

Abstract
In normal tissue, cell division is carefully regulated to maintain the correct proliferative balance. Abnormal cell division underlies many hypoproliferative and hyperproliferative disorders, including cancer, and a better understanding of the mechanisms involved could lead to new strategies for treatment and prevention. Cellular senescence, a state of irreversible growth arrest, was first described as a limit to the replicative life span of somatic cells after serial cultivation in vitro. Recently, however, it has also been shown to be triggered prematurely by potentially oncogenic stimuli such as oncogene expression, oxidative stress, and DNA damage in cell culture studies. These data suggest that cellular senescence is therefore acting as a tumor‐protective fail‐safe mechanism. However, the significance of cellular senescence has remained an issue of debate over the years, with the possibility that it might be a cell culture‐related artifact. Recent reports on oncogene‐induced senescence detected in premalignant tumors have provided evidence to validate its role as a physiological response to prevent oncogenesis in vivo. In this review, we discuss the mechanisms for cellular senescence and its roles in vivo. (Cancer Sci 2009; 100: 792–797)
In contrast to germ cells and certain stem cells, most somatic cells permanently stop dividing after a finite number of cell divisions in culture and enter a state termed cellular or replicative senescence. These cells are irreversibly arrested in the G1 phase of the cell cycle and are no longer able to divide despite remaining viable and metabolically active for long periods of time, thereby distinguishing senescence from apoptotic cell death. The finite replicative life span of normal cells in culture was first described approximately 50 years ago by Hayflick, and is often termed the ‘Hayflick limit’.( 1 ) Most tumors contain cells that appear to have bypassed this limit and evaded senescence. Immortality, or even an extended replicative lifespan, greatly increases susceptibility to malignant progression because it permits the extensive cell divisions that might acquire successive mutations. Therefore, cellular senescence may act as a barrier to cancer and play an important role in tumor suppression (Fig. 1).( 2 ) It could also be responsible for aspects of the aging process.( 2 )
Figure 1.
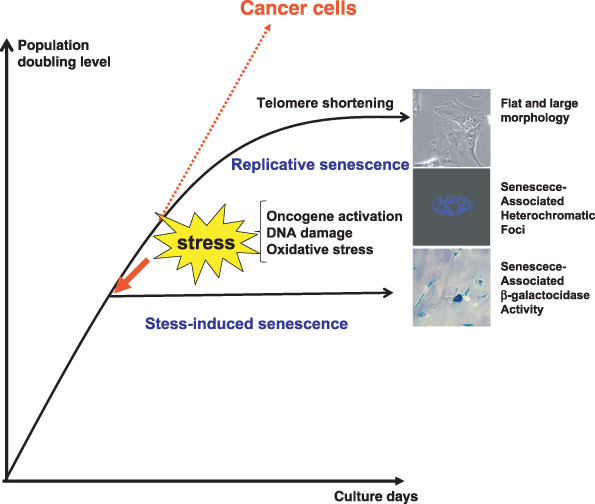
Cellular senescence. Normal human fibroblasts enter a state of irreversible growth arrest after a finite number of cell divisions in vitro caused by telomere shortening but cancer cells appear to bypass this replicative limit and proliferate indefinitely. Recent reports have shown that cellular senescence can also be induced prematurely by a number of cellular stresses such as oncogenic stimuli, oxidative stress, and DNA damage, before reaching their limits of replicative life span. This type of cellular senescence is called ‘stress‐induced senescence’. Senescent cells are characterized by a large and flat morphology, senescence‐associated acidic β‐galactosidase activity, and senescence‐associated heterochromatic foci.
