

Abstract
Fibrin clots in non‐malignant conditions form only at the onset or during the active stage of disease and disappear within a few weeks as a result of plasmin digestion or replacement with collagen. In contrast, fibrin clot formation and subsequent replacement with collagen in cancer persist for as long as the cancer cells survive in the body. We developed an anti‐fibrin chimeric antibody that reacts with fibrin only, and not fibrinogen (the precursor of fibrin), and then attached an anticancer agent (ACA) to the antibody. Thus, the immunoconjugate did not create an immune complex in the blood stream and was selectively accumulated to fibrin clots in the tumor stroma to create a scaffold, from which effective sustained release of the ACA occurred. In a mouse model, the ACA diffused throughout the tumor tissue to damage both tumor cells and vessels, resulting in potent antitumor activity in stroma‐rich spontaneous tumors. This new cancer stroma‐targeting therapy may result in an increased duration of drug exposure and be a highly effective new therapy, particularly for refractory, stroma‐rich cancers. (Cancer Sci 2011; 102: 1396–1402)
Low molecular weight (LMW) anticancer agents (ACA), including molecular targeting agents, are very efficient cytocidal agents in the closed space of a monolayer culture dish. The antitumor effects of these agents are determined using subcutaneous tumor xenografts, the pathophysiological features of which are far removed from those of general human cancers. Although there have been numerous reports of genetic and phenotype changes in tumors, there are no pivotal changes in tumor cells that distinguish them from normal dividing cells.( 1 , 2 , 3 , 4 , 5 , 6 ) Unlike the situation in culture, following the administration of LMW ACA to patients, these agents are cleared quickly from tumors in the body. In addition, the ACA are distributed throughout the body, resulting in serious side effects.( 7 ) To overcome off‐target effects caused by LMW ACA, immunoconjugate therapy was developed in which an ACA or toxin is conjugated to a cancer cell‐specific mAb, which is too large to pass through a normal vessel wall but can extravasate from leaky tumor vessels and accumulate selectively in tumor tissue.( 8 , 9 , 10 , 11 , 12 ) The kinetics of drug distribution within tumors are considered to be a function of interstitial conductivity, which is determined by the quantity and density of the extracellular matrix (e.g. proteoglycan, fibronectin) and fibrosis (e.g. fibrin, collagen fibers) in the stroma.( 13 , 14 , 15 , 16 ) Most human solid tumors have abundant stroma that hinders the distribution of high molecular weight (HMW) agents, including ACA‐conjugated antibodies. Consequently, the tissue becomes a barrier preventing the immunoconjugates from attacking cancer cells.( 14 , 15 , 16 ) This led us to design a novel alternative antitumor strategy that turned this apparent handicap into an asset.
In the 19th century, French surgeon Armand Trousseau described, for the first time, thrombophlebitis in patients with stomach cancer.( 17 ) Today, a large body of clinical evidence supports the conclusion that abnormal coagulation followed by fibrin formation occurs in a variety of cancers.( 18 , 19 ) Different types of tumor cells express the tissue factor that is known to be a cell surface membrane protein and a trigger of the extrinsic coagulation pathway.( 18 , 20 ) Above all, any malignant tumor erodes adjacent normal or tumor vessels, resulting in microscopic hemorrhages within or adjacent to cancer tissues; fibrin clots should form immediately in situ to stop the bleeding. Although fibrin clot formation also occurs in non‐malignant disorders, such as cardiac or brain infarction, injuries, and active rheumatoid arthritis, these fibrin clots form only at the onset or during the active stage of the disease. Moreover, these fibrin clots disappear within a few weeks as a result of plasmin digestion or replacement with collagen fibers. In tumor tissues, the fibrin clots are replaced by collagenous stroma in a process similar to that in normal wound healing and other non‐malignant diseases.( 13 ) However, unlike non‐malignant conditions, fibrin clot formation in cancer tissues lasts for as long as the cancer cells survive in the body. Therefore, unlike growth factors and tyrosine kinases, fibrin clots are pathophysiologically specific for tumors. In that context, we developed an mAb against fibrin to target the tumor stroma. In addition, we exploited the newly developed specialized immunoconjugate linker to conjugate the anti‐fibrin mAb with an ACA.
Materials and Methods
Antibodies. A hybridoma producing anti‐fibrinogen (mouse IgG clone K88‐3) or anti‐fibrin antibody (mouse IgM clone 102‐10) was established using myeloma cells (P3U1) and lymph node cells from the mouse with immunizing human fibrinogen (Sigma, St. Louis, MO, USA) or fibrin, with the latter converted from fibrinogen by thrombin (Sigma) cleavage. Heavy chain variable and kappa light chain variable cDNAs were cloned into the vector for human IgG1 expression. The vectors were transfected into Chinese hamster ovary (CHO) cells (Riken Bioresource Center, Tsukuba, Japan) and a stable clone (humanized IgG, clone 102‐10) was isolated.
Camptothecin‐11 (CPT‐11; irinotecan) and SN‐38 were purchased from Tokyo Chemical Industry (Tokyo, Japan) and Yakult (Tokyo, Japan), respectively.
In vivo imaging and immunohistochemistry. Antibodies were conjugated with Alexa‐647 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. In vivo fluorescence imaging was performed using a confocal fluorescence microscope (A1R; Nikon, Tokyo, Japan) or a small animal imaging system (OV110; Olympus, Tokyo, Japan). For immunohistochemistry, anti‐fibrin IgM was incubated with Alexa 488‐labeled anti‐mouse IgG (Invitrogen) as a secondary antibody. The unbound antibodies were blocked with mouse serum (Dako, Glostrup, Denmark). First, samples were incubated for 30 minutes at room temperature in the dark with the immunocomplex (anti‐fibrin IgM and Alexa 488 secondary Ab), anti‐cytokeratin (Abcam, Cambridge, UK), or anti‐CD31 (Becton Dickinson, Franklin Lakes, NJ, USA). Then, samples were incubated for 60 minutes at room temperature in the dark with Alexa 488‐, 555‐ or 647‐labeled anti‐rabbit IgG (Invitrogen) or phycoerythrin (PE)‐labeled anti‐rat IgG (Jackson ImmunoResearch, West Grove, PA, USA). Fluorescence images were obtained using a digital high‐definition microscopic system (BZ‐9000; Keyence, Osaka, Japan).
Synthesis of the SN‐38 derivative linker. A detailed description of the synthesis of the linker is provided online as supplementary material for this paper (Data S1). Briefly, PEG was purchased from Quanta BioDesign (Powell, OH, USA) and Merck (Whitehouse Station, NJ, USA). Reagents and solvents were purchased from Sigma and Kanto (Tokyo, Japan). The final structure was composed of one maleimide for the attachment of mAb, one PEG12 spacer and three PEG27 ester bonds for attachment of three SN‐38 molecules. The structure and purity of the chemically synthesized materials were determined by 400‐MHz 1H‐NMR and 13C‐NMR (NMR ECX‐400 or NMR AL; JEOL Ltd., Tokyo, Japan) and mass spectrometry (MS AXIMA; Shimadzu, Kyoto, Japan). The derivatives were resolved in DMSO (Sigma).
Immunoconjugate. The interchain disulfides of the Abs were first reduced with 10 mM DTT (Sigma). The number of free thiols was quantified using 5,5′‐dithiobis 2‐nitrobenzoic acid (DTNB; Wako Pure Chemical Industries, Osaka, Japan). Reduced Abs were purified by gel filtration (Amicon Ultra Centrifugal Filter Devices; Millipore, Billerica, MA, USA) and reacted with linker SN‐38 derivative in PBS containing 5 mM EDTA (pH 6) at room temperature for 1 h, then at 4°C overnight. The SN‐38‐conjugated Abs were purified by gel filtration (Millipore). The concentration of Ab–prodrug conjugates was determined using the Bradford method (Bio‐Rad Protein Assay, 500‐0006JA; Bio‐Rad, Hercules, CA, USA). The number of residual thiols was quantified with DTNB. The ratio of each drug (SN‐38)/Ab was determined by comparison of the number of free and residual thiols.
High‐performance liquid chromatography. The kinetics of release of SN‐38 from the immunoconjugate was investigated in vitro at 37°C in 5% glucose (pH 4.6), PBS (pH 7.4), or mouse serum. Whole tumor tissues were mixed with 0.1 M glycine–HCl buffer (pH 3.0)/methanol (5 w/w%) and then homogenized. Samples (100 μL) were then mixed with 20 μL of 1 mM phosphoric acid/methanol (1:1), 40 μL ultrapure water, and 60 μL camptothecin solution (Sigma) as an internal standard. Reaction solutions and plasma were mixed with 0.1 M HCl at 50% (w/w). The samples (50 μL) were then mixed with 20 μL of 1 mM phosphoric acid in methanol (1:1) and 100 μL camptothecin as an internal standard. Samples were vortexed for 10 s and filtered through an Ultrafree‐MC (Millipore). To detect immunoconjugated SN‐38, samples (20 μL plasma, 100 μL tumor) were diluted with 20 μL methanol (50% w/w) and 20 μL NaOH (0.7 M). After incubation for 15 minutes at room temperature, 20 μL HCl (0.7 M) and 60 μL internal standard solution were added to the samples, and the hydrolysate was then filtered. Reverse‐phase HPLC was performed at 35°C on a Mightysil RP‐18 GP column (150 × 4.6 mm; Kanto). Samples were injected into an Alliance Waters 2795 HPLC system (Waters, Milford, MA, USA) equipped with a Waters 2475 multi λ fluorescence detector at excitation and emission wavelengths of 365 and 540 nm, respectively, for SN‐38 or 365 and 430 nm, respectively, for CPT‐11.
Animal model and antitumor effects. Chemical skin carcinogenesis was induced in female FVB/N mice (CLEA, Tokyo, Japan) as described previously.( 21 , 22 ) Briefly, a single application of 7,12‐dimethylbenz[α]anthracene (DMBA; 250 μg/mL in acetone; Sigma) was applied to the shaved dorsal skin of mice. After 1 week, phorbol 12‐myristate 13‐acetate (PMA); 25 μg/mL in acetone; Sigma) was applied weekly (for a total of 32 times). Carcinoma was determined on the basis of the clinical appearance of characteristic features, according to the previous report.( 22 ) Histological analysis was performed to confirm the diagnosis of carcinoma based on clinical appearance (Fig. 1A). Tumor volume was calculated as (length × width2)/2. When the mean size of tumors that grew continuously over a period of 3 weeks reached approximately 70 mm3, the tumors were randomly divided into three groups comprising five tumors in each group such that there was no significant difference in tumor size among them. Immunoconjugates were administered on Days 0, 7, 14, and 21 by tail vein injection. An injection dose of antibody–SN‐38 prodrug equal to an SN‐38 dose of 13.3 mg/kg was determined by calculations based on the drug (SN‐38)/antibody ratio for each drug. Statistical analyses of both antitumor effects and changes in body weight were performed using anova.
Figure 1.

Preparation and characterization of the anti‐fibrin antibody. (A) Chemical skin carcinogenesis. A mouse bearing a tumor (left) and hematoxylin‐eosin staining (right) of the tumor. (B) Both anti‐fibrin (Fib) IgM and its chimeric IgG (AFCA) were shown to recognize human (Hu) and mouse (Ms) fibrin, but not their fibrinogen (Fng), by ELISA. OD, optimal density. (C) In vivo confocal microscope imaging showing leakage of Alexa 488‐labeled anti‐fibrin IgM (left; green) and Alexa 647‐labeled AFCA (middle; red) from tumor vessels 1 h after injection. Arrows indicate the area into which the AFCA has leaked (right; red in Merge). Scale bar, 100 μm. (D) In vivo systemic imaging analysis of Alexa 647‐labeled anti‐fibrin IgM (upper), AFCA (middle), or anti‐fibrinogen mAb (lower) on Days 1, 3, and 7 after injection. Arrows indicate each tumor position. (E) Intratumoral distribution of injected fluorescent AFCA (lower left; red) was examined 24 h after injection. Immunohistochemistry with anti‐cytokeratin (upper left; light‐blue) or anti‐fibrin (upper right; green). Yellow indicates the overlap of injected anti‐fibrin IgG and deposited fibrin (lower right). Scale bar, 100 μm.
All animal procedures were performed in compliance with the Guidelines for the Care and Use of Experimental Animals established by the Committee for Animal Experimentation of the National Cancer Center. These guidelines meet the ethical standards required by law and also comply with the guidelines for the use of experimental animals in Japan (http://www.scj.go.jp/ja/info/kohyo/pdf/kohyo‐20‐k16‐2.pdf).
In vivo fiber confocal fluorescence microscopy. For visualization of tumor vessels, 400 μg FITC–dextran (250 kDa; Sigma) was injected into tumor‐bearing mice before and 5 days after treatment with the immunoconjugate using the imaging system Cellvizio (Mauna Kea Technologies, Paris, France).( 23 ) Mean vessel diameter, total vessel length, and total area were estimated at six different sites within the tumor before and after treatment using ImageCell software (Mauna Kea Technologies). The length of functional capillary density (FCD) was calculated as (total vessel length/total area). Statistical analyses were performed using Student’s t‐test.
Results
Preparation of anti‐fibrin chimeric IgG for stroma targeting. We developed an mAb against fibrin, which is abundant in the stroma of human solid tumors. After extensive screening using two ELISA sets, one for human fibrinogen (the precursor of fibrin that is present physiologically) and the other for human fibrin (which is formed only in some abnormal conditions), we successfully developed an mAb that reacted with human fibrin only and not with human fibrinogen. However, because the mAb obtained was IgM, it was converted into the human IgG format for clinical application using an antibody engineering technique. Another advantage of the mAb was that the mAb cross‐reacted with mouse fibrin and not with mouse fibrinogen (Fig. 1B). Chemically induced mouse cutaneous cancer was selected as an appropriate experimental model in which to evaluate the therapeutic effects of our immunoconjugate chemotherapy because this spontaneous carcinogenetic model has marked fibrin deposition and abundant interstitial tissue, as in human cancer (Fig. 1A) and unlike human tumor xenografts, which have fewer fibrin clots and less interstitial tissue.( 24 , 25 ) In addition, the spontaneous tumors exhibit very slow growth, which is similar to the condition in general human cancer but not in the xenografts.
Using real‐time in vivo confocal microscope imaging, the anti‐fibrin chimeric IgG (AFCA) was found to be distributed in the extravascular component 1 h after injection, whereas the leakage of anti‐fibrin IgM from the vessels was so restricted that obvious extravascular distribution was not observed over the same period (Fig. 1C). Using systemic in vivo imaging, anti‐fibrin IgM, anti‐fibrin IgG, and anti‐fibrinogen IgG were delivered and retained in the tumor until Day 3, utilizing leaky tumor vessels.( 10 , 11 ) The accumulation of anti‐fibrin IgM and anti‐fibrinogen IgG was weak and they were eliminated by Day 7; however, on Day 7, AFCA was still highly retained within the tumor (Fig. 1D). Therefore, we used AFCA as a vehicle for drug delivery, providing high accumulation and long‐term retention in the tumor. Investigating the intratumoral distribution of AFCA, we found that it was observed mainly in fibrin‐positive stroma and was rarely seen in cytokeratin‐positive tumor cell areas (Fig. 1E). Thus, we succeeded in developing an anti‐fibrin IgG for targeting the tumor stroma.
Drug design, anti‐tumor activity, and pharmacokinetic study of the anti‐fibrin immunoconjugate. Another special feature of our design is the conjugation of highly cytotoxic SN‐38 with each mAb, using a specially produced linker composed mainly of PEG, which provided both increased payload capacity and efficient, sustained drug release within the tumor. The branched composition had one maleimide for the attachment of the mAb, one PEG12 spacer, and three PEG27 ester bonds for the attachment of three SN‐38 molecules (Fig. 2A). There were approximately eight thiol residues able to react with the maleimide in the reduced mAb. The calculated drug (SN‐38)/mAb ratio of the immunoconjugate was approximately 24. Previous studies have reported drug/mAb ratios in the range 3–8.( 26 , 27 , 28 ) Therefore, the ratio of 24 obtained in the present study is so far the highest, which means that the drug can be carried with the minimum amount of antibody, resulting in a reduction of both undesirable drug effects in the body and production costs.
Figure 2.
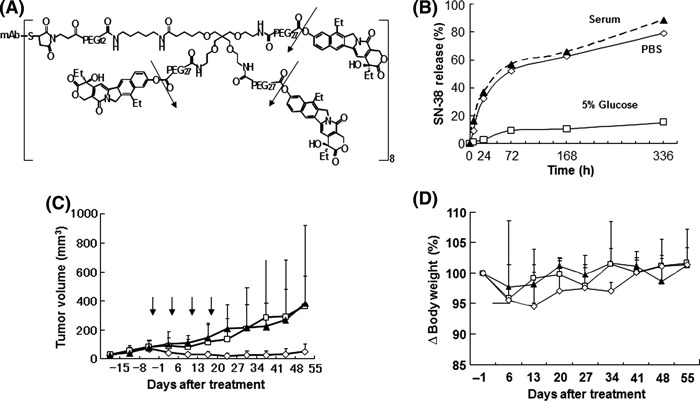
Drug design, anti‐tumor effect and pharmacokinetics of the anti‐fibrin immunoconjugate. (A) Drug design of the immunoconjugate: mAb–PEG–three‐branched PEG–(SN‐38)3 linked via an ester bond. One antibody bears 24 molecules of SN‐38. The arrows indicate the cleavage sites to release free active SN‐38. (B) Release of SN‐38 from immunoconjugates over time in mouse serum, PBS (pH 7.4), or 5% glucose at 37°C. Data show single values determined at each time point. (C) Antitumor activity was examined in vivo. Immunoconjugates (⋄), camptothecin‐11 (CPT‐11; □), or saline (), were administered to mice bearing chemical‐induced cutaneous tumors via intravenous injection on Days 0, 7, 14, and 21. Arrows indicate the day of administration and the curves illustrate the effect of treatment on tumor size. Data are the mean ± SD (n = 5 in each group). P = 0.0005 for CPT‐11 compared with immunoconjugate; P < 0.0001 for saline compared with immunoconjugate. (D) Changes in the relative body weight of mice injected with Immunoconjugates (⋄), CPT‐11 (□), or saline () on Days 0, 7, 14, and 21. Data are the mean ± SD. There were no significant differences between the three groups (P = 0.09 for CPT‐11 versus immunoconjugate; P = 0.0866 for saline versus immunoconjugate).
The conjugated SN‐38 had no cytotoxic effect, which means that the cytotoxic immunoconjugate itself is a prodrug. Consequently, we succeeded in producing a cytotoxic immunoconjugate, namely AFCA‐branched‐PEG‐(SN‐38)3, hereafter abbreviated to AFCA‐B‐P‐(SN‐38)3 (Fig. 2A). The immunoconjugate, via the ester bond, was stable in 5% glucose (pH 4.6) because the phenyl ester bond is stable under acidic conditions and labile under mild alkaline conditions.( 29 ) Under physiological conditions (PBS pH 7.4 and serum) such as in the extracellular environment, the immunoconjugate, via the ester bond, can release SN‐38 enzyme independently, gradually, and effectively (Fig. 2B). This bond has already been introduced into clinical use, such as in NK012.( 30 ) The antitumor activity of AFCA‐B‐P‐(SN‐38)3 was evaluated in vivo and, following its administration four times weekly at an equivalent SN‐38 dose of 13.3 mg/kg per day, showed significant antitumor activity compared with findings in mice treated with either saline or CPT‐11 (40 mg/kg per day at the maximum tolerated dose [MTD], equivalent to a dose of 23.2 mg/kg per day SN‐38). Although tumors continued to increase in mice treated with CPT‐11, the growth of tumors in mice treated with AFCA‐B‐P‐(SN‐38)3 was significantly suppressed for more than 1 month (Fig. 2C). Thus, AFCA‐B‐P‐(SN‐38)3 exerted strong antitumor activity compared with CPT‐11 (Fig. 2C). Although treatment‐related body weight loss was observed in mice treated with each of the drugs, there was no significant difference in body weight loss between the control group and the CPT‐11‐ and AFCA‐B‐P‐(SN‐38)3‐treated groups (Fig. 2D). Blood tests revealed no significant bone marrow toxicity or liver and kidney dysfunction in any of the treatment groups (data not shown).
Release of SN‐38 from the anti‐fibrin immunoconjugate induces damage to both tumor cells and tumor vessels. After injection of AFCA‐B‐P‐(SN‐38)3, the concentration of total SN‐38 (antibody‐bound and unbound forms) and free SN‐38 (unbound form) in plasma declined gradually within a week, whereas CPT‐11 exhibited rapid clearance (Fig. 3A). We then examined the intratumoral distribution of SN‐38 released from the immunoconjugate using HPLC. Significantly high concentrations of total and free SN‐38 were detected in tumor tissues treated with the immunoconjugate for 168 hours compared with CPT‐11 (Fig. 3B). The second significant observation following treatment with AFCA‐B‐P‐(SN‐38)3 was a change in gross tumor color from reddish to white (Fig. 3C). To clarify the cause for this change in color, the histopathological features of the tumor stroma after immunoconjugate therapy were examined. There was no clear change in fibroblasts or macrophages, which play an important role in tumor progression( 31 , 32 ) (data not shown). We then evaluated stromal vascular changes both qualitatively and quantitatively using immunohistochemistry and in vivo fluorescence endomicroscopy. Using these techniques, discontinuation and irregularity comprising a mixture of narrowness and enlargement of tumor vessels were manifested following treatment with AFCA‐B‐P‐(SN‐38)3 (Fig. 3D,E).
Figure 3.
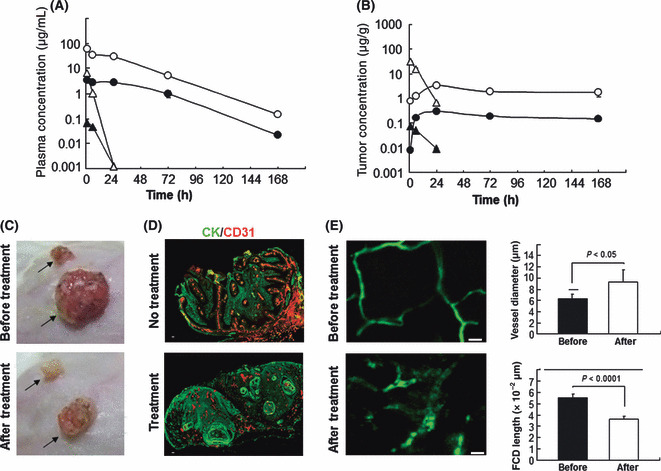
Drug distribution and antivascular activity of the immunoconjugate. (A,B) Plasma (A) and tumor (B) concentrations of total SN‐38 (bound and unbound form; ○), camptothecin‐11 (CPT‐11; △), and free SN‐38 (unbound form) released from the immunoconjugate (•) or converted from CPT‐11 () were determined by HPLC 1, 6, 24, 72, and 168 h after injection. (C) The color of the tumor changed from reddish to white 5 days after injection of the immunoconjugate, but not CPT‐11. The arrows indicate the position of each tumor. (D) Tumor vessels after injection of the immunoconjugate (Treatment) were examined using CD31 (red) and cytokeratin (CK; green). Untreated mice (No treatment) were used as a control. Scale bar, 100 μm. (E) The left‐hand figures show the change of tumor vessels visualized with FITC‐dextran before and after the injection of the immunoconjugate, visualized using FITC–dextran by in vivo fiber confocal fluorescence microscopy. Quantified vessel diameter and functional capillary density (FCD) length are shown on the right. Scale bars, 20 μm.
Discussion
It is known that LMW ACA, including molecular targeting agents, can easily extravasate from normal blood vessels, resulting in various adverse effects (Fig. 4A). To overcome the off‐target effects caused by LMW ACA, an immunoconjugate therapy was developed in which ACA or a toxin is conjugated to a cancer cell‐specific mAb (Fig. 4B).
Figure 4.
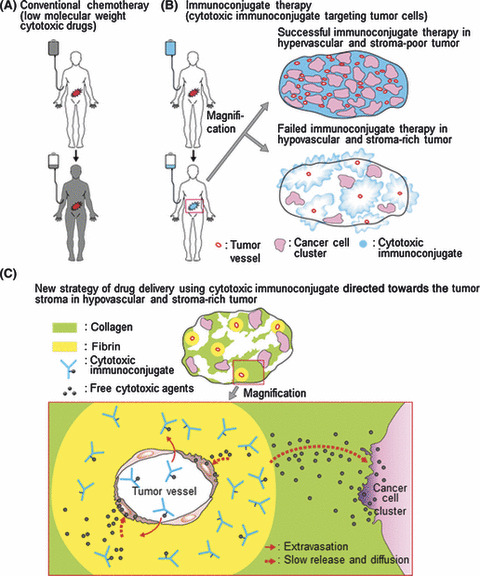
Diagram of the background and the new concept of drug delivery using tumor stroma as a ligand. (A) Low molecular weight anticancer agents (black) can be distributed throughout the entire body, resulting in serious side effects. (B) Cytotoxic immunoconjugates (blue) accumulate selectively in the tumor tissue. Successful (upper) and failed (lower) immunoconjugate therapy is shown. (C) The newly developed immunoconjugate (i.e. anti‐fibrin chimeric IgG–branched‐PEG‐(SN‐38)3) extravasates selectively from leaky tumor vessels, binds specifically to the fibrin network around the tumor vessels to create a scaffold, and then allows the effective, sustained release of SN‐38, an anticancer agent with time‐dependent effects, from the scaffold. Because this released anticancer agent is of a low molecular weight, it is subsequently distributed throughout the entire tumor stroma, which normally acts as a barrier, and induces damage not only to tumor cells, but also to tumor vessels.
The immunoconjugates selectively extravagate from leaky tumor vessels and this is an advantage in cancer treatment. However, to date, immunoconjugate therapies for common solid tumors have not been successful in clinical practice because of the heterogeneity of the target antigens.( 1 , 2 , 3 , 4 , 5 , 6 , 8 , 9 ) Moreover, most human tumors have abundant stroma that hinders the distribution of the immunoconjugate. Our basic strategy for overcoming the stromal barrier as a protective shield for cancer cells is to make use of the fibrin in the stroma as a scaffold assembly of the immunoconjugates, followed by the release of SN‐38 to the cancer cells. This free SN‐38 can easily reach the cancer cells by diffusion through the stromal barrier. Another important finding of the present study is that SN‐38 released from the immunoconjugates and located in the fibrin networks around the tumor vasculature may attack vascular endothelial cells.
During the process of blood coagulation, extrinsically activated thrombin cleaves fibrinopeptides A and B from the α‐ and β‐chains of fibrinogens, generating soluble fibrin monomers. Thereafter, an insoluble fibrin clot forms following enzymatic and non‐enzymatic processing.( 33 ) Therefore, it is speculated that the epitope of our anti‐fibrin mAb is adjacent to the thrombin cutting site of the fibrinogens.
Another feature of our anti‐fibrin mAb is the conversion of human chimeric IgG from mouse IgM using an antibody engineering technique. The use of human chimeras is beneficial for clinical applications to avoid human anti‐mouse neutralizing antibodies and allergic reactions in humans. In addition, because of the rapid blood clearance and low penetration of IgM compared with IgG, based on the faster elimination of IgM from the liver and its larger molecular size,( 34 ) IgM is not suitable as a drug delivery vehicle.
The anti‐fibrin mAb was conjugated with SN‐38 using newly designed linker assembly. SN‐38 is a topoisomerase I inhibitor, with time‐dependent antitumor activity, and is an active component of CPT‐11, which is used clinically in the treatment of colorectal, lung, and other cancers.( 35 ) Linker technology is an important part of immunoconjugate chemotherapy, and various linkers have been exploited to date. Of these, acid labile hydrazone linkage, thiol reduction of disulfide linkers, and enzymatic proteolysis of peptide linkers have been used successfully to ensure stability in plasma.( 26 , 27 , 28 ) For these types of linkers, cell‐mediated endocytosis and intracellular processing of the immunoconjugates are indispensable to make the active agent work. In our newly deployed linker construct, an ester bond can release SN‐38 gradually, independent of enzymes, under physiological conditions such as in the extracellular environment. In our design, PEG was combined close to this ester bond. It is known that PEG evades non‐specific capture by the reticuloendothelial system (RES) in the body, with the steric structure around the bond protecting against immunoconjugate degradation in the blood. Furthermore, PEG has already been used for this purpose.( 11 , 12 ) Moreover, the unique branched composition enables the attachment of three SN‐38 molecules, rather than only one as in standard linear types of linkers.
As mentioned above, asymptomatic fibrin formation occurs only during cancer invasion and metastasis. Patients with advanced cancer are candidates for treatment with systemic ACA and almost all such patients are fibrin free, except for in the cancer tissue in the body. In addition, HMW proteins, including IgG, cannot extravasate from normal blood vessels to cause unwanted side effects in non‐neoplastic organs (Fig. 4C). In spontaneous mouse tumors characterized by abundant stroma, this cancer stroma‐targeting therapy using tumor‐induced fibrin clots has succeeded, for the first time, in producing conditions that achieve drug exposure levels specifically in tumor cells that are similar to those in monolayer culture dishes and it is thus a highly effective new strategy for treating solid tumors, especially stroma‐rich cancers, which are refractory to conventional therapy. In the 1960s, and 131I‐Ab that targeted fibrin was suggested as a potential cancer therapy.( 36 ) However, that Ab was polyclonal and reacted with fibrinogen, which is the physiological precursor of fibrin and abundant in the blood stream. The polyclonal Ab against fibrin bound to fibrinogen in the blood stream and easily changed to a circulating immune complex. This immune complex was eliminated more rapidly from the liver than intact IgG, resulting in rapid blood clearance.( 34 , 37 ) In fact, the present study demonstrated that the accumulation of the anti‐fibrinogen mAb was weaker and that the anti‐fibrinogen mAb was eliminated more rapidly than the anti‐fibrin chimeric mAb.
This linker technology can be applied to many other ACA, including molecular targeting agents. Thus, this present discovery, the development of which was based on cancer pathophysiology and organic chemistry, may change current therapy with ACA and open a new fields of medical science, consequently producing many useful treatment modalities in the field of oncology, cardiovascular disease, and inflammation.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting information
Data S1. Detailed information regarding the synthesis of branched linker, branched PEG‐linked SN‐38, and NMR spectra for each compound.
Supporting info item
Acknowledgments
This work was supported by the Third Term Comprehensive Control Research for Cancer from the Ministry of Health, Labour and Welfare of Japan (YM), a Grant‐in‐Aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology, the Princess Takamatsu Cancer Research Fund (YM), the Japanese Foundation for Multidisciplinary Treatment of Cancer (YM), and a Grant‐in‐Aid for Scientific Research from Japan Society for the Promotion of Science (MY). The authors thank Drs. D Tarin (Department of Pathology, University of California, La Jolla, San Deigo, CA, USA), J Kuroda (Investigative Treatment Division, Research Center for Innovative Oncology, National Cancer Center Hospital East, Kashiwa, Japan), and T Sugino (Department of Basic Pathology, Fukushima Medical University, Fukushima, Japan) for their helpful discussions. The authors also thank Mrs. H Koike and Mrs. M Mizoguchi‐Araake for their technical assistance.
References
- 1. Heng HH, Bremer SW, Stevens JB et al. Genetic and epigenetic heterogeneity in cancer: a genome‐centric perspective. J Cell Physiol 2009; 220: 538–47. [DOI] [PubMed] [Google Scholar]
- 2. Hayden E. Cancer complexity slows quest for cure. Nature 2006; 455: 148. [DOI] [PubMed] [Google Scholar]
- 3. Grizzi F, DiIeva A, Russo C et al. Cancer initiation and progression: an unsimplifiable complexity. Theor Biol Med Model 2006; 3: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koenders PG, Peters WH, Wobbes T et al. Epidermal growth factor receptor levels are lower in carcinomatous than in normal colorectal tissue. Br J Cancer 1992; 65: 189–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Messersmith W, Oppenheimer D, Peralba J et al. Assessment of epidermal growth factor receptor (EGFR) signaling in paired colorectal cancer and normal colon tissue samples using computer‐aided immunohistochemical analysis. Cancer Biol Ther 2005; 4: 1381–6. [DOI] [PubMed] [Google Scholar]
- 6. Keshava Prasad TS, Goel R, Kandasamy K et al. Human protein reference database: 2009 update. Nucleic Acids Res 2009; 37: D767–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berthiaume JM, Wallace KB. Adriamycin‐induced oxidative mitochondrial cardiotoxicity. Cell Biol Toxicol 2007; 23: 15–25. [DOI] [PubMed] [Google Scholar]
- 8. Imai K, Takaoka A. Comparing antibody and small‐molecule therapies for cancer. Nat Rev Cancer 2006; 6: 714–27. [DOI] [PubMed] [Google Scholar]
- 9. Ricart AD, Tolcher AW. Technology insight: cytotoxic drug immunoconjugates for cancer therapy. Nat Clin Pract Oncol 2007; 4: 245–55. [DOI] [PubMed] [Google Scholar]
- 10. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 1986; 46: 6387–92. [PubMed] [Google Scholar]
- 11. Matsumura Y. Poly (amino acid) micelle nanocarriers in preclinical and clinical studies. Adv Drug Deliv Rev 2008; 22: 899–914. [DOI] [PubMed] [Google Scholar]
- 12. Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer 2006; 6: 688–701. [DOI] [PubMed] [Google Scholar]
- 13. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 1986; 315: 1650–9. [DOI] [PubMed] [Google Scholar]
- 14. Ghajar CM, Bissell MJ. Extracellular matrix control of mammary gland morphogenesis and tumorigenesis: insights from imaging. Histochem Cell Biol 2008; 130: 1105–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Minchinton AI, Tannock IF. Drug penetration in solid tumors. Nat Rev Cancer 2006; 6: 583–92. [DOI] [PubMed] [Google Scholar]
- 16. Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst 2007; 99: 1441–54. [DOI] [PubMed] [Google Scholar]
- 17. Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007; 110: 1723–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shoji M, Hancock WW, Abe K, Micko C. Activation of coagulation and angiogenesis in cancer: immunohistochemical localization in situ of clotting proteins and vascular endothelial growth factor in human cancer. Am J Pathol 1998; 152: 399–411. [PMC free article] [PubMed] [Google Scholar]
- 19. Stein PD, Beemath A, Meyers FA, Skaf E. Incidence of venous thromboembolism in patients hospitalized with cancer. Am J Med 2006; 119: 60–8. [DOI] [PubMed] [Google Scholar]
- 20. Belting M, Ahamed J, Ruf W. Signaling of the tissue factor coagulation pathway in angiogenesis and cancer. Arterioscler Thromb Vasc Biol 2005; 25: 1545–50. [DOI] [PubMed] [Google Scholar]
- 21. Hirakawa S, Kodama S, Kunstfeld R et al. VEGF‐A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J Exp Med 2005; 201: 1089–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Filler RB, Roberts SJ, Girardi M. Cutaneous two‐stage chemical carcinogenesis. CSH Protoc 2007; 2007: doi:10.1101/pdb.prot4837. [DOI] [PubMed] [Google Scholar]
- 23. Lin KY, Maricevich M, Bardeesy N et al. In vivo quantitative microvasculature phenotype imaging of healthy and malignant tissues using a fiber‐optic confocal laser microprobe. Transl Oncol 2008; 1: 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ellis LM, Fidler IJ. Finding the tumor copycat. Therapy fails, patients don’t. Nat Med 2010; 16: 974–5. [DOI] [PubMed] [Google Scholar]
- 25. Hawighorst T, Velasco P, Streit M et al. Thrombospondin‐2 plays a protective role in multistep carcinogenesis: a novel host anti‐tumor defense mechanism. EMBO J 2001; 20: 2631–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Doronina SO, Toki BE, Torgov MY et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol 2003; 21: 778–84. [DOI] [PubMed] [Google Scholar]
- 27. Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol 2005; 23: 1137–46. [DOI] [PubMed] [Google Scholar]
- 28. Lewis Phillips GD, Li G, Dugger DL et al. Targeting HER2‐positive breast cancer with trastuzumab‐DM1, an antibody‐cytotoxic drug conjugate. Cancer Res 2008; 68: 9280–90. [DOI] [PubMed] [Google Scholar]
- 29. Koizumi F, Kitagawa M, Negishi T et al. Novel SN‐38‐incorporating polymeric micelles, NK012, eradicate vascular endothelial growth factor‐secreting bulky tumors. Cancer Res 2006; 66: 10. [DOI] [PubMed] [Google Scholar]
- 30. Hamaguchi T, Doi T, Eguchi‐Nakajima T et al. Phase I study of NK012, a novel SN‐38‐incorporating micellar nanoparticle, in adult patients with solid tumors. Clin Cancer Res 2010; 16: 5058–66. [DOI] [PubMed] [Google Scholar]
- 31. Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblast in cancer initiation and progression. Nature 2004; 432: 332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alderton GK. Tumor microenvironment: macrophages lead the way. Nat Rev Cancer 2010; 10: 162–3. [Google Scholar]
- 33. Mosesson MW. Fibrinogen and fibrin structure and functions. J Thromb Haemost 2005; 3: 1894–904. [DOI] [PubMed] [Google Scholar]
- 34. Rehlaender BN, Cho MJ. Antibodies as carrier proteins. Pharm Res 1998; 15: 1652–6. [DOI] [PubMed] [Google Scholar]
- 35. Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer 2006; 6: 789–802. [DOI] [PubMed] [Google Scholar]
- 36. Bale WF, Spar IL, Goodland RL. Experimental radiation therapy of tumors with I131‐carrying antibodies to fibrin. Cancer Res 1960; 20: 1488–94. [PubMed] [Google Scholar]
- 37. Finbloom DS, Magilavy DB, Harford JB, Rifai A, Plotz PH. Influence of antigen on immune complex behavior in mice. J Clin Invest 1981; 68: 214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Detailed information regarding the synthesis of branched linker, branched PEG‐linked SN‐38, and NMR spectra for each compound.
Supporting info item