

Abstract
Epstein–Barr virus (EBV) nuclear antigen (EBNA)1 is expressed in every EBV‐infected cell, regardless of the state of EBV infection. Although EBNA1 is thought to be a promising antigen for immunotherapy of all EBV‐associated malignancies, it is less clear whether EBNA1‐specific CD4+ T cells can act as direct effectors. Herein, we investigated the ability of CD4+ T‐cell clones induced with overlapping peptides covering the C‐terminal region of EBNA1, and identified minimal epitopes and their restricted major histocompatibility complex class II molecules. Of these, a novel epitope, EYHQEGGPD, was found to be presented by DRB1*0401, 0403 and 0406. Five CD4+ T‐cell clones recognized endogenously processed and presented antigens on EBV‐transformed lymphoblastoid cell lines (LCL) and one example proved capable of killing EBV‐carrying natural killer (NK) and T‐cell lines derived from patients with chronic active EBV infection (CAEBV). Identification of minimal epitopes facilitates design of peptide‐based vaccines and our data suggest that EBNA1‐specific CD4+ T cells may play roles as direct effectors for immunotherapy targeting EBV‐carrying NK and T‐cell malignancies. (Cancer Sci 2008; 99: 1633–1642)
The Epstein–Barr virus (EBV) is involved in development of many malignancies, including Burkitt's lymphoma (BL), Hodgkin's disease (HD) and the nasopharyngeal carcinoma, as well as post‐transplant lymphoproliferative disorder.( 1 ) It is also related to natural killer (NK) and T lymphomas and causes chronic active EBV infection (CAEBV).( 1 , 2 ) Only EBV nuclear antigen (EBNA)1 is expressed in most BL, referred to as latency I. In addition to EBNA1, latent membrane protein (LMP)1 and/or 2 are expressed in HD, nasopharyngeal carcinomas, NK and T lymphomas, and CAEBV (latency II). All EBV latent antigens, EBNA1, 2, 3A, 3B, 3C and the leader protein, and LMP1 and 2 are expressed in post‐transplant lymphoproliferative disorder (latency III). EBNA1 is expressed in common in all these diseases,( 3 ) and may be present diffusely bound to mitotic chromosomes.( 4 ) EBV has a cis‐acting element, termed OriP, that enables the persistence of episomes in EBV‐infected cells. EBNA1 also binds to OriP,( 5 ) and is essential for EBV episome maintenance.( 6 ) Thus, the existence of EBV DNA is tightly associated with EBNA1 expression in EBV‐infected cells.
Evidence for the significance of EBV‐specific T cells for control of EBV infection has been obtained from both in vitro and in vivo studies.( 7 , 8 ) Human leukocyte antigen (HLA) class I‐restricted CD8+ cytotoxic T lymphocytes (CTL) recognize latent and lytic EBV antigens,( 8 ) and latent EBV antigen‐specific CTL kill not only lymphoblastoid cell lines (LCL) expressing the full spectrum of latent viral proteins but also tumor cells with limited viral proteins.( 9 , 10 , 11 ) EBV‐specific CTL responses have been extensively studied, and CTL are thought to be the main effectors. EBNA1‐specific CTL were long believed to be immunologically silent because EBNA1 contains an internal G‐A repeat (GAr) domain which has an immune evasion function endowing resistance to proteasomal degradation in the antigen presentation pathway.( 12 , 13 ) However, it has been reported that having GAr EBNA1 does not completely evade major histocompatibility complex (MHC) class I presentation.( 10 , 14 , 15 , 16 ) There are thus some EBNA1 antigen epitopes, though the importance of EBNA1‐specific CTL for EBV‐positive tumors remains unclear.
Accumulating current evidence indicates that CD4+ T cells, as well as CTL, are required for effective antitumor immunity,( 17 , 18 , 19 ) for example, playing an essential role in generation of CD8+ T memory cells.( 20 , 21 , 22 ) In addition, there are reports of cytotoxic action of CD4+ T effector cells in vitro ( 23 ) and in vivo.( 23 , 24 ) This has drawn attention to the possibility that EBV‐specific CD4+ T cells are able to recognize EBV‐infected cells. There is increasing interest in CD4+ T‐cell responses to EBV as direct effectors. HLA class II‐restricted CD4+ T cells specific for EBV latent and lytic antigens have been explored,( 25 , 26 , 27 ) with the focus on EBV‐infected B cells, mainly LCL, in which the HLA class II pathway of antigen presentation is active. Because immunoglobulin (Ig) isotype switching requires T‐cell help, the presence of IgG antibodies to antigens implies that the latter are also targets of CD4+ T‐cell responses.( 28 , 29 ) Actually, healthy virus carriers are consistently positive for anti‐EBNA1 IgG antibodies,( 1 ) implying the existence of EBNA1‐specific CD4+ T cells in vivo, because CD4+ T cells indeed recognize EBNA1 in healthy EBV carriers.( 30 , 31 ) EBNA1 has been considered a promising antigen for T‐helper (Th) cells. Moreover, it is clear that CD4+ T cells specific for EBNA1 can act as direct effectors for lysing BL cells and HD cells in vitro.( 32 ) Furthermore, in a mouse model of BL, EBNA1‐specific CD4+ T cells could suppress BL tumor growth in vivo.( 33 )
We report here the identification of five EBNA1‐specific CD4+ T‐cell clones recognizing LCL and their minimal epitopes. One HLA‐DR4‐restricted example is novel and the other four appear to be parts of longer peptides,( 25 , 31 ) some of which are presented by other HLA class II molecules than those determined in this report. Of particular interest is a CD4+ T‐cell clone killing EBV‐infected T cells positive for DR51 and EBV‐infected NK cells transduced with DRB5*0101. Because these EBV‐infected NK and T cells express DR, DP and DQ, they could be potential targets of CD4+ T cells. These results imply that EBNA1‐specific CD4+ T cells may also act as direct effectors in vivo.
Materials and Methods
Donors and cell lines. The study design and purpose, which had been approved by the institutional review board of Aichi Cancer Center, were fully explained and informed consent was obtained from all blood donors. HLA typing was carried out at the HLA Laboratory (Kyoto, Japan). The HLA class II genotype from donors X and W, and cell lines are shown in Table 1.
Table 1.
Human leukocyte antigen (HLA) class II genotype of the donors and cell lines
| Donors or cell lines | DRβ1 | Other DR alleles | DPβ1 | DQβ1 |
|---|---|---|---|---|
| Donor X | *0401, *1501 | DRβ4*0102, DRβ5*0101 | *0201, *1401 | *0301, *0602 |
| Donor W | *0803, *1401 | DRβ3*0202, NA | *0202, *0501 | *0601, *0503 |
| A1 LCL | *0101, *1401 | DRβ3*0202, NA | *0402, *0501 | *0501, *0502 |
| A2 LCL | *0901, *0901 | DRβ4*0103, DRβ4*0103 | *0201, *0901 | *0303, *0303 |
| SNK10 | *0901, *0901 | DRβ4*0103, DRβ4*0103 | *0201, *0402 | *0303, *0303 |
| SNT15 | *0101, *0406 | DRβ4*0103, NA | *0201, *0402 | *0302, *0501 |
| SNT16 | *1201, *1502 | DRβ3*0101, DRβ5*0102 | *0501, *0901 | *0303, *0601 |
NA, not assayed. LCL, lymphoblastoid cell lines.
Epstein–Barr virus‐transformed B‐LCL were established as described previously,( 34 ) and cultured in RPMI‐1640 medium supplemented with 10% fetal calf serum, 2 mM L‐glutamine, 50 U/mL penicillin, 50 µg/mL streptomycin and 50 µg/mL kanamycin.
An EBV‐carrying NK cell line, SNK10, and an EBV‐carrying γδ T‐cell line, SNT15, and an EBV‐carrying αβ T‐cell line, SNT16, were kindly provided by Dr Shimizu (Tokyo Medical and Dental University, Tokyo, Japan). All three were derived from different CAEBV patients and cultured as previously described,( 35 , 36 ) along with HEK‐293 T cells. CD40‐activated B (CD40‐B) cells were generated as detailed earlier,( 37 , 38 ) using NIH/3T3‐human CD40 ligand cells, kindly provided by Dr Freeman (Dana‐Farber Cancer Institute, Boston, MA, USA).
Phoenix‐GALV cells kindly provided by Dr Kiem (Fred Hutchinson Cancer Research Center, Seattle, WA, USA) and Dr Nolan (Stanford University School of Medicine, Stanford, CA, USA) were cultured as previously described.( 39 ) Retroviral transduction of HLA genes was performed as reported earlier.( 37 )
Plasmid construction. HLA‐DRA cDNA were amplified by polymerase chain reaction (PCR) using a sense primer, 5′‐ggatccgccaccATGGCCATAAGTGGAGTCCCTG‐3′, and an antisense primer, 5′‐gcggccgcTTACAGAGGCCCCCTGCGTTC‐3′. The following primer pairs were used for PCR amplification of HLA‐DRB1*0401 cDNA, HLA‐DRB1*1501 cDNA, HLA‐DRB4*0102 cDNA and HLA‐DRB5*0101 cDNA: DRB1*0401 sense primer, 5′‐ggatccgccaccATGGTGTGTCTGAAGTTCC‐3′; DRB1*0401 antisense primer, 5′‐gcggccgcTCAGCTCAGGAATCCTGTTG‐3′. DRB1*0403, 0405 and 0406 cDNA were amplified by PCR with the DRB1*0401 sense primer and an antisense primer: DRB1*1501 sense primer, 5′‐ggatccgccaccATGGTGTGTCTGAAGCTCC‐3′ (fwd‐1); DRB1*1501 antisense primer, 5′‐gcggccgcTCAGCTCAGGAATCCTGTTG‐3′; DRB4*0102 sense primer, fwd‐1; DRB4*0102 antisense primer, 5′‐gcggccgcTCAGCTCAAGAGTCCTGTTG‐3′; DRB5*0101 sense primer, fwd‐1; and DRB5*0101 antisense primer, 5′‐gcggccgcTCAGCTCACGAGTCCTGTTG‐3′. The resultant individual DNA fragments were cloned into pcDNA 3.1(+) using its BamHI and NotI sites.
Full‐length EBNA1 cDNA was cloned into pcDNA 3.1(+) (pcDNA/EBNA1).( 16 ) EBNA1 without GAr (referred to as ΔGA‐EBNA1) was constructed from pcDNA/EBNA1, as described previously.( 40 ) The resulting construct was deleted from the GAr domain (EBNA1 codons 92–323).
Synthetic peptides. C‐terminal EBNA1 polypeptides covering its 402–624 amino acids (a.a.) were deduced from the prototype B95‐8 (National Center for Biotechnology Information accession no. V01555) DNA sequence. A total of thirty 20‐mer peptides overlapping by 13 a.a. were designed, and purchased from Bio‐Synthesis (Lewisville, TX, USA). A six‐subpool was constructed from four to five peptides, excluding peptides including HLA class I‐restricted epitopes. For example, HLA‐B35‐seropositive donors might be expected to respond to HPVGEADYFEY, and epitope‐specific CD8+ T cells might be expanded. Other known epitopes were excluded as well. To identify the core epitope sequence, 13‐ and 11‐mer peptides were further designed and purchased from Bio‐Synthesis. The one‐letter a.a. code is used throughout the article.
Generation of CD4+ T‐cell lines and clones. Peripheral blood mononuclear cells (PBMC) from two donors were stimulated with individual peptide pools of 500 nM of each peptide in 2 mL RPMI‐1640 medium supplemented with 10% human serum, 2 mM L‐glutamine, 50 U/mL penicillin, 50 µg/mL streptomycin and 50 µg/mL kanamycin at 5% CO2 in a humidified incubator. On days 8 and 15, T cells were restimulated with peptide‐pulsed γ‐irradiated (33 Gy) autologous PBMC. One day after each restimulation, interleukin (IL)‐2, kindly provided by Ms Sawada (Shionogi, Osaka, Japan), was added to a final concentration of 10 U/mL. After four rounds of stimulation, CD4+ T cells were isolated with CD4 Microbeads (Miltenyi Biotec, Tokyo, Japan).
To establish T‐cell clones, limiting dilution of isolated CD4+ T cells was performed as previously described,( 41 ) with slight modifications. Where necessary, EBNA1 peptide‐reactive CD4+ T cells were enriched from restimulated CD4+ T cells using an γ‐interferon (IFN‐γ) Secretion Assay (Miltenyi Biotec), according to the manufacturer's instructions. Then, the purified CD4+ T cells were seeded at 1 cell/well in round‐bottomed 96‐well plates in culture medium containing anti‐CD3 monoclonal antibodies (mAb) (30 ng/mL), IL‐2 (200 U/mL), γ‐irradiated (33 Gy) 1 × 105 PBMC and γ‐irradiated (55 Gy) 2 × 104 LCL. After 14–16 days of culture, the specificity of growing cells was examined with enzyme‐linked immunosorbent spot (ELISPOT) assays of EBNA1 peptide‐pulsed CD40 B cells and autologous LCL. Positive wells were transferred into flasks and expanded with anti‐CD3 mAb, IL‐2 (30 U/mL) and γ‐irradiated feeders.
ELISPOT assays. ELISPOT assays were performed as previously described,( 37 , 41 ) with minor modifications. Briefly, CD4+ T cells were co‐cultured with various stimulators for 20 h in AIM‐V medium (Invitrogen, Carlsbad, CA, USA) in wells of Multiscreen‐HA plates (MAHA S4510; Millipore, Billerica, MA, USA) coated with antihuman IFN‐γ mAb (M700A; Pierce Biotechnology, Philadelphia, PA, USA). As stimulators, LCL (1 × 105 cells/well), HEK‐293 T cells (5 × 104 cells/well) transfected with plasmids encoding HLA‐DRA and DRB1 cDNA, and/or ΔGA‐EBNA1 or EGFP,( 37 ) cDNA with Lipofectamin 2000 (Invitrogen) 36 h earlier were seeded into each well. For peptide titration assays, serial concentrations of synthetic peptides were pulsed to HLA‐DR‐expressing HEK‐293T (referred to as DR‐293T) cells for 1 h at room temperature. In blocking assays, anti‐HLA‐DR mAb (L243; BD Bioscience, San Jose, CA, USA), anti‐HLA‐DQ mAb (TÜ169; BD Bioscience), and anti‐HLA‐DP mAb (BRA‐FB6; MorphoSys, Kingston, NH, SUA) were added at the indicated concentrations, and incubated for 1 h prior to co‐cultivation with T cells. After probing with polyclonal antirabbit IFN‐γ antibody (P700; Pierce Biotechnology), and following exposure to horseradish peroxidase‐labeled antirabbit IgG antibody and spot visualization, the plates were washed and dried. IFN‐γ spots were enumerated using a dissecting microscope. In all experiments using CD4+ T cell clones, results from ELISPOT assays are shown as the mean of two duplicate values.
CTL assays. Target cells were labeled with 1.85 MBq chromium (51Cr) for 1.5 h at 37°C, washed and mixed with CTL at the indicated effector to target ratios in 96‐well plates. After incubation for 14 h at 37°C, the radioactivity in the supernatants was counted in a γ‐counter. The minimal release was less 30% of maximal release in all experiments. The percentage‐specific 51Cr release was calculated as follows: 100 × (experimental release –spontaneous release)/(maximum release – spontaneous release). All assays were done in triplicate wells. Standard deviations were calculated from each data.
Results
Induction of EBNA1‐specific CD4+ T cells. It has been reported that CD4+ T‐cell epitopes are concentrated within the C‐terminal of the EBNA1 protein,( 25 , 31 , 32 ) and we designed 30 peptides (20‐mers overlapping by 13 residues) covering a.a. 402–624. To generate EBNA1‐specific CD4+ T cells, PBMC from two EBV‐seropositive donors were stimulated with 6‐peptide pools. After four rounds of stimulation, CD4+ T cells were isolated using CD4 Microbeads and ELISPOT assays were performed to test the specificity of the CD4+ T‐cell lines. As shown in Fig. 1(a,b), X1, X3 and X4 CD4+ T‐cell lines and W4 and W5 CD4+ T‐cell lines specifically secreted IFN‐γ in response to cognate peptide pool‐pulsed autologous CD40‐B cells, but not to irrelevant peptide pool‐pulsed CD40‐B cells.
Figure 1.
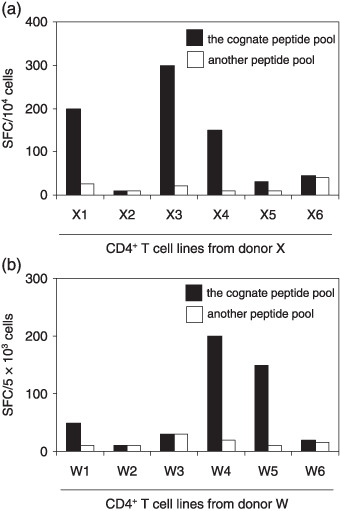
Establishment of CD4+ T‐cell lines specific for Epstein–Barr virus nuclear antigen (EBNA)1 peptide pools. (a,b) Aliquots of peripheral blood mononuclear cells from donors X and W were stimulated in vitro with synthetic peptide pools four times. After CD4‐positive selection, the responder cells were evaluated for their reactivity to the cognate peptide pools by enzyme‐linked immunosorbent spot assays using autologous CD40‐B cells as antigen‐presenting cells. Another EBNA1 peptide pool that had not used in stimulation was tested for control. Data are numbers of spots per 10 000 CD4+ T cells in (a) and 5000 CD4+ T cells in (b). SFC, spot forming units.
To further assess IFN‐γ secretion against each peptide, CD4+ T‐cell lines were tested against peptides belonging to each pool. As illustrated in Fig. 2(a), production of IFN‐γ by the X1 CD4+ T‐cell line from donor X was raised by two peptides (residues 409–428 and 416–435). Interestingly, the X3 CD4+ T‐cell line recognized three peptides (residues 472–491, 479–498 and 486–505) (Fig. 2b). Fig. 2(c) shows that the X4 CD4+ T‐cell line responded to two peptides (residues 514–533 and 521–540). As shown in Fig. 2(d, e), the W4 CD4+ T‐cell line recognized one peptide (residues 514–533) and the W5 CD4+ T‐cell line recognized two (residues 563–582 and 570–589).
Figure 2.
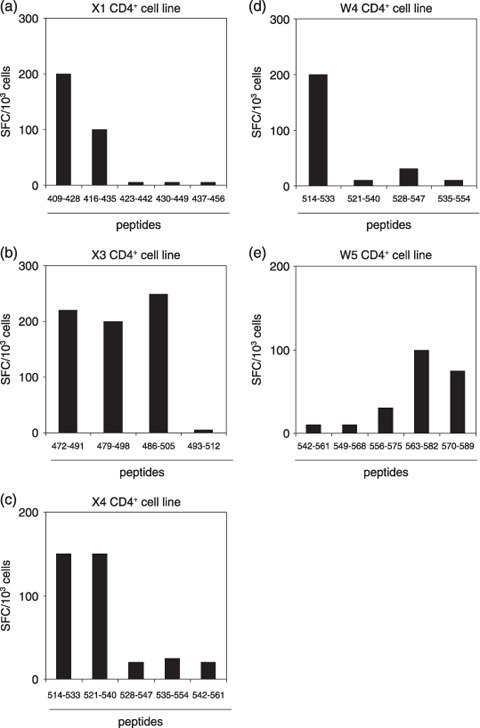
Reactivity of CD4+ T‐cell lines with individual peptides belonging to each pool. (a–e) Each CD4+ T‐cell line was tested for its reactivity with individual peptides by enzyme‐linked immunosorbent spot assay. Data are numbers of spots/1000 CD4+ T cells.
Identification of the EBNA1 epitope. We established five CD4+ T‐cell clones by limiting‐dilution culture of CD4+ T‐cell lines. As expected from the specificity of the X3 CD4+ T‐cell line, the derived clones X3‐11D1 and X3‐3G2 responded to two distinct peptides. ELISPOT assays were performed to map the recognized epitope regions (3, 4). Because clone X1‐12B12 secreted IFN‐γ in response to two overlapping peptides (residues 409–428 and 416–435, Fig. 2a), the epitope was speculated to be located around a.a. residues 416–428. We attempted to identify minimal epitopes despite the fact that they may not be as definitive as those recognized by CD8+ T cells because MHC class II molecules can present peptides with various lengths.( 42 , 43 ) To this end, 13‐ and 11‐mer peptides spanning the overlapped regions were synthesized and tested in ELISPOT assays to explore the N‐ and C‐terminal a.a. which are indispensable for recognition. As shown in Fig. 3(a), deletion of E at position 416 (referred to as E416) from the 13‐mer peptide abolished the IFN‐γ production from clone X1‐12B12. At the C‐terminus, loss of G425 from the 11‐mer peptide had the same effect. Accordingly we infer that the a.a. sequence, EYHQEGGPDG, is a putative minimal epitope for the clone recognition. Fig. 4(a) additionally demonstrates that a 13‐mer peptide, (F)EYHQEGGPDG(EP), containing the minimal epitope sequence was better recognized than the 11‐mer, (F)EYHQEGGPDG, and 13‐mer, EYHQEGGPDG(EPD), peptides suggesting elongation of both N‐ and C‐termini may augment the antigenicity.
Figure 3.

Mapping of core regions recognized by CD4+ T‐cell clones. (a–e) Peptides of 13‐ and 11‐mer spanning overlapping regions were synthesized and tested for antigenicities in enzyme‐linked immunosorbent spot assays. Numbers represent Epstein–Barr virus nuclear antigen 1 amino acid residues. All results are the mean of two duplicate values. SFC, spot forming units.
Figure 4.
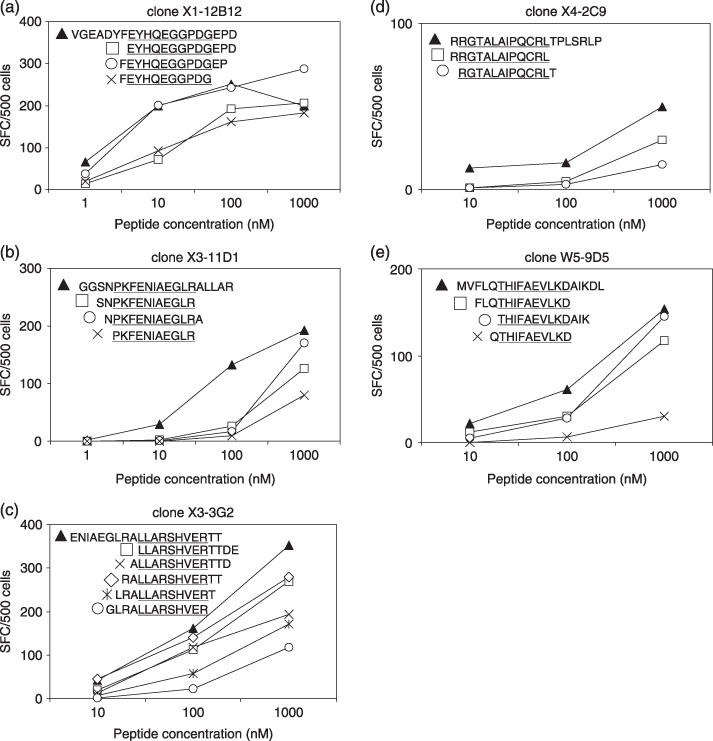
Peptide dilution assay for CD4+ T‐cell clones. (a–e) Each peptide at serial concentrations was tested in enzyme‐linked immunosorbent spot assays. Data are numbers of spots/500 CD4+ T‐cell clones. The core peptide sequences are underlined. All results are the mean of two duplicate values. SFC, spot forming units.
As illustrated in Fig. 3(b), removal of P476 from 13‐ and 11‐mer peptides completely abolished clone X3‐11D1 recognition. The 13‐ and 11‐mer peptides having R486 at the C‐terminal stimulated the clone X3‐11D1 to secrete IFN‐γ. Thus, clone X3‐11D1 responded to PKFENIAEGLR as the putative minimal epitope. Moreover, it recognized (SN)PKFENIAEGLR and (N)PKFENIAEGLR(A) more efficiently than the putative minimal epitope, PKFENIAEGLR (Fig. 4b).
As demonstrated in Fig. 3(c), deletion of L488 from the 13‐mer peptide abolished the clone X3‐3G2 recognition and IFN‐γ secretion was also eliminated by deletion of R496 from the 13‐ and 11‐mer peptides. This implies that LLARSHVER is the putative minimal epitope for the clone recognition. Furthermore, clone X3‐3G2 could secrete IFN‐γ in response to (RA)LLARSHVER(TT) and LLARSHVER(TTDE) as well as a 20‐mer peptide (Fig. 4c).
As shown in Fig. 3(d), lack of R522 and L533 from 13‐mer peptides abolished the clone X4‐2C9 recognition. Accordingly, RGTALAIPQCRL is the putative minimal epitope required for IFN‐γ secretion by clone X4‐2C9, which did not respond to any 11‐mer peptides. Moreover, clone X4‐2C9 responded to 20‐mer better than 13‐mer peptides (3, 4).
As shown in Fig. 3(e), deletion of T568 from the 13‐mer peptide abolished the IFN‐γ production by clone W5‐9D5, as did loss of the C‐terminal residues K576 and D577 from the 20‐mer peptide. In consequence, THIFAEVLKD is the putative minimal epitope. Furthermore, removal of F 565 from the 13‐ and 11‐mer peptides resulted in decrease of IFN‐γ secretion. As illustrated in Fig. 4(e), clone W5‐9D5 could produce IFN‐γ in response to (FLQ)THIFAEVLKD and THIFAEVLKD(AIK) as well as the 20‐mer peptide. In conclusion, as listed in Table 2, the minimal peptide sequences recognized by CD4+ T‐cell clones were identified. Four out of five established clones recognized minimal epitopes that are parts of epitope regions previously reported.( 25 , 31 )
Table 2.
Identification of epitopes recognized by five clones
| Clone | Minimal epitope † | Identified HLA‐restriction | Known epitope sequence | Reported HLA‐restriction | REF |
|---|---|---|---|---|---|
| X1‐12B12 | (F)EYHQEGGPDG(EP) | DR4 | |||
| X3‐11D1 | (SN)PKFENIAEGLR(A) | DP2, DP5 | NPKFENIAEGLRALL | DP, DR11 | 25 |
| X3‐3G2 | (RA)LLARSHVER(TTDE) | DQ6 | LRALLARSHVERTTD | ND | 25 |
| X4‐2C9 | (R)RGTALAIPQCRL(T) | DR51 | NLRRGTALAIPQCRL | DR4, DQ3 | 25 |
| W5‐9D5 | (FLQ)THIFAEVLKD(AIK) | DP5 | MVFLQTHIFAEVLKD | DR15 | 25 |
| VFLQTHIFAEVLKDLV | DP5 | 31 |
The minimal epitope represents the shortest fragment where any further truncation may result in drastic reduction of antigenicity. Addition of amino acids in parentheses may augment the antigenicity. HLA, human leukocyte antigen; ND, not determined; REF, reference.
HLA restriction of the EBNA1‐specific clones. To identify the HLA molecule presenting EBNA1 to the CD4+ T‐cell clones, ELISPOT assays were performed in the presence of antibodies against HLA‐DR, DQ and DP molecules. Epitope‐specific IFN‐γ secretion by clones X1‐12B12 and X4‐2C9 was inhibited in the presence of the mAb against HLA‐DR (Fig. 5a,b, top panel). The other mAb were without effect. To determine the restricting HLA‐DR molecule, HEK‐293 T cells expressing each donor HLA‐DR allele were pulsed with their cognate peptides and used as stimulators in the ELISPOT assays. The cognate peptide was presented by HLA‐DR4 for clone X1‐12B12, and by HLA‐DR51 for clone X4‐2C9 (Fig. 5a,b bottom panel, respectively). As shown in Fig. 5(c,e), the HLA‐DP‐specific mAb blocked recognition by clones X3‐11D1 and W5‐9D5. No block was evident using mAb against HLA‐DR and HLA‐DQ. To verify the restriction molecule, ELISPOT assays were carried out using HLA‐matched LCL pulsed with cognate or irrelevant peptides. Clones X3‐11D1 and W5‐9D5 recognized their cognate peptides in the context of HLA‐DP2 and HLA‐DP5 molecules, respectively. Accordingly, clone X3‐11D1 could also recognize the epitope presented by HLA‐DP5 (data not shown). As demonstrated in Fig. 5(d), clone X3‐3G2 recognized its cognate peptide on the HLA‐DQ6 molecule. The identified HLA restriction alleles are listed in Table 2.
Figure 5.
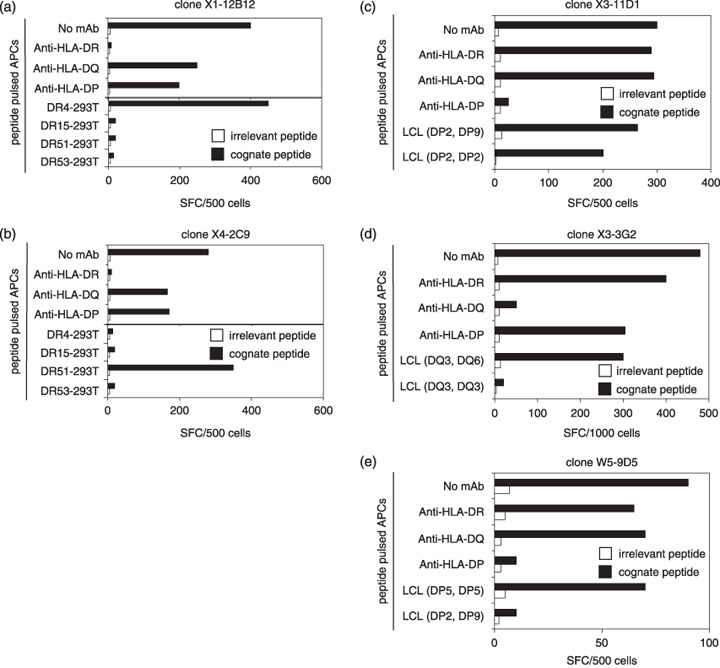
Identification of human leukocyte antigen (HLA) class II restriction molecules. (a,b, top panel) Enzyme‐linked immunosorbent spot (ELISPOT) assays were performed using autologous lymphoblastoid cell lines (LCL) pulsed with peptides in the presence of monoclonal antibody (mAb) against HLA‐DR, DQ and DP. (bottom panel) CD4+ T‐cell clone recognition of HEK‐293T cells transfected with each donor HLA‐DR allele and pulsed with peptide was determined by ELISPOT assay. (c–e) ELISPOT assays were performed using autologous LCL pulsed with peptides in the presence of mAb against HLA‐DR, DQ, DP or HLA‐typed allogeneic LCL pulsed with peptides. All results are the mean of two duplicate values. SFC, spot forming units.
Recognition of LCL and DGA‐transfected 293T by EBNA1‐specific CD4+ T‐cell clones. It has been reported that a CD4+ T‐cell clone specific for the EBNA1515–527 epitope fails to recognize LCL presenting naturally processed peptides on their surfaces.( 44 ) To test whether established clones can recognize LCL without exogenous EBNA1 peptides, ELISPOT assays were performed. As shown in Fig. 6(a), all clones could secrete IFN‐γ in response to autologous LCL. To identify their restricted molecules, HEK‐293 T cells expressing the restricted HLA‐DR alleles and ΔGA‐EBNA1 protein were used in the ELISPOT assay (Fig. 6b,c). Clone X1‐12B12 could recognize ΔGA‐EBNA1‐expressing HEK‐293 T cells transfected with HLA‐DRB1*0401, 0403 or 0406, but not DRB1*0405 (Fig. 6b). Clone X4‐2C9 could recognize DR51‐293 T cells transfected with ΔGA‐EBNA1 (Fig. 6c).
Figure 6.
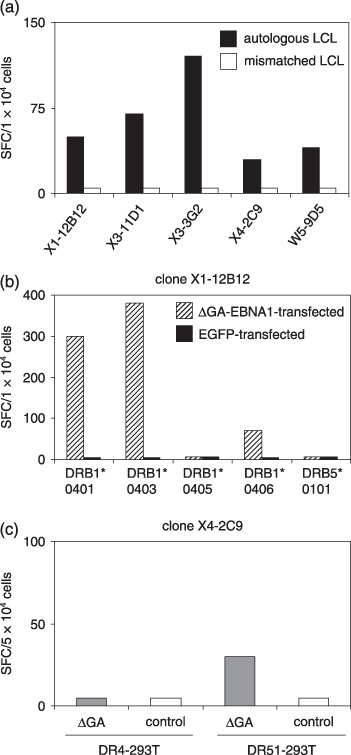
Recognition of endogenously processed Epstein–Barr virus nuclear antigen (EBNA)1 by CD4+ T‐cell clones. (a) γ‐interferon spot production with CD4+ T‐cell clones was estimated by enzyme‐linked immunosorbent spot (ELISPOT) assays using lymphoblastoid cell lines (LCL)as antigen‐presenting cells. Human leukocyte antigen (HLA)‐mismatched LCL was A1 for clones from donor X and A2 for a clone from donor W. (b) Recognition by clone X1‐12B12 of DR4‐ (DRB1*0401, 0403, 0405, or 0406) or DR51 (DB5*0101)‐293T cells transfected with ΔGA‐EBNA1 or a control construct was tested by ELISPOT assays. (c) Recognition by clone X4‐2C9 of DR4‐ or DR51‐293 T cells transfected with ΔGA‐EBNA1 or control construct was tested by ELISPOT assays. All results are the mean of two duplicate values. EGFP, enhanced green fluorescent protein; SFC, spot forming units.
Cytotoxic activity of EBNA1‐specific CD4+ T‐cell clones against EBV‐positive NK and T‐cell lines. It is reported that EBNA1‐specific CD4+ T‐cell clones can kill EBV‐positive BL and HD cells. We tested the lytic activity of clones against EBV‐carrying NK and T‐cell lines established from patients with CAEBV as representative of EBV latency II malignancies and retaining characteristics of the original tumors, such as identical EBV clonality.( 35 ) All clones with one exception (clone W5‐9D5) had lytic activity against peptide‐pulsed autologous CD40‐B cells within 4‐h incubation (data not shown). Clone X4‐2C9, with the highest killing activity against peptide‐pulsed targets, showed killing activity after 14‐h incubation with HLA‐matched EBV‐carrying SNT16 cells (Fig. 7a) and HLA‐DR51‐transfected SNK10 cells (Fig. 7b).
Figure 7.
Cytotoxic activity of CD4+ T cell clone X4‐2C9 against EBV‐carrying NK and T cell lines. (a) Fourteen‐hour cytotoxic T lymphocytes assays were performed using Epstein–Barr virus‐carrying T‐cell lines as targets. Data for specific lysis of an human leukocyte antigen (HLA)‐DR51‐positive T‐cell line (SNT16;  ) and an HLA‐DR51‐negative T‐cell line (SNT15;
) and an HLA‐DR51‐negative T‐cell line (SNT15;  ) are shown. (b) Cytolytic activity of the CD4+ T‐cell clone X4‐2C9 was assessed against HLA‐DR51 (
) are shown. (b) Cytolytic activity of the CD4+ T‐cell clone X4‐2C9 was assessed against HLA‐DR51 ( ) or HLA‐DR1‐retrovirally transduced (
) or HLA‐DR1‐retrovirally transduced (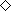 ) SNK10 cells. All assays were done in triplicate wells. Bars indicate standard deviations calculated from each data. The minimal release was less 30% of maximal release in all experiments.
) SNK10 cells. All assays were done in triplicate wells. Bars indicate standard deviations calculated from each data. The minimal release was less 30% of maximal release in all experiments.
Discussion
Previous studies revealed a DR4‐ or DQ3‐restricted promiscuous epitope (NLRRGTALAIPQCRL), a DR4‐ or DR15‐restricted promiscuous epitope (AEGLRALLARSHVER), a DR15‐restricted epitope (MVFLQTHIFAEVLKD), a DP5‐restricted (VFLQTHIFAEVLKDAIKDL) and a DP‐restricted promiscuous epitope (NFKFENIAEGLRALL).( 25 , 31 , 45 ) Our donors had HLA alleles that could present the above epitopes, but there were no T‐cell responses to the HLA/peptide complexes with two exceptions (DP5‐ and DP‐restricted epitopes). The DP5‐restricted epitope (THIFAEVLKD) and the DP2‐ and DP5‐restricted epitope (PKFENIAEGLR) may have strong immunogenicity. These results are in line with a report by Tsang et al.( 31 ) who detected DP5‐restricted epitope‐specific CD4+ T cells in all donors positive for EBV and DP5. The HLA‐DPB1*0501 allele is the most frequent HLA‐DPB1 allele in the Japanese population and more than 60% of Japanese people are positive for this allele. It would be useful for vaccine development to further investigate which epitopes are immunodominant in vivo.
In the present study, all clones with one exception (clone W5‐9D5) demonstrated killing activity (30–80%) against peptide‐pulsed targets when assayed after 4 h. The clone X4‐2C9 had the highest killing activity; although it showed less lytic activity against an EBV‐carrying T‐cell line in 4‐h cytotoxic assay (data not shown), cytolysis was found with a DR51‐carrying EBV‐positive T‐cell line and a DR51‐transfecting EBV‐positive NK cell line after 14‐h incubation. The present data confirm that EBNA1‐specific CD4+ T cells work as direct effectors in vitro, in line with the data reported by Pauldan et al.( 32 ) They documented that EBNA1‐specific CD4+ T cells recognize and kill BL cells and Hodgkin's lymphoma cells in vitro.
Long et al.( 46 ) established CD4+ T‐cell clones specific for a single epitope of EBNA2276–295 presented by four different MHC. Among them, only the HLA‐DR52b‐restricted clone could kill autologous LCL, though all four clones secreted IFN‐γ against LCL. Two groups reported CD4+ T‐cell clones specific for another single epitope of EBNA2280–290 presented by several MHC.( 47 , 48 ) Similarly, DQ2‐, DQ7‐ and DR52‐restricted clones could kill LCL, while the others could only secrete IFN‐γ against LCL. Long et al.( 46 ) mentioned that it was important for immunotherapeutic applications to identify combinations of epitopes and restriction molecules that have the capability for more efficient antigen presentation. In our study, only the clone X4‐2C9 showed killing activity against EBV‐positive NK and T‐cell lines (Fig. 7a,b), underscoring the importance of particular combinations of epitopes and restriction molecules.
We reported earlier that EBV‐infected NK cells established from patients with lymphomas or CAEBV are susceptible to LMP1‐specific CTL‐mediated lysis.( 11 ) For immunotherapy against EBV‐associated malignancies, it might be efficient to combine CD4+ T cells and CTL as direct effectors, but further studies are needed to clarify their synergistic effects in vitro and in vivo. For the present, we can conclude that the CD4+ T‐cell‐mediated lysis of EBV‐carrying NK and T‐cell lines demonstrated here provides clues to immunotherapy targeting EBV‐associated malignancies.
Acknowledgments
The authors of this paper thank Ms K. Nishida and Ms F. Ando for their technical expertise, and Ms H. Tamaki and Ms Y. Matsudaira for secretarial assistance. A. O. is an awardee of a Research Resident Fellowship from the Foundation for Promotion of Cancer Research (Japan) for the Third Team Comprehensive Control Research for Cancer. This work was supported in part by Grants‐in‐Aid for Scientific Research (C) (nos. 17590428 and 18591212) from the Japan Society for the Promotion of Science, Grants‐in‐Aid for Scientific Research on Priority Areas (nos. 17016089 and 18015053), from the Ministry of Education, Culture, Science and Sports, and Grants‐in‐Aid for Cancer Research (nos. 16–17 and 18S‐1) from the Ministry of Health, Labor, Welfare and Technology, Japan, the Third Team Comprehensive Control Research for Cancer (no. 26) from the Ministry of Health, Labor and Welfare, Japan, and a Research Grant from the Princess Takamatsu Cancer Research Fund.
References
- 1. Rickinoson AB, Kieff E. Epstein–Barr virus. In: Fields BN, Knipe DM, Howley PM, eds. Fields Virology, 5th edn. Philadelphia: Lippincott, 2006; 2655–700. [Google Scholar]
- 2. Kimura H, Hoshino Y, Kanegane H et al . Clinical and virologic characteristics of chronic active Epstein–Barr virus infection. Blood 2001; 98: 280–6. [DOI] [PubMed] [Google Scholar]
- 3. Kieff E, Rickinoson AB. Epstein–Barr virus and its replication. In: Fields BN Knipe DM, Howley PM, eds. Fields Virology, 5th edn. Philadelphia: Lippincott, 2006; 2655–700. [Google Scholar]
- 4. Ohno S, Luka J, Lindahl T, Klein G. Identification of a purified complement‐fixing antigen as the Epstein–Barr‐virus determined nuclear antigen (EBNA) by its binding to metaphase chromosomes. Proc Natl Acad Sci USA 1977; 74: 1605–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Frappier L, O'Donnell M. Overproduction, purification, and characterization of EBNA1, the origin binding protein of Epstein–Barr virus. J Biol Chem 1991; 266: 7819–26. [PubMed] [Google Scholar]
- 6. Lee MA, Diamond ME, Yates JL. Genetic evidence that EBNA‐1 is needed for efficient, stable latent infection by Epstein–Barr virus. J Virol 1999; 73: 2974–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Davis JE, Moss DJ. Treatment options for post‐transplant lymphoproliferative disorder and other Epstein–Barr virus‐associated malignancies. Tissue Antigens 2004; 63: 285–92. [DOI] [PubMed] [Google Scholar]
- 8. Rickinson AB, Moss DJ. Human cytotoxic T lymphocyte responses to Epstein–Barr virus infection. Annu Rev Immunol 1997; 15: 405–31. [DOI] [PubMed] [Google Scholar]
- 9. Khanna R, Burrows SR, Nicholls J et al . Identification of cytotoxic T cell epitopes within Epstein–Barr virus (EBV) oncogene latent membrane protein 1 (LMP1): evidence for HLA A2 supertype‐restricted immune recognition of EBV‐infected cells by LMP1‐specific cytotoxic T lymphocytes. Eur J Immunol 1998; 28: 451–8. [DOI] [PubMed] [Google Scholar]
- 10. Voo KS, Fu T, Wang HY et al . Evidence for the presentation of major histocompatibility complex class I‐restricted Epstein–Barr virus nuclear antigen 1 peptides to CD8+ T lymphocytes. J Exp Med 2004; 199: 459–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Demachi‐Okamura A, Ito Y, Akatsuka Y et al . Epstein–Barr virus (EBV) latent membrane protein‐1‐specific cytotoxic T lymphocytes targeting EBV‐carrying natural killer cell malignancies. Eur J Immunol 2006; 36: 593–602. [DOI] [PubMed] [Google Scholar]
- 12. Levitskaya J, Coram M, Levitsky V et al . Inhibition of antigen processing by the internal repeat region of the Epstein–Barr virus nuclear antigen‐1. Nature 1995; 375: 685–8. [DOI] [PubMed] [Google Scholar]
- 13. Levitskaya J, Sharipo A, Leonchiks A, Ciechanover A, Masucci MG. Inhibition of ubiquitin/proteasome‐dependent protein degradation by the Gly‐Ala repeat domain of the Epstein–Barr virus nuclear antigen 1. Proc Natl Acad Sci USA 1997; 94: 12616–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee SP, Brooks JM, Al‐Jarrah H et al . CD8 T cell recognition of endogenously expressed Epstein–Barr virus nuclear antigen 1. J Exp Med 2004; 199: 1409–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tellam J, Connolly G, Green KJ et al . Endogenous presentation of CD8+ T cell epitopes from Epstein–Barr virus‐encoded nuclear antigen 1. J Exp Med 2004; 199: 1421–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ito Y, Demachi‐Okamura A, Ohta R et al . Full‐length EBNA1 mRNA‐transduced dendritic cells stimulate cytotoxic T lymphocytes recognizing a novel HLA‐Cw*0303‐ and ‐Cw*0304‐restricted epitope on EBNA1‐expressing cells. J General Virol 2007; 88: 770–80. [DOI] [PubMed] [Google Scholar]
- 17. Castellino F, Germain RN. Cooperation between CD4+ and CD8+ T cells: when, where, and how. Annu Rev Immunol 2006; 24: 519–40. [DOI] [PubMed] [Google Scholar]
- 18. Ostrand‐Rosenberg S. CD4+ T lymphocytes: a critical component of antitumor immunity. Cancer Invest 2005; 23: 413–9. [PubMed] [Google Scholar]
- 19. Su Z, Dannull J, Yang BK et al . Telomerase mRNA‐transfected dendritic cells stimulate antigen‐specific CD8+ and CD4+ T cell responses in patients with metastatic prostate cancer. J Immunol 2005; 174: 3798–807. [DOI] [PubMed] [Google Scholar]
- 20. Bevan MJ. Helping the CD8 (+) T‐cell response. Nat Rev Immunol 2004; 4: 595–602. [DOI] [PubMed] [Google Scholar]
- 21. Ito D, Albers A, Zhao YX et al . The wild‐type sequence (wt) p53 (25–35) peptide induces HLA‐DR7 and HLA‐DR11‐restricted CD4+ Th cells capable of enhancing the ex vivo expansion and function of anti‐wt p53 (264‐272): peptide CD8+ T cells. J Immunol 2006; 177: 6795–803. [DOI] [PubMed] [Google Scholar]
- 22. Gao FG, Khammanivong V, Lie WJ, Leggatt GR, Frazer IH, Fernando GJ. Antigen‐specific CD4+ T‐cell help is required to activate a memory CD8+ T cell to a fully functional tumor killer cell. Cancer Res 2002; 62: 6438–41. [PubMed] [Google Scholar]
- 23. Guo Y, Niiya H, Azuma T et al . Direct recognition and lysis of leukemia cells by WT1‐specific CD4+ T lymphocytes in an HLA class II‐restricted manner. Blood 2005; 106: 1415–8. [DOI] [PubMed] [Google Scholar]
- 24. Jellison ER, Kim SK, Welsh RM. MHC class II‐restricted killing in vivo during viral infection. J Immunol 2005; 174: 614–18. [DOI] [PubMed] [Google Scholar]
- 25. Leen A, Meij P, Redchenko I et al . Differential immunogenicity of Epstein–Barr virus latent‐cycle proteins for human CD4 (+) T‐helper 1 responses. J Virol 2001; 75: 8649–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Landais E, Saulquin X, Scotet E et al . Direct killing of Epstein–Barr virus (EBV)‐infected B cells by CD4 T cells directed against the EBV lytic protein BHRF1. Blood 2004; 103: 1408–16. [DOI] [PubMed] [Google Scholar]
- 27. Adhikary D, Behrends U, Moosmann A, Witter K, Bornkamm GW, Mautner J. Control of Epstein–Barr virus infection in vitro by T helper cells specific for virion glycoproteins. J Exp Med 2006; 203: 995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kelsoe G, Zheng B. Sites of B‐cell activation in vivo . Curr Opin Immunol 1993; 5: 418–22. [DOI] [PubMed] [Google Scholar]
- 29. MacLennan IC, Gulbranson‐Judge A, Toellner KM et al . The changing preference of T and B cells for partners as T‐dependent antibody responses develop. Immunol Rev 1997; 156: 53–66. [DOI] [PubMed] [Google Scholar]
- 30. Munz C, Bickham KL, Subklewe M et al . Human CD4(+) T lymphocytes consistently respond to the latent Epstein–Barr virus nuclear antigen EBNA1. J Exp Med 2000; 191: 1649–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tsang CW, Lin X, Gudgeon NH et al . CD4+ T‐cell responses to Epstein–Barr virus nuclear antigen EBNA1 in Chinese populations are highly focused on novel C‐terminal domain‐derived epitopes. J Virol 2006; 80: 8263–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Paludan C, Bickham K, Nikiforow S et al . Epstein–Barr nuclear antigen 1‐specific CD4 (+) Th1 cells kill Burkitt's lymphoma cells. J Immunol 2002; 169: 1593–603. [DOI] [PubMed] [Google Scholar]
- 33. Fu T, Voo KS, Wang RF. Critical role of EBNA1‐specific CD4+ T cells in the control of mouse Burkitt lymphoma in vivo . J Clin Invest 2004; 114: 542–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kuzushima K, Hoshino Y, Fujii K et al . Rapid determination of Epstein–Barr virus‐specific CD8 (+) T‐cell frequencies by flow cytometry. Blood 1999; 94: 3094–100. [PubMed] [Google Scholar]
- 35. Zhang Y, Nagata H, Ikeuchi T et al . Common cytological and cytogenetic features of Epstein–Barr virus (EBV)‐positive natural killer (NK) cells and cell lines derived from patients with nasal T/NK‐cell lymphomas, chronic active EBV infection and hydroa vacciniforme‐like eruptions. Br J Haematol 2003; 121: 805–14. [DOI] [PubMed] [Google Scholar]
- 36. Oyoshi MK, Nagata H, Kimura N et al . Preferential expansion of Vgamma9‐JgammaP/Vdelta2‐Jdelta3 gammadelta T cells in nasal T‐cell lymphoma and chronic active Epstein–Barr virus infection. Am J Pathol 2003; 162: 1629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kondo E, Topp MS, Kiem HP et al . Efficient generation of antigen‐specific cytotoxic T cells using retrovirally transduced CD40‐activated B cells. J Immunol 2002; 169: 2164–71. [DOI] [PubMed] [Google Scholar]
- 38. Schultze JL, Michalak S, Seamon MJ et al . CD40‐activated human B cells: an alternative source of highly efficient antigen presenting cells to generate autologous antigen‐specific T cells for adoptive immunotherapy. J Clin Invest 1997; 100: 2757–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Akatsuka Y, Goldberg TA, Kondo E et al . Efficient cloning and expression of HLA class I cDNA in human B‐lymphoblastoid cell lines. Tissue Antigens 2002; 59: 502–11. [DOI] [PubMed] [Google Scholar]
- 40. Blake N, Lee S, Redchenko I et al . Human CD8+ T cell responses to EBV EBNA1: HLA class I presentation of the (Gly‐Ala)‐containing protein requires exogenous processing. Immunity 1997; 7: 791–802. [DOI] [PubMed] [Google Scholar]
- 41. Kuzushima K, Hayashi N, Kimura H, Tsurumi T. Efficient identification of HLA‐A*2402‐restricted cytomegalovirus‐specific CD8 (+) T‐cell epitopes by a computer algorithm and an enzyme‐linked immunospot assay. Blood 2001; 98: 1872–81. [DOI] [PubMed] [Google Scholar]
- 42. Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee HG. Pool sequencing of natural HLA‐DR, DQ, and DP ligands reveals detailed peptide motifs, constraints of processing, and general rules. Immunogenetics 1994; 39: 230–42. [DOI] [PubMed] [Google Scholar]
- 43. Rudensky Ayu Preston‐Hurlburt P, Hong SC, Barlow A, Janeway CA Jr. Sequence analysis of peptides bound to MHC class II molecules. Nature 1991; 353: 622–7. [DOI] [PubMed] [Google Scholar]
- 44. Khanna R, Burrows SR, Steigerwald‐Mullen PM, Thomsom SA, Kurilla MG, Moss DJ. Isolation of cytotoxic T lymphocytes from healthy seropositive individuals specific for peptide epitopes from Epstein–Barr virus nuclear antigen 1: implications for viral persistence and tumor surveillance. Virology 1995; 214: 633–7. [DOI] [PubMed] [Google Scholar]
- 45. Kruger S, Schroers R, Rooney CM, Gahn B, Chen SY. Identification of a naturally processed HLA‐DR‐restricted T‐helper epitope in Epstein–Barr virus nuclear antigen type 1. J Immunother 2003; 26: 212–21. [DOI] [PubMed] [Google Scholar]
- 46. Long HM, Haigh TA, Gudgeon NH et al . CD4+ T‐cell responses to Epstein–Barr virus (EBV) latent‐cycle antigens and the recognition of EBV‐transformed lymphoblastoid cell lines. J Virol 2005; 79: 4896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Omiya R, Buteau C, Kobayashi H, Paya CV, Celis E. Inhibition of EBV‐induced lymphoproliferation by CD4 (+) T cells specific for an MHC class II promiscuous epitope. J Immunol 2002; 169: 2172–9. [DOI] [PubMed] [Google Scholar]
- 48. Khanna R, Burrows SR, Thomsom SA et al . Class I processing‐defective Burkitt's lymphoma cells are recognized efficiently by CD4+ EBV‐specific CTLs. J Immunol 1997; 158: 3619–25. [PubMed] [Google Scholar]