A number of hypotheses have been proposed to explain the mechanisms of cellular senescence, and they can be grouped into two categories: one set proposes that the loss of proliferative potential is due to random accumulation of damage or stress, for example through inappropriate culture conditions, whereas the other proposes that it is a genetically programmed process. Although much evidence suggests that senescence in human cells is genetically controlled through a cell division counting mechanism caused by telomere shortening,( 3 ) recent reports strongly suggest that cellular senescence can also be induced by inappropriate tissue culture stresses such as continuous mitogenic stimulation, exposure to high oxygen concentration, and DNA damage.( 4 ) However, recent evidence has begun to indicate that cellular senescence does in fact also take place in vivo, playing important roles in tumor suppression, aging, vascular diseases, and fibrosis.( 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 ) We are just at the beginning of understanding the physiological roles of cellular senescence in vivo. In this review, we will focus on the current knowledge of the molecular mechanisms leading to cellular senescence and its roles in vivo. ( 2 , 14 )
Telomere‐dependent and telemore‐independent senescence
At the end of eukaryotic chromosomes, special DNA structures that consist of repetitive DNA elements can be found. These are called telomeres and protect the DNA ends from degradation and recombination. Mammalian telomeres are associated with six core proteins forming the shelterin complex that serves to protect telomeric ends.( 15 ) Telomere length is maintained by a specific enzyme called telomerase, which is not expressed in most normal human somatic cells. Due to the nature of the DNA replication process and the lack of telomerase, telomeres become progressively shorter with every round of cell division.( 16 ) Critical telomere shortening or uncapping of telomere binding proteins result in telomere dysfunction and this is thought to initiate DNA damage response signals to activate p53‐dependent checkpoints that contribute to either cellular senescence or apoptosis.( 15 , 17 ) Telomere length therefore functions as a mitotic clock, counting the number of cell divisions that human somatic cells undergo. In this regard, introduction of telomerase extends the replicative life span of normal human cells.( 18 ) By using telomerase‐deficient mice,( 19 ) telomere shortening has been shown to be involved in a disease of progeria,( 20 ) and telomerase also appears to be essential for the maintenance of self‐renewing of stem cells.( 21 ) In contrast, mice genetically overexpressing telomerase reverse transcriptase have been shown to possess anti‐aging activity, illustrating the importance of telomerase in organismal aging.( 22 ) Although telomerase expression is strictly regulated in normal human somatic cells, in tumor cells its expression is frequently deregulated. Although the mean length of telomeres is reported to be reduced in various cancer cells, the length of telomeres is maintained either by telomerase expression or by a mechanism known as alternative lengthening of telomeres in cancer cells.( 23 , 24 ) It is clear that telomere maintenance is essential for cellular immortality and thus telomerase offers a possible therapeutic target in human cancer.
In contrast to human cells, there is no strong evidence in rodent cells that replicative senescence is dependent on telomere erosion.( 3 , 25 ) Telomeres in rodent cells are quite long, and many somatic rodent tissues and cultured cells have telomerase activity. Moreover, the proliferative block in rodent cells occurs without detectable telomere shortening. This telomere‐independent proliferation block, which can also occur in human cells, may reflect a cell cycle checkpoint response to inappropriate culture conditions rather than an intrinsic limitation imposed by a cell division counting mechanism. In this regard, it is interesting to note that primary mouse embryonic fibroblasts have been shown to proliferate indefinitely if maintained in appropriate culture conditions, such as low oxygen conditions.( 26 ) Also, rat oligodendrocyte precursor cells and rat Schwann cells do not senesce in serum‐free medium, but serum addition induces senescence.( 27 ) Even in human cells, substantial extended replicative life span was observed when cells were cultured in non‐serum‐based medium,( 28 ) or under low oxygen conditions.( 29 ) These findings clearly demonstrate that cellular senescence can be induced without apparent telomere shortening when cells are exposed to non‐physiological circumstances in vitro.
Moreover, aberrant growth signaling from activated Ras signaling pathways is known to rapidly induce a senescence‐like proliferative arrest in normal human fibroblasts.( 30 ) There are also reports that oxidative stress( 2 ) and DNA damage( 31 ) accelerate the onset of cellular senescence in human fibroblasts. Taken together, this evidence suggests that senescence can be induced by a variety of physiological stresses, via a process that is now called ‘stress‐induced senescence’.( 4 ) It is plausible that the elevated level of reactive oxygen species (ROS) triggers culture stress‐derived cellular senescence because treatment of antioxidants to cultured cells reduces the induction of Ras‐induced cellular senescence,( 32 ) and culturing cells in low oxygen conditions is known to extend their replicative life span in both human cells and mouse cells.( 26 , 29 )
Role of cyclin‐dependent kinase inhibitor proteins in cellular senescence
In both human and rodent cells, the retinoblastoma (Rb) and p53 tumor‐suppressor proteins are crucial gate keepers of cellular senescence.( 33 , 34 ) The activities of Rb and p53 are highly regulated by phosphorylation, protein–protein interactions, and protein stability.( 35 , 36 ) The cyclin‐dependent kinases (CDKs) CDK4, CDK6, and CDK2 play critical roles in controlling Rb activity. When Rb is phosphorylated by these CDKs, it loses its ability to bind to and repress the functions of the E2F family of transcription factors, resulting in gene transactivation allowing the initiation of DNA replication (2, 3).( 37 , 38 ) This process requires strict regulation of the CDK in a cell cycle‐dependent manner. p16Ink4a( 39 ) and the p53 target p21Waf1/Cip1( 40 ) are CDKs inhibitor proteins that have crucial functions in regulating CDKs and inducing the onset of cellular senescence.( 30 , 41 , 42 ) Simultaneous activation of p21Waf1/Cip1, and p16Ink4a constitutively activates the Rb protein,( 43 , 44 ) and cell cycle progression is irreversibly arrested, thereby allowing the state of cellular senescence( 45 ) (Fig. 2). In fact, both the p16Ink4a–Rb and p53–p21Waf1/Cip1 pathways are known to be frequently inactivated in human cancers,( 46 , 47 ) illustrating the importance of disrupting this cell cycle arrest to the tumorigenic process.
Figure 2.
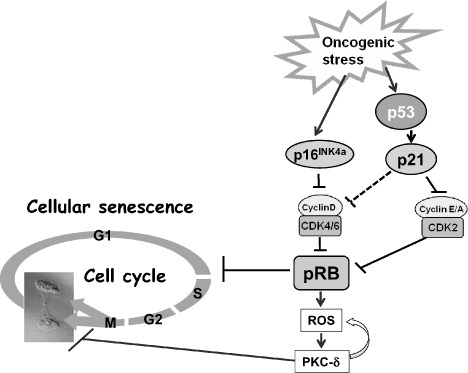
Molecular mechanisms of cellular senescence. Oncogenic stress induces p16 and the p53‐target p21. When protein retinoblastoma (pRb) is fully activated by high‐level expression of p16INK4a, mitogenic signals, in turn, increase the level of reactive oxygen species (ROS) and elicit a positive feedback activation of the ROS–PKC‐δ signaling pathway. Elevated levels of p16INK4a therefore establish the autonomous activation of ROS–PKC‐δ signaling, leading to an irrevocable block to cytokinesis in human senescent cells. CDK, cyclin‐dependent kinase.
Figure 3.
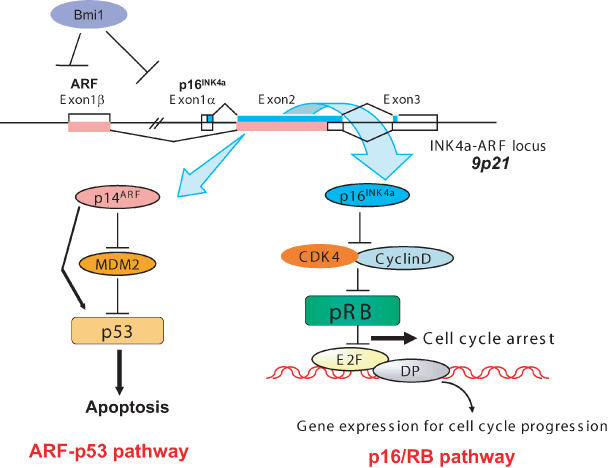
Ink4a/Arf locus. The p16INK4a gene is located in the Ink4a/Arf locus in human chromosome 9p21, and this locus encodes not only p16INK4a but also Arf via a shared second exon using a different translational reading frame. These two protein products participate in major tumor‐suppressor networks that are inactivated in human cancer. p16INK4a binds directly to and inhibits the activity of cyclin‐dependent kinase (CDK) 4 and CDK6 and hence activates the retinoblastoma (RB) tumor‐suppressor protein whereas ARF binds directly to mouse double minutes (MDM2) resulting in the stabilization and activation of the p53 tumor suppressor. This locus is repressed by polycomb proteins such as Bmi1 (B lymphoma Mo‐MLV insertion region 1). DP, DRTP1‐polypeptide‐1.
Unlike mouse embryonic fibroblasts,( 48 , 49 ) in human somatic cells, once Rb is fully engaged, particularly by its activator p16Ink4a, senescent cell cycle arrest becomes irreversible and is no longer revoked by subsequent inactivation of Rb and p53.( 50 , 51 ) Interestingly, subsequent inactivation of Rb and p53 enables human senescent cells to reinitiate DNA synthesis, but they subsequently fail to complete the cell cycle, suggesting that these cells may be arrested in G2 or M phase.( 50 ) Recently, we have found that cytokinesis is irreversibly blocked after Rb and p53 are subsequently inactivated in p16Ink4a‐expressing human senescent cells.( 52 ) We have also shown that a strongly activated p16Ink4a–Rb pathway cooperates with mitogenic signals to enforce an irreversible cytokinesis block through causing the production of ROS. In this setting, mitogenic signaling, in turn, increases ROS production, thereby activating protein kinase C (PKC)δ, a critical downstream mediator of the ROS signaling pathway.( 52 , 53 ) Importantly moreover, once PKCδ is activated by ROS, PKCδ promotes further generation of ROS, thus establishing a positive feedback loop to sustain ROS–PKCδ signaling.( 52 ) In senescent human cells, sustained activation of ROS–PKCδ signaling irreversibly blocks cytokinesis, at least partly through reducing the level of WARTS (also known as large tumor suppressor (LATS)1), a mitotic exit network kinase required for cytokinesis.( 52 , 54 ) Thus, elevated levels of p16Ink4a establish autonomous activation of ROS–PKCδ signaling, leading to an irrevocable block to cytokinesis in human senescent cells. These findings demonstrate that multiple cell cycle check points function to irreversibly arrest cell cycle entry in senescent cells (Fig. 2). Recently, the Akt signaling pathway has been reported to induce premature senescence,( 55 , 56 ) with elevated levels of ROS under mitogenic stimulation.( 56 ) Their findings are consistent with previous reports that the ROS level is upregulated in cellular senescence,( 4 , 26 , 32 ) and could function as a tumor suppressor.( 52 , 57 )
Ink4a/Arf locus
The p16Ink4a gene is located in the Ink4a/Arf locus in human chromosome 9 (chromosome 4 for mouse),( 39 , 58 , 59 ) and this locus encodes not only the p16Ink4a gene but also the Arf gene (p19 for mouse and p14 for human) by reading a shared second exon in a different translational reading frame.( 60 , 61 ) These two products participate in the major tumor‐suppressor networks that are frequently inactivated in human cancer. p16Ink4a binds directly to and inhibits the activity of CDK4 and CDK6, thereby activating the Rb tumor‐suppressor protein, whereas Arf binds directly to and inhibits mouse double minutes (MDM2), resulting in the stabilization and activation of the p53 tumor suppressor (Fig. 3).( 61 , 62 )
p16Ink4a appears to be more important in cellular senescence and tumor suppression in human cells, whereas Arf is likely to have a more prominent role in mouse cells. For mice, the most convincing evidence has come from the characterization of knockout mice in which individual exons within the Ink4a/Arf locus have been specifically knocked out by homologous recombination. Mice that are defective for either of the genes are cancer prone, although the degrees of tumorigenesis are different.( 63 , 64 , 65 ) The case for Arf as a tumor suppressor is perhaps the strongest in mice, but comparison of the phenotype of the knockout mice in the same genetic background has shown that whereas p16Ink4a‐null and Arf‐null mice are both tumor prone, the effects are strongest in mice that lack both genes.( 66 ) The reason why the cancer‐prone phenotype is milder in p16Ink4a‐null mice than in Arf‐null mice( 66 ) could be because other Ink4 family proteins such as p15Ink4b and p18Ink4c could compensate for the lack of functional p16Ink4a.( 67 , 68 , 69 ) On the other hand, the studies of point mutations in human cancers, including melanoma families, implicate the p16Ink4a gene as being more important than the Arf gene.( 47 , 70 ) Also, p16Ink4a is highly induced in primary human cells by oncogenic Ras but Arf is not, whereas both genes are induced in murine senescent cells.( 71 ) These results suggest that overall, the p16Ink4a gene is more responsible in inducing cellular senescence and tumor suppression in human cells.
Interestingly, the Ink4a/Arf locus is known to be repressed by the polycomb proteins B lymphoma Mo‐MLV insertion region 1 (Bmi‐1) and related family members in both human and mouse cells.( 72 ) Overexpression of Bmi‐1 in human primary cells appears to extend the replicative life span of primary cells by inhibiting p16Ink4a function.( 73 ) Recently it was reported that repression of the Ink4a/Arf locus by polycomb proteins is mediated by Rb protein and histone H3K27 methylation at the Ink4a/Arf locus.( 74 , 75 )
Cellular senescence as a barrier to tumorigenesis in vivo
Much of our current knowledge of p16Ink4a gene regulation derives from tissue culture studies. However, because emerging evidence has begun to indicate that p16Ink4a gene expression is also induced in response to tissue culture stress, it is difficult to understand the physiological relevance of p16Ink4a expression using such a system; in vivo studies are likely to provide far greater insight. Recently, a number of reports have shown that cellular senescence is induced in premalignant tumors, but is rare in more advanced malignant tumors.( 5 , 6 , 7 , 8 ) These observations strongly argue in favor of cellular senescence being an important in vivo physiological response to prevent tumor development and provide evidence that it is not simply a cell culture artifact. In addition, these reports clearly rebut the view that oncogene‐induced senescence may occur only when the driving mitogenic oncogene is expressed at supraphysiological levels. Thus, these results open the possibility of using senescence markers as diagnostic and prognostic tools and illustrate the potential for senescence‐inducing drugs as anticancer agents.
One additional intriguing piece of evidence highlights a link between oncogene‐induced senescence and inflammation and its potential link to cancer. Recent studies have revealed an unexpected function of the inflammatory pathways in inducing cellular senescence.( 76 , 77 , 78 ) These studies used function‐based screens and demonstrated that cells undergoing oncogene‐induced senescence produce inflammatory cytokines such as interleukin (IL)‐6 and IL‐8. These inflammatory cytokines appear to play an essential role in the induction and maintenance of cellular senescence. On the other hand, Campisi's group reported that senescence‐associated secretory phenotypes, which involve secretion of the same cytokines, promote oncogenesis and the epithelial to mesenchymal transition, a response characteristic of metastatic tumors.( 79 ) They speculate that an inflammatory microenvironment promotes the development of cancer.( 79 ) Altogether, these findings imply that inflammation acts as a double‐edged sword in preventing and promoting oncogenesis.
Cellular senescence in aging in vivo
Several groups have suggested a role for the senescence machinery in mammalian aging in vivo. For example, excess p53 activity has been shown to induce premature aging in mice in multiple tissue types.( 80 ) Sharpless and his colleagues reported that the expression of p16Ink4a and Arf is markedly increased in almost all rodent tissues with aging, whereas there is little or no change in the expression of other INK4 family members.( 81 ) In addition, the age‐related increase in p16Ink4a/Arf expression is accompanied by the strongly correlated expression of v‐ets erythroblastosis virus E26 oncogene homolog (Ets)‐1,( 81 ) a known p16Ink4a transcriptional activator.( 82 ) They suggest that expression of the Ink4a/Arf tumor‐suppressor locus is a robust biomarker and a possible effector of mammalian aging. Also, there is a report that Ets‐1 isoform‐deficient mice demonstrate lymphocyte maturation defects associated with downregulation of p16Ink4a expression.( 83 ) It is interesting to note that other Ets family transcription factors, such as Ets‐2( 82 ) and ESE‐3, an enterocyte‐specific Ets transcription factor,( 84 ) also upregulate p16Ink4a gene expression during the process of cellular senescence, suggesting that Ets family transcription factors play an important role in controlling p16Ink4a gene expression.
Recently, Baker et al. showed that hypomorphic budding uninhibited by benzimidazoles 1‐related kinase (BubR1) mutant mice have high levels of p16Ink4a and Arf in prematurely aged skeletal muscle and fat,( 85 ) confirming that the expression of p16Ink4a and Arf are biomarkers of aging. However, surprisingly, inactivation of p16Ink4a in BubR1‐insufficient mice attenuates both cellular senescence and premature aging in these tissues but, conversely, Arf inactivation exacerbates senescence and aging in BubR1 mutant mice. These results show that BubR1 insufficiency is a trigger for activation of the Ink4a/Arf locus and that p16Ink4a is an effector and Arf an attenuator of senescence and aging in these tissues although the molecular mechanisms underlying this remain unclear.( 85 )
Several lines of evidence suggest that higher eukaryotes age, in part, because our self‐renewing stem cells become senescent due to p16Ink4a upregulation. Ito et al. reported that self‐renewal of hematopoietic stem cells is severely impaired due to elevated ROS in ataxia telangiectasia mutated (ATM) knockout mice, resulting in the upregulation of p16Ink4a.( 86 ) Moreover, recent papers have shown that self‐renewal of stem cells is highly retained in p16Ink4a‐deficient mice, whereas it is severely attenuated in wild‐type mice in advancing age.( 9 , 10 , 11 ) These genetic data support the view that an age‐induced increase in p16Ink4a expression limits the regenerative capacity of tissue‐specific stem cells with aging.
Even though the expression of p53 or p16Ink4a has the effect of accelerating aging, these genes are beneficial with respect to tumor suppression. Recently, Serrano's group generated bacterial artificial chromosome‐based transgenic mice with a single extra dose of chromosomal fragment containing the p53 and Ink4a/Arf loci.( 87 ) The expression of both tumor suppressors from these transgenes under physiological regulation protects mice against cancer and these mice also showed delayed aging.( 87 ) These data indicate that the naturally regulated response from the extra dose of these tumor suppressors is tumor protective and anti‐aging.
Cellular senescence of non‐cancerous pathologies in vivo
It is now clear that cellular senescence is important as a potent mechanism of tumor suppression in vivo. Recently, links between cellular senescence and non‐cancerous pathologies have also been emerging. Minamino and his colleagues reported that senescent cells were detected in human atherosclerotic plaques in coronary arteries obtained from patients who had ischemic heart disease.( 88 ) Yao et al. recently published that pulmonary damage by cigarette smoke is reduced in p21 knockout mice, suggesting that p21 expression by cigarette smoke in pulmonary epithelium mediates inflammatory damage‐induced senescence that could cause chronic obstructive pulmonary diseases.( 89 ) Also in the liver, Krizhanovsky et al. showed that senescent cells accumulated in murine livers treated to produce fibrosis, a precursor pathology to cirrhosis.( 13 ) The senescent cells are derived primarily from activated hepatic stellate cells that initially proliferate in response to liver damage and produce the extracellular matrix deposited in the fibrotic scar. In mice lacking key senescence regulators, such as p53 and INK4a/Arf, stellate cells continue to proliferate, leading to excessive liver fibrosis. Therefore, the cellular senescence in liver stellate cells seems to limit the fibrogenic response to acute tissue damage.( 13 )
Conclusions
In summary, it is now accepted that cellular senescence is induced by a number of cellular stresses such as oncogene activation, oxidative stress, and DNA damage in vitro and in vivo through elevated levels of ROS. Unlike programmed differentiation, cellular senescence is likely to be a stochastic event that is induced by a variety of genotoxic stresses. Recently, we developed a real‐time in vivo imaging system for visualizing the expression of senescence‐related genes, such as p21Waf1/Cip1 in mice (Fig. 4).( 90 ) Visualizing the dynamics of cellular senescence responses in vivo in the context of living animals is likely to be a useful tool in the identification of the location and timing of gene expression and hence their likely roles in cellular senescence in vivo.
Figure 4.
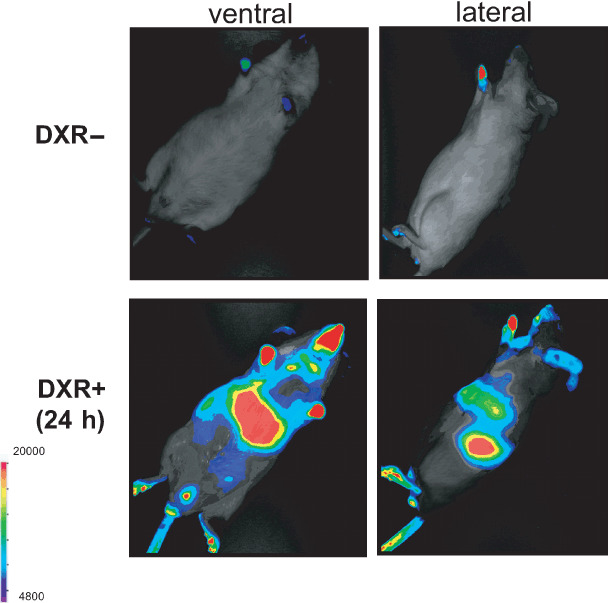
Real‐time in vivo imaging of p21Waf1/Cip1 gene expression after doxorubicin (DXR) treatment. We established a transgenic mouse line (p21‐p‐luc) expressing firefly luciferase under control of the p21Waf1/Cip1 gene promoter. The 8‐week‐old p21‐p‐luc mouse was injected intraperitoneally with DXR (20 mg/kg) and was subjected to non‐invasive bioluminescene imaging 24 h after DXR treatment under anesthesia. DXR treatment (lower panels) and its control (untreated mice) (upper panels). The color bar indicates photons with minimal and maximal threshold values.
References
- 1. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 1961; 25: 585–621. [DOI] [PubMed] [Google Scholar]
- 2. Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 9: 729–40. [DOI] [PubMed] [Google Scholar]
- 3. Wright WE, Shay JW. Telomere dynamics in cancer progression and prevention: fundamental differences in human and mouse telomere biology. Nat Med 2000; 6: 849–51. [DOI] [PubMed] [Google Scholar]
- 4. Serrano M, Blasco MA. Putting the stress on senescence. Curr Opin Cell Biol 2001; 13: 748–53. [DOI] [PubMed] [Google Scholar]
- 5. Collado M, Gil J, Efeyan A et al . Tumor biology: senescence in premalignant tumors. Nature 2005; 436: 642. [DOI] [PubMed] [Google Scholar]
- 6. Braig M, Lee S, Loddenkemper C et al . Oncogene‐induced senescence as an initial barrier in lymphoma development. Nature 2005; 436: 660–5. [DOI] [PubMed] [Google Scholar]
- 7. Michaloglou C, Vredeveld LC, Soengas MS et al . BRAFE600‐associated senescence‐like cell cycle arrest of human naevi. Nature 2005; 436: 720–4. [DOI] [PubMed] [Google Scholar]
- 8. Chen Z, Trotman LC, Shaffer D et al . Crucial role of p53‐dependent cellular senescence in suppression of Pten‐deficient tumorigenesis. Nature 2005; 436: 725–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Janzen V, Forkert R, Fleming HE et al . Stem‐cell ageing modified by the cyclin‐dependent kinase inhibitor p16INK4a . Nature 2006; 443: 421–6. [DOI] [PubMed] [Google Scholar]
- 10. Molofsky AV, Slutsky SG, Joseph NM et al . Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 2006; 443: 448–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krishnamurthy J, Ramsey MR, Ligon KL et al . p16INK4a induces an age‐dependent decline in islet regenerative potential. Nature 2006; 443: 453–7. [DOI] [PubMed] [Google Scholar]
- 12. Minamino T, Komuro I. Vascular aging: insights from studies on cellular senescence, stem cell aging, and progeroid syndromes. Nat Clin Pract Cