

Abstract
Oncogenic mutation of epidermal growth factor receptor kinase domain is strongly associated with clinical response to tyrosine kinase inhibitors in non‐small‐cell lung carcinoma. Despite an initial encouraging response, patients eventually develop drug resistance and relapse. Great efforts have been made to identify the molecular mechanisms of drug resistance. With the recognition of cancer as a whole complex system, here it is proposed that cancer may evolve drug resistance in a cancer‐cell‐autonomous manner as well as a non‐cancer‐cell‐autonomous manner. The former mainly arises at three levels: the robustness of the epidermal growth factor receptor signaling network; cancer epigenetic changes; or cancer genetic alteration, which may be dependent on the therapeutics methods and treatment duration. As cancer stroma plays an essential role in lung cancerigenesis, we further discuss the potential mechanisms for drug resistance development in a non‐cancer‐cell‐autonomous manner, which may arise from the interaction between cancer cells and cancer stroma, including stromal cells and extracellular matrix. (Cancer Sci 2010)
Lung cancer, pathologically divided into small‐cell lung carcinoma and non‐small cell lung carcinoma (NSCLC), is the leading cause of cancer‐related death worldwide.( 1 ) Three main sub‐types of NSCLC, squamous cell lung carcinoma, adenocarcinoma, and large cell lung carcinoma, are grouped together because their prognosis and management are similar. The EGFR signal pathway, essential for normal epithelial cell proliferation and survival,( 2 ) is frequently deregulated in lung adenocarcinoma and squamous cell carcinoma. For example, EGFRvIII mutation with a partial deletion of the extracellular domain encoded by exons 2–7 exists exclusively in lung squamous cell carcinoma.( 3 ) In contrast, EGFR kinase domain mutation is observed in 30–60% of lung adenocarcinomas with features similar to that of bronchioloalveolar carcinoma that arises in the distal bronchioles or alveoli that initially show a specific non‐invasive growth pattern,( 4 , 5 , 6 ) sometimes in adenosquamous carcinoma, but seldom in pure squamous or large cell carcinoma.( 7 ) EGFR kinase domain mutation is also preferentially observed in females, non‐smokers, and those of East Asian ethnicity.( 8 ) Interestingly, the gender bias of EGFR kinase domain mutation is largely due to the high prevalence of tobacco smoking in men in East Asia.( 9 , 10 , 11 )
Tyrosine kinase inhibitors, including gefitinib and erlotinib, have been developed to target EGFR in lung cancer treatment. The EGFR kinase domain mutation is strongly associated with clinical response to TKIs.( 4 , 5 , 6 ) Although multiple molecular testing methods, including immunohistochemistry, FISH, and mutational analyses, were used to assess the EGFR status, EGFR mutation remains one of the most important determinants for the prediction of clinical response and survival benefit.( 12 ) This association may be explained by the different biological functions of wild‐type or mutated EGFR in contributing to cancer maintenance.( 13 ) From the biological standpoint, EGFR‐targeted therapy in those NSCLC patients with EGFR‐activating mutations is targeting the key oncogene that is directly involved in tumorigenesis and is essential for tumor maintenance, thus frequently correlated with effective clinical outcome.( 13 )
Despite impressive and durable clinical responses, those patients with EGFR mutant NSCLCs almost invariably develop drug resistance after approximately 1 year of TKI treatment.( 14 ) In the past several years, studies have revealed certain molecular mechanisms that contribute to drug resistance, including secondary EGFR mutation T790M and tyrosine protein kinase MET amplification.( 15 , 16 , 17 , 18 , 19 , 20 ) With the recognition of cancer as a whole complex system, here we propose that cancer may evolve TKI resistance in a cancer‐cell‐autonomous manner as well as a non‐cancer‐cell‐autonomous manner. We further discuss the cancer‐cell‐autonomous mechanisms from the robustness of the EGFR signaling network, to epigenetic changes, to genetic alterations in cancer cells, and the potential mechanisms for drug resistance in a non‐cancer‐cell‐autonomous manner, which may arise from the interaction between cancer cells and cancer stroma (Fig. 1).
Figure 1.
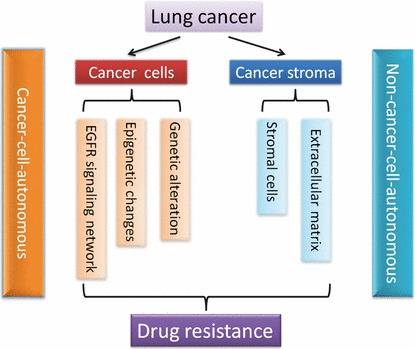
Schematic illustration of the potential molecular mechanisms of drug resistance development in epidermal growth factor receptor (EGFR) mutation‐targeted lung cancer therapy in a cancer‐cell‐autonomous and non‐cancer‐cell‐autonomous manner. The robustness of the EGFR signaling network, epigenetic changes, and genetic alterations in cancer cells contribute to drug resistance in a cancer‐cell‐autonomous manner. The interaction between cancer cells and cancer stroma, including stromal cells and ECM, contributes to drug resistance in a non‐cancer‐cell‐autonomous manner.
Drug Resistance Developed in a Cancer‐Cell‐Autonomous Manner
Lung cancer is a robust system featured with extreme complexity and high heterogeneity. The EGFR pathway is located at the center of converging signals for cell proliferation, motility, and other cancer cell behaviors.( 2 ) Stress or perturbation may modulate multiple signaling pathways and trigger epigenetic changes. Genetic alteration frequently occurs during lung cancerigenesis due to genomic instability. Although these three factors might contribute to drug resistance development at different timepoints during cancer treatment, they work together to confer cancer cells with the ability to maintain stable function against drug treatment. It will be discussed how the drug resistance develops during EGFR‐targeted therapy in a cancer‐cell‐autonomous manner.
Epidermal growth factor receptor signaling network in drug resistance development.
The EGFR signal pathway contributes to multiple biological events, including normal cell proliferation, differentiation, and survival, and is frequently deregulated under pathological conditions such as in NSCLC.( 2 , 21 ) Epidermal growth factor receptor belongs to the ERBB family, which consists of four closely related receptor tyrosine kinases: HER1/ERBB1; HER2/NEU/ERBB2; HER3/ERBB3; and HER4/ERBB4. After ligand stimulation, EGFR is activated through homodimerization or heterodimerization and transmits signals to downstream substrates such as PI3K/AKT, RAS/RAF/MAPK, and STAT3/5 pathways, which contributes significantly to cell proliferation and cell survival (Fig. 2).
Figure 2.
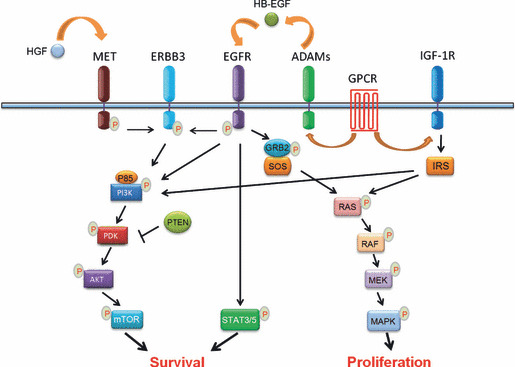
Simplified schematic illustration of the epidermal growth factor receptor (EGFR) signaling network. After ligand stimulation, EGFR is activated and directly transmits signals to downstream pathways, including the phosphatidylinositol‐3‐kinase (PI3K)/3‐phosphoinositide‐dependent protein kinase (PDK)/v‐akt murine thymoma viral oncogene homolog (AKT)/mammalian target of rapamycin (mTOR) and signal transducer and activator of transcription 3/5 (STAT3/5) pathways for cell survival, and the rat sarcoma viral oncogene homolog (RAS)/v‐raf‐1 murine leukemia viral oncogene homolog (RAF)/MAPK/ERK kinase (MEK)/MAPK pathway for cell proliferation. Phosphatase and tensin homolog (PTEN) inhibits AKT activity through PDK. The EGFR pathway cross‐talks with other signaling pathways: either hepatocyte growth factor receptor (MET) activation by hepatocyte growth factor (HGF) stimulation or EGFR activation is able to transmit signals through erythroblastic leukemia viral (v‐erb‐b) oncogene homolog 3 (ERBB3) and results in the activation of the PI3K/AKT pathway; and G‐protein coupled receptor (GPCR) activation enhances EGFR signaling through a disintegrin and metalloproteinases (ADAMs) that shed pro‐heparin‐binding EGF‐like growth factor (HB‐EGF) and increase the amount of HB‐EGF. As well as the cross‐talk with GPCR signaling, insulin‐like growth factor‐1 receptor (IGF‐1R) signaling through insulin receptor substrate (IRS) activates the PI3K/PDK/AKT/mTOR and RAS/RAF/MEK/MAPK pathways, which are commonly shared by EGFR signaling. GRB2, growth factor receptor‐bound 2; P, phosphorylation; SOS, son of sevenless homolog; P85, phosphatidylinositol 3‐kinase regulatory subunit.
Several EGFR downstream substrates have been indicated to be responsible for drug resistance despite the current lack of clinical evidence. Activation of the EGFR downstream signal PI3K/AKT pathway is essential for cancer cell survival (Fig. 2). An effective TKI treatment in NSCLC cell lines usually inhibits PI3K/AKT pathway activation.( 14 , 22 ) Conversely, the PI3K/AKT pathway remains activated in most models of acquired drug resistance.( 19 , 23 , 24 , 25 ) Oncogenic mutations in the PI3K catalytic subunit alpha (PIK3CA) have been found in 1–5% of NSCLC samples.( 26 , 27 ) In contrast to other oncogenic drivers such as KRAS, BRAF, or HER2 mutations, the PIK3CA mutations is not mutually exclusive with EGFR kinase domain mutations.( 26 , 27 ) Thus, the coexistence of PIK3CA mutation may confer TKI resistance in those cases of NSCLC with EGFR mutations. This was confirmed by an in vitro study.( 24 ) In breast cancer, the presence of PIK3CA mutations predicts a lack of response of HER2 amplified tumors to trastuzumab treatment.( 28 ) Similar findings have been reported in colon cancer.( 29 ) Loss of phosphatase and tensin homolog (PTEN), which activates AKT, may confer a TKI resistance during NSCLC and colon cancer treatment.( 28 , 29 , 30 ) In addition, increased internalization of ligand‐activated EGFR and altered EGFR trafficking was previously found to confer drug resistance through unknown mechanisms.( 31 ) Further investigation using lung cancer specimens may improve our understanding about whether and/or how the EGFR downstream signaling, including the PI3K/AKT pathway, contributes to drug resistance development.
The cross‐talk of EGFR signaling with other receptor partners is of potential interest in the development of TKI resistance. Multiple membrane proteins including G‐protein coupled receptors (GPCR), IGF‐1R, and MET cross‐talk with EGFR or other members of the EGFR family, such as HER2 and ERBB3.( 32 ) For example, GPCR activation results in G‐protein‐mediated activation of ADAM family members, which shed pro‐HB‐EGF (Fig. 2). The increase in the HB‐EGF amount in turn enhances EGFR signaling.( 33 , 34 , 35 ) The IGF‐1R protein plays an important role in tumor formation and progression through its multiple biological functions including cell proliferation, apoptosis, and angiogenesis.( 36 ) Both in vitro and in vivo studies have indicated that the cross‐talk between EGFR and IGF‐1R contributes to drug resistance in EGFR‐targeted therapy.( 37 ) Multiple levels of interaction between EGFR and IGF‐1R have been proposed, either through a direct association between these two receptors or through common interaction partners such as GPCR or downstream signaling molecules( 38 ) (Fig. 2). A recent study has shown that simultaneous targeting of EGFR and IGF‐1R significantly decreased the possibility of TKI resistance development in PC9 cells, an NSCLC cell line which has EGFR kinase domain mutation and is initially sensitive to TKI treatment.( 39 )
The importance of MET in contributing to TKI resistance was initially documented by Engelman et al. ( 19 ) In approximately 20% of patients with acquired TKI resistance, lung cancer cells display MET gene amplification.( 19 , 20 ) Interestingly, a recent study has identified the existence of subpopulations of lung cancer cells with MET amplification prior to TKI treatment that may be responsible for drug resistance development.( 40 ) In addition, autocrine hepatocyte growth factor production, which activates MET, also induces drug resistance.( 40 , 41 ) MET contributes to EGFR TKI resistance through transactivation of ERBB3.( 19 ) Interestingly, the PI3K/AKT pathway is the main downstream pathway activated by MET through ERBB3( 19 ) (Fig. 2), which allows the cancer cells to maintain survival signaling even in the presence of EGFR inhibitors. Interestingly, MET‐resistant cancer cells can shift back the survival‐dependent oncogene to EGFR,( 42 ) highlighting the importance of the cross‐talk between MET and ERBB3. Concomitant inhibition of both EGFR and MET is very important for overcoming TKI resistance and may provide a survival benefit for NSCLC patients. Interestingly, this is somehow supported by recently released phase II trial data showing that ARQ 197, a MET inhibitor, when used in combination with erlotinib, showed a 66% improvement in median progression‐free survival in patients with advanced, refractory NSCLC.
Epigenetic changes in drug resistance development.
Epigenetic changes, referring to changes of the cell “epigenome” without the direct involvement of DNA sequence alteration, not only contribute significantly to cancer development and progression but also provide cancer cells with another means to escape from various targeted therapy. In response to various perturbations, the epigenome may change correspondingly and turn on or off genes or pathways that may confer survival benefit. Similar to genetic alterations, epigenetic changes can be transmitted to the next generation at least for a certain period. DNA cytosine methylation, gene imprinting, and chromatin remodeling are key events involved in epigenetic changes. Inhibitors towards DNA methylation and histone deacetylase (HDAC) have been developed as two main classes of drugs over past years. Here the main focus is on the potential role of HDACs in drug resistance development.
Histone deacetylases play an important role in cellular proliferation and survival.( 43 ) HDACs actively regulate gene expression through chromatin modification in the opposing action of histone acetyltransferases (HATs) in normal cells.( 44 ) Histone acetyltransferases transfer acetyl groups to the aminoterminal lysine residues in histones, which causes local chromatin expansion and allows proteins to access and activate gene transcription.( 44 ) Conversely, HDACs catalyze the removal of acetyl groups, which results in chromatin condensation and the repression of gene transcription.( 45 ) The well‐balanced action of HATs and HDACs in normal cells is frequently disrupted during the cancerigenesis process, which therefore confers tumor cells with the ability to efficiently silence tumor suppressor genes and genes related to cell cycle arrest, differentiation, and apoptosis.( 46 ) In addition, HDACs also modify several non‐histone proteins such as TP53, E2F1, HIF‐1α, and HSP90.( 47 ) The inhibition of HDAC activity reactivates silenced genes and induces growth arrest and apoptosis in numerous tumor cell lines.( 48 ) Several HDAC inhibitors, including vorinostat, LBH‐589, PDX‐101, and MS‐275, have been developed for NSCLC treatment. Interestingly, treatment of LBH‐589 in lung cancer cells with EGFR mutations proves to be effective in triggering apoptosis, mainly through HSP90 acetylation and downregulation of EGFR and other key survival signaling proteins.( 49 ) Inhibition of HDAC activity by LBH‐589 also synergizes with erlotinib for treatment of lung cancer cell lines.( 49 )
Interestingly, an essential role of HDACs in a “reversible drug resistance” has been recently proposed.( 39 ) While modeling the acute response of PC9 cells to TKI treatment, Sharma and coworkers consistently detected a small subpopulation of reversibly “drug‐tolerant” cells, which was significantly associated with an altered chromatin state.( 39 ) Both HDACs and the histone demethylase RBP2/KDM5A/Jarid1A are involved in TKI resistance.( 39 ) Interestingly, continuous propagation for approximately 30 passages resensitizes these drug‐tolerant cells to TKI, further supporting the view that the epigenetic alteration is dynamically involved in drug resistance and may be inheritable, at least for a certain period.( 39 )
Genetic alteration in drug‐resistance development.
Genomic instability remains one of most important obstacles for lung cancer therapy. Genetic alterations in lung cancer, especially oncogenic driver mutations of EGFR, KRAS, BRAF, HER2, and ALK fusion,( 50 , 51 ) provide the driving force for cancer development and progression. Yet, it also works as the basis for the development of molecular targeted strategies in the clinic. At the same time, the genomic instability behind these essential genetic alterations enables cancer cells to evolve and find ways to escape molecular targeting.
Despite the initial encouraging response to EGFR TKI in certain lung cancer patients, genetic alteration such as MET amplification, found in approximately 20% of relapsed patients, sustains the normal function of cancer cells through cross‐talk with the EGFR pathways. Studies have revealed that approximately half of these relapsed patients have a secondary EGFR mutation, the substitution of a threonine for methionine at position 790 (T790M).( 15 , 16 , 17 , 18 ) The EGFR T790M mutation occurs at the “gatekeeper” residue in tyrosine kinases and confers a drug resistance analogous to BCR‐ABL and KIT in imatinib‐resistant CML and gastrointestinal stromal tumors, respectively.( 52 , 53 )
In contrast to primary EGFR mutations such as L858R and exon 19 LREA deletion mutations, EGFR T790M mutation cannot be inhibited by either gefitinib or erlotinib treatment in in vitro studies, and maintains downstream PIK3/AKT activation.( 16 , 31 , 54 ) This has been further confirmed by in vivo animal model studies.( 55 , 56 ) Ectopic expression of the EGFR T790M mutant is sufficient to render EGFR mutant cancer cell lines resistant to gefitinib.( 24 ) When NSCLC cell lines such as PC‐9 and H3255, which have the highly sensitive EGFR mutant, were exposed to gefitinib for a long period, they eventually acquired drug resistance and a T790M mutation.( 23 , 24 ) Interestingly, a rather small percentage of the EGFR alleles within one single TKI‐resistant cell are observed to harbor T790M mutation in H3255 cell drug resistant model, which is termed “allelic dilution”.( 24 )
In contrast to the reversible TKIs like gefitinib and erlotinib, the second generation EGFR inhibitors, the irreversible TKIs such as CL387,785, EKB‐569, PF00299804, BIBW2992, and HKI‐272, seem to effectively inhibit EGFR T790M and block the growth of NSCLC cell lines harboring T790M mutations.( 16 , 24 , 31 , 54 , 57 )
Preclinical work in mice suggests that irreversible EGFR inhibitors such as HKI‐272 might not be potent enough to completely block EGFR T790M signaling in vivo, and the combinational therapy with inhibitors blocking downstream signaling, such as rapamycin, improves efficacy.( 55 ) Interestingly, a novel EGFR TKI, WZ4002, efficiently inhibits EGFR phosphorylation and induces significant tumor regression in murine models of EGFR T790M.( 58 ) Additionally, HSP90 inhibitors may effectively target EGFR mutants for degradation and thus overcome the T790M mutation.( 59 ) As T790M remains as a good target, a better designed drug or combinational therapeutic strategies are necessary to overcome drug resistance.
Drug Resistance Development in a Non‐Cancer‐Cell‐Autonomous Way
The complexity of cancer exists not only in cancer cells but also in tumor stroma, which is composed of stromal cells such as fibroblasts, inflammatory cells, and endothelial cells and extracellular matrix (ECM). The interplay between cancer cells and the microenvironment strongly impacts upon the cancer development and progression.( 60 , 61 , 62 ) Cancer‐associated infiltrated inflammatory cells or ECM has been shown to contribute to tumor progression.( 60 , 61 , 62 , 63 ) Additionally, blood vessels provide cancer cells with oxygen and nutrition. Targeting both the cancer cells and the new blood vessel genesis, so called “angiogenesis”, are under development now. The use of dual EGFR/vascular endothelial growth factor inhibitors, which target both EGFR and vascular endothelial growth factor receptor signaling pathways, may be more effective than the inhibition of one single pathway and help to overcome resistance to EGFR inhibition.( 64 )
Additionally, genetic alterations in tumor stroma may contribute to tumor formation. For example, the genetic inactivation of Pten in stromal fibroblasts of mouse mammary glands accelerated the initiation, progression, and malignant transformation of mammary epithelial tumours.( 65 ) Similarly, LKB1 loss in mesenchymal cells has also been shown to cause the formation of gastrointestinal polyps.( 66 ) The interaction between stromal cells with Scribble loss and RAS mutant tumor cells results in JNK pathway activation and cancer metastasis into distant organs.( 67 ) Although the above mechanisms have not been identified in clinic specimens yet, genetic analyses have revealed that TP53 mutations occur in non‐neoplastic stromal cells.( 68 ) Targeting the cancer cells alone without concomitant treatment towards stromal cells in this case may be ineffective in blocking drug resistance development. A comprehensive understanding of the interaction between cancer cells and cancer stroma may provide the basis for development of better therapeutic strategies for lung cancer treatment and overcoming drug resistance.
Conclusion and Future Directions
Cancers have integrated several essential hallmarks, including uncontrolled growth, limitless proliferation, insensitivity to antigrowth signaling, sustained angiogenesis, and metastasis.( 69 ) Cancer robustness is achieved through the interplay between cancer cells and the microenvironment, which provides cancer the ability to maintain normal function against various perturbations and to eventually develop drug resistance( 70 , 71 ) The view of cancer as a whole is helpful for studies on drug resistance mechanisms and clinical therapeutic strategy development.
To overcome drug resistance in cancer therapy still remains a big challenge. Deregulation of the EGFR pathway through oncogenic mutations is well recognized as an important cause of lung cancerigenesis. Several positive and negative feedback loops are located in the EGFR signaling pathway.( 71 ) For example, two positive feedback loops have been proposed: first, ADAMs activated by proline‐rich tyrosine kinase 2/v‐src sarcoma viral oncogene homolog shed pro‐HB‐EGF and increase the HB‐EGF amount, which further enhances the EGFR signaling; second, phospholipase Cγ activation produces diacylglycerol from PI4,5‐P2, which consecutively activates downstream PKC, phospholipase D, and phosphatidylinositol‐5‐kinase (PI5K). Activation of PI5K produces PI4,5‐P2 from PI4‐P and thus enhances the EGFR signaling (Fig. 3a). In addition, there exist six negative feedback loops: the activation of protein tyrosine phosphatase either SHP‐1 or SHP‐2 inhibits EGFR signaling; the activation of ERK1 or ERK2, or ribosomal protein S6 kinase inhibits the son of sevenless homolog; and recruitment of Casitas B‐lineage lymphoma proto‐oncogene by growth factor receptor‐bound protein mediates EGFR degradation (Fig. 3b). Additionally, multiple membrane proteins such as IGF‐1R and MET acting as either direct or indirect partners for EGFR or EBB3 contribute to the transactivation of EGFR signaling (Fig. 2). The partners of MET, including integrin, the adhesive molecule CD44, class B plexins, FAS, and other tyrosine kinase receptors such as RON, are also involved in cancer initiation, progression, and malignant conversion. All of these feedback loops and signaling cross‐talk strengthen and solidify the robustness of the EGFR signal pathway, which may dampen the efficiency of EGFR‐targeted therapy by a single agent and trigger drug resistance sooner or later. Additionally, cancer cells harbor far more complex genetic alteration, which may make the single agent treatment unsuccessful. PIK3CA oncogenic mutations occur simultaneously with EGFR mutations.( 26 ) Why do cancer cells need two such oncogenes to tangle and how do these two synergize in cancerigenesis? The answers to these questions may improve current therapeutic strategies. Furthermore, research into the role of stromal cells or ECM in drug resistance is still in its infancy. However, there is no doubt that future work will uncover mechanisms related to how the interaction between cancer cells and their microenvironment works together to resist the drug treatment. It is our hope that we could one day overcome drug resistance with a comprehensive view of cancer.
Figure 3.
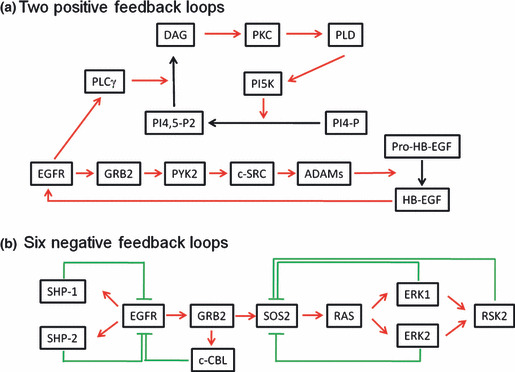
Positive and negative feedback loops in the epidermal growth factor receptor (EGFR) signaling pathway. (a) Two positive feedback loops. First, the activation of proline‐rich tyrosine kinase 2 (PYK2)/v‐src sarcoma viral oncogene homolog (c‐Src) activates a disintegrin and metalloproteinases (ADAMs) which shed pro‐heparin‐binding EGF‐like growth factor (HB‐EGF) and results in an increase in HB‐EGF and enhancement of EGFR signaling. Second, phospholipase Cγ (PLCγ) activation produces diacylglycerol (DAG) from PI4,5‐P2, which consecutively activates downstream PKC, phospholipase D (PLD), and phosphatidylinositol‐5‐kinase (PI5K). Activation of PI5K produces PI4,5‐P2 from PI4‐P and thus enhances the signaling. (b) Six negative feedback loops. The activation of protein tyrosine phosphatases (SHP‐1 and SHP‐2) inhibits EGFR signaling; the activation of ERK1 and ERK2, or ribosomal protein S6 kinase (RSK2) inhibits the son of sevenless homolog (SOS); recruitment of Casitas B‐lineage lymphoma proto‐oncogene (c‐Cbl) by growth factor receptor‐bound protein (GRB2) mediates EGFR degradation. RAS, rat sarcoma viral oncogene homolog.
Abbreviations
- ADAM
a disintegrin and metalloprotease
- AKT
v‐akt murine thymoma viral oncogene homolog
- EGFR
epidermal growth factor receptor
- ERBB
erythroblastic leukemia viral (v‐erb‐b) oncogene
- GPCR
G‐protein coupled receptor
- HB‐EGF
heparin‐binding EGF‐like growth factor
- IGF‐1R
insulin‐like growth factor‐1 receptor
- MET
hepatocyte growth factor receptor
- NSCLC
non‐small‐cell lung carcinoma
- PI3K
phosphatidylinositol‐3‐kinase
- RAF
v‐raf‐1 murine leukemia viral oncogene homolog
- RAS
rat sarcoma viral oncogene homolog
- STAT3/5
signal transducer and activator of transcription 3/5
- TKI
tyrosine kinase inhibitor
Acknowledgments
The author thanks Yijun Gao for manuscript preparation. H. J. is a scholar of the Hundred Talents Program of the Chinese Academy of Sciences and receives funding from the Chinese Academy of Sciences (KSCX1‐YW‐22).
References
- 1. Jemal A, Siegel R, Ward E et al. Cancer statistics, 2008. CA Cancer J Clin 2008; 58: 71–96. [DOI] [PubMed] [Google Scholar]
- 2. Reinehr R, Haussinger D. Epidermal growth factor receptor signaling in liver cell proliferation and apoptosis. Biol chem 2009; 390: 1033–7. [DOI] [PubMed] [Google Scholar]
- 3. Ji H, Zhao X, Yuza Y et al. Epidermal growth factor receptor variant III mutations in lung tumorigenesis and sensitivity to tyrosine kinase inhibitors. Proc Natl Acad Sci USA 2006; 103: 7817–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lynch TJ, Bell DW, Sordella R et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–39. [DOI] [PubMed] [Google Scholar]
- 5. Paez JG, Janne PA, Lee JC et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Sci (New York, NY) 2004; 304: 1497–500. [DOI] [PubMed] [Google Scholar]
- 6. Pao W, Miller V, Zakowski M et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 2004; 101: 13306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Toyooka S, Yatabe Y, Tokumo M et al. Mutations of epidermal growth factor receptor and K‐ras genes in adenosquamous carcinoma of the lung. Int J Cancer 2006; 118: 1588–90. [DOI] [PubMed] [Google Scholar]
- 8. Haber DA, Bell DW, Sordella R et al. Molecular targeted therapy of lung cancer: EGFR mutations and response to EGFR inhibitors. Cold Spring Harb Symp Quant Biol 2005; 70: 419–26. [DOI] [PubMed] [Google Scholar]
- 9. Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res 2004; 64: 8919–23. [DOI] [PubMed] [Google Scholar]
- 10. Sun YH, Fang R, Gao B et al. Comparable rate of EGFR kinase domain mutation in lung adenocarcinomas from Chinese male and female never‐smokers. Acta Pharmacol Sin 2010; 31: 647–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tanaka T, Matsuoka M, Sutani A et al. Frequency of and variables associated with the EGFR mutation and its subtypes. Int J Cancer 2010; 126: 651–5. [DOI] [PubMed] [Google Scholar]
- 12. Eberhard DA, Giaccone G, Johnson BE. Biomarkers of response to epidermal growth factor receptor inhibitors in Non‐Small‐Cell Lung Cancer Working Group: standardization for use in the clinical trial setting. J Clin Oncol 2008; 26: 983–94. [DOI] [PubMed] [Google Scholar]
- 13. Ji H, Sharpless NE, Wong KK. EGFR targeted therapy: view from biological standpoint. Cell cycle (Georgetown, Tex) 2006; 5: 2072–6. [DOI] [PubMed] [Google Scholar]
- 14. Engelman JA, Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr Opin Genet Dev 2008; 18: 73–9. [DOI] [PubMed] [Google Scholar]
- 15. Pao W, Wang TY, Riely GJ et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2005; 2: e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kobayashi S, Boggon TJ, Dayaram T et al. EGFR mutation and resistance of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2005; 352: 786–92. [DOI] [PubMed] [Google Scholar]
- 17. Balak MN, Gong Y, Riely GJ et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor‐mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res 2006; 12: 6494–501. [DOI] [PubMed] [Google Scholar]
- 18. Kosaka T, Yatabe Y, Endoh H et al. Analysis of epidermal growth factor receptor gene mutation in patients with non‐small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res 2006; 12: 5764–9. [DOI] [PubMed] [Google Scholar]
- 19. Engelman JA, Zejnullahu K, Mitsudomi T et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Sci (New York, NY) 2007; 316: 1039–43. [DOI] [PubMed] [Google Scholar]
- 20. Bean J, Brennan C, Shih JY et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA 2007; 104: 20932–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Govindan R. A review of epidermal growth factor receptor/HER2 inhibitors in the treatment of patients with non‐small‐cell lung cancer. Clin Lung Cancer 2010; 11: 8–12. [DOI] [PubMed] [Google Scholar]
- 22. Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib‐sensitizing EGFR mutations in lung cancer activate anti‐apoptotic pathways. Sci (New York, NY) 2004; 305: 1163–7. [DOI] [PubMed] [Google Scholar]
- 23. Ogino A, Kitao H, Hirano S et al. Emergence of epidermal growth factor receptor T790M mutation during chronic exposure to gefitinib in a non small cell lung cancer cell line. Cancer Res 2007; 67: 7807–14. [DOI] [PubMed] [Google Scholar]
- 24. Engelman JA, Mukohara T, Zejnullahu K et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR‐amplified lung cancer. J Clin Invest 2006; 116: 2695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yamasaki F, Johansen MJ, Zhang D et al. Acquired resistance to erlotinib in A‐431 epidermoid cancer cells requires down‐regulation of MMAC1/PTEN and up‐regulation of phosphorylated Akt. Cancer Res 2007; 67: 5779–88. [DOI] [PubMed] [Google Scholar]
- 26. Kawano O, Sasaki H, Endo K et al. PIK3CA mutation status in Japanese lung cancer patients. Lung cancer (Amsterdam, Netherlands). 2006; 54: 209–15. [DOI] [PubMed] [Google Scholar]
- 27. Yamamoto H, Shigematsu H, Nomura M et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res 2008; 68: 6913–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Berns K, Horlings HM, Hennessy BT et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007; 12: 395–402. [DOI] [PubMed] [Google Scholar]
- 29. Sartore‐Bianchi A, Martini M, Molinari F et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR‐targeted monoclonal antibodies. Cancer Res 2009; 69: 1851–7. [DOI] [PubMed] [Google Scholar]
- 30. Noro R, Gemma A, Miyanaga A et al. PTEN inactivation in lung cancer cells and the effect of its recovery on treatment with epidermal growth factor receptor tyrosine kinase inhibitors. Int J Oncol 2007; 31: 1157–63. [PubMed] [Google Scholar]
- 31. Kwak EL, Sordella R, Bell DW et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA 2005; 102: 7665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shepard HM, Brdlik CM, Schreiber H. Signal integration: a framework for understanding the efficacy of therapeutics targeting the human EGFR family. J Clin Invest 2008; 118: 3574–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G‐protein‐coupled receptors. Nature 1996; 379: 557–60. [DOI] [PubMed] [Google Scholar]
- 34. Ohtsu H, Dempsey PJ, Eguchi S. ADAMs as mediators of EGF receptor transactivation by G protein‐coupled receptors. Am J Physiol 2006; 291: C1–10. [DOI] [PubMed] [Google Scholar]
- 35. Rozengurt E. Mitogenic signaling pathways induced by G protein‐coupled receptors. J Cell Physiol 2007; 213: 589–602. [DOI] [PubMed] [Google Scholar]
- 36. Dziadziuszko R, Camidge DR, Hirsch FR. The insulin‐like growth factor pathway in lung cancer. J Thorac Oncol. 2008; 3: 815–8. [DOI] [PubMed] [Google Scholar]
- 37. Desbois‐Mouthon C, Baron A, Blivet‐Van Eggelpoel MJ et al. Insulin‐like growth factor‐1 receptor inhibition induces a resistance mechanism via the epidermal growth factor receptor/HER3/AKT signaling pathway: rational basis for cotargeting insulin‐like growth factor‐1 receptor and epidermal growth factor receptor in hepatocellular carcinoma. Clin Cancer Res 2009; 15: 5445–56. [DOI] [PubMed] [Google Scholar]
- 38. Sangha R, Lara PN Jr, Mack PC, Gandara DR. Beyond antiepidermal growth factor receptors and antiangiogenesis strategies for nonsmall cell lung cancer: exploring a new frontier. Curr Opin Oncol 2009; 21: 116–23. [DOI] [PubMed] [Google Scholar]
- 39. Sharma SV, Lee DY, Li B et al. A chromatin‐mediated reversible drug‐tolerant state in cancer cell subpopulations. Cell 2010; 141: 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Turke AB, Zejnullahu K, Wu YL et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010; 17: 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yano S, Wang W, Li Q et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor‐activating mutations. Cancer Res 2008; 68: 9479–87. [DOI] [PubMed] [Google Scholar]
- 42. McDermott U, Pusapati RV, Christensen JG, Gray NS, Settleman J Acquired resistance of non‐small cell lung cancer cells to MET kinase inhibition is mediated by a switch to epidermal growth factor receptor dependency. Cancer Res 2010; 70: 1625–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 2009; 27: 5459–68. [DOI] [PubMed] [Google Scholar]
- 44. Selvi RB, Kundu TK. Reversible acetylation of chromatin: implication in regulation of gene expression, disease and therapeutics. Biotechnol J 2009; 4: 375–90. [DOI] [PubMed] [Google Scholar]
- 45. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev 2006; 5: 769–84. [DOI] [PubMed] [Google Scholar]
- 46. Mai A, Massa S, Rotili D et al. Histone deacetylation in epigenetics: an attractive target for anticancer therapy. Med Res Rev 2005; 25: 261–309. [DOI] [PubMed] [Google Scholar]
- 47. Spange S, Wagner T, Heinzel T, Kramer OH. Acetylation of non‐histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol 2009; 41: 185–98. [DOI] [PubMed] [Google Scholar]
- 48. Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst 2000; 92: 1210–6. [DOI] [PubMed] [Google Scholar]
- 49. Edwards A, Li J, Atadja P, Bhalla K, Haura EB. Effect of the histone deacetylase inhibitor LBH589 against epidermal growth factor receptor‐dependent human lung cancer cells. Mol Cancer Ther 2007; 6: 2515–24. [DOI] [PubMed] [Google Scholar]
- 50. Ding L, Getz G, Wheeler DA et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008; 455: 1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Horn L, Pao W. EML4‐ALK: honing in on a new target in non‐small‐cell lung cancer. J Clin Oncol 2009; 27: 4232–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Antonescu CR, Besmer P, Guo T et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res 2005; 11: 4182–90. [DOI] [PubMed] [Google Scholar]
- 53. Gorre ME, Mohammed M, Ellwood K et al. Clinical resistance to STI‐571 cancer therapy caused by BCR‐ABL gene mutation or amplification. Sci (New York, NY) 2001; 293: 876–80. [DOI] [PubMed] [Google Scholar]
- 54. Kobayashi S, Ji H, Yuza Y et al. An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res 2005; 65: 7096–101. [DOI] [PubMed] [Google Scholar]
- 55. Li D, Shimamura T, Ji H et al. Bronchial and peripheral murine lung carcinomas induced by T790M‐L858R mutant EGFR respond to HKI‐272 and rapamycin combination therapy. Cancer Cell 2007; 12: 81–93. [DOI] [PubMed] [Google Scholar]
- 56. Ji H, Li D, Chen L et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR‐targeted therapies. Cancer Cell 2006; 9: 485–95. [DOI] [PubMed] [Google Scholar]
- 57. Engelman JA, Zejnullahu K, Gale CM et al. PF00299804, an irreversible pan‐ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res 2007; 67: 11924–32. [DOI] [PubMed] [Google Scholar]
- 58. Zhou W, Ercan D, Chen L et al. Novel mutant‐selective EGFR kinase inhibitors against EGFR T790M. Nature 2009; 462: 1070–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shimamura T, Lowell AM, Engelman JA, Shapiro GI. Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res 2005; 65: 6401–8. [DOI] [PubMed] [Google Scholar]
- 60. Johansson M, Denardo DG, Coussens LM. Polarized immune responses differentially regulate cancer development. Immunol Rev 2008; 222: 145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol 2006; 1: 119–50. [DOI] [PubMed] [Google Scholar]
- 62. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev 2006; 6: 392–401. [DOI] [PubMed] [Google Scholar]
- 63. Xu R, Boudreau A, Bissell MJ. Tissue architecture and function: dynamic reciprocity via extra‐ and intra‐cellular matrices. Cancer Metastasis Rev 2009; 28: 167–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tabernero J. The role of VEGF and EGFR inhibition: implications for combining anti‐VEGF and anti‐EGFR agents. Mol Cancer Res 2007; 5: 203–20. [DOI] [PubMed] [Google Scholar]
- 65. Trimboli AJ, Cantemir‐Stone CZ, Li F et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 2009; 461: 1084–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Katajisto P, Vaahtomeri K, Ekman N et al. LKB1 signaling in mesenchymal cells required for suppression of gastrointestinal polyposis. Nat Genet 2008; 40: 455–9. [DOI] [PubMed] [Google Scholar]
- 67. Wu M, Pastor‐Pareja JC, Xu T. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature 2010; 463: 545–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet 2002; 32: 355–7. [DOI] [PubMed] [Google Scholar]
- 69. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 70. Kitano H. Computational systems biology. Nature 2002; 420: 206–10. [DOI] [PubMed] [Google Scholar]
- 71. Kitano H. Cancer as a robust system: implications for anticancer therapy. Nat Rev 2004; 4: 227–35. [DOI] [PubMed] [Google Scholar]