter R et al . PIK3CA and PTEN mutations in uterine endometrioid carcinoma and complex atypical hyperplasia. Clin Cancer Res 2006; 12: 5932–5. [DOI] [PubMed] [Google Scholar]
- 13. Mizuuchi H, Nasim S, Kudo R, Silverberg SG, Greenhouse S, Garrett CT. Clinical implications of K‐ras mutations in malignant epithelial tumors of the endometrium. Cancer Res 1992; 52: 2777–81. [PubMed] [Google Scholar]
- 14. Enomoto T, Fujita M, Inoue M et al . Alterations of the p53 tumor suppressor gene and its association with activation of the c‐K‐ras‐2 protooncogene in premalignant and malignant lesions of the human uterine endometrium. Cancer Res 1993; 53: 1883–8. [PubMed] [Google Scholar]
- 15. Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006; 441: 424–30. [DOI] [PubMed] [Google Scholar]
- 16. Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal‐regulated kinase activation. Cell 1999; 80: 179–85. [DOI] [PubMed] [Google Scholar]
- 17. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature 2001; 410: 37–40. [DOI] [PubMed] [Google Scholar]
- 18. Mizumoto Y, Kyo S, Mori N et al . Activation of ERK1/2 occurs independently of KRAS or BRAF status in endometrial cancer and is associated with favorable prognosis. Cancer Sci 2007; 98: 652–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mizumoto Y, Kyo S, Ohno S et al . Creation of tumorigenic human endometrial epithelial cells with intact chromosomes by introducing defined genetic elements. Oncogene 2006; 25: 5673–82. [DOI] [PubMed] [Google Scholar]
- 20. Katabuchi H, Van Rees B, Lambers AR et al . Mutations in DNA mismatch repair genes are not responsible for microsatellite instability in most sporadic endometrial carcinomas. Cancer Res 1995; 55: 5556–60. [PubMed] [Google Scholar]
- 21. Boland CR, Thibodeau SN, Hamilton SR et al . A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998; 58: 5248–57. [PubMed] [Google Scholar]
- 22. Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer 2006; 6: 184–92. [DOI] [PubMed] [Google Scholar]
- 23. Kanamori Y, Kigawa J, Itamochi H et al . Correlation between loss of PTEN expression and Akt phosphorylation in endometrial carcinoma. Clin Cancer Res 2001; 7: 892–5. [PubMed] [Google Scholar]
- 24. Panigrahi AR, Pinder SE, Chan SY, Paish EC, Robertson JFR, Ellis IO. The role of PTEN and its signalling pathways, including AKT, in breast cancer: an assessment of relationships with other prognostic factors and with outcome. J Pathol 2004; 204: 93–100. [DOI] [PubMed] [Google Scholar]
- 25. Osman I, Dai J, Mikhail M et al . Loss of neural endopeptidase and activation of protein kinase B (Akt) is associated with prostate cancer progression. Cancer 2006; 107: 2628–36. [DOI] [PubMed] [Google Scholar]
- 26. Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3‐kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci USA 2005; 102: 802–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu G, Xing M, Mambo E et al . Somatic mutation and gain of copy number of PIK3CA in human breast cancer. Breast Cancer Res 2005; 7: 609–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li VS, Wong CW, Chan TL et al . Mutations of PIK3CA in gastric adenocarcinoma. BMC Cancer 2005; 5: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Y, Helland A, Holm R, Kristensen GB, Borresen‐Dale AL. PIK3CA mutations in advanced ovarian carcinomas. Hum Mutat 2005; 25: 322. [DOI] [PubMed] [Google Scholar]
- 30. Wang D, Konishi I, Koshiyama M et al . Expression of c‐erbB‐2 protein and epidermal growth receptor in endometrial carcinomas: Correlation with clinicopathologic and sex steroid receptor status. Cancer 1993; 72: 2628–37. [DOI] [PubMed] [Google Scholar]
- 31. Khalifa MA, Mannel RS, Haraway SD, Walker J, Min KW. Expression of EGFR, HER‐2/neu, P53, and PCNA in endometrioid, serous papillary, and clear cell endometrial adenocarcinomas. Gynecol Oncol 1994; 53: 84–92. [DOI] [PubMed] [Google Scholar]
- 32. Sinha D, Bannergee S, Schwartz JH, Lieberthal W, Levine JS. Inhibition of ligand‐independent ERK1/2 activity in kidney proximal tubular cells deprived of soluble survival factors up‐regulates Akt and prevents apoptosis. J Biol Chem 2004; 279: 10 962–72. [DOI] [PubMed] [Google Scholar]
- 33. Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M. Regulation of Raf–Akt cross‐talk. J Biol Chem 2002; 277: 31 099–106. [DOI] [PubMed] [Google Scholar]
- 34. Mirza AM, Gysin S, Malek N, Nakayama K, Roberts JM, McMahon M. Cooperative regulation of the cell division cycle by the protein kinases RAF and AKT. Mol Cell Biol 2004; 24: 10 868–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dai DL, Martinka M, Li G. Prognostic significance of activated Akt expression in melanoma: a clinicopathologic study of 292 cases. J Clin Oncol 2005; 23: 1473–82. [DOI] [PubMed] [Google Scholar]
- 36. David O, Jett J, LeBeau H et al . Phospho‐Akt overexpression in non‐small cell lung cancer confers significant stage‐independent survival disadvantage. Clin Cancer Res 2004; 10: 6865–71. [DOI] [PubMed] [Google Scholar]
- 37. Kreisberg JI, Malik SN, Prihoda TJ et al . Phosphorylation of Akt (Ser473) is an excellent predictor of poor clinical outcome in prostate cancer. Cancer Res 2004; 64: 5232–6. [DOI] [PubMed] [Google Scholar]
- 38. Schmitz KJ, Otterbach F, Callies R et al . Prognostic relevance of activated Akt in node‐negative breast cancer: a clinicopathological study of 99 cases. Mod Pathol 2004; 17: 15–21. [DOI] [PubMed] [Google Scholar]
- 39. Castellvi J, Garcia A, Rojo F et al . Phosphorylated 4E binding protein 1: a hallmark of cell signaling that correlates with survival in ovarian cancer. Cancer 2006; 107: 1801–11. [DOI] [PubMed] [Google Scholar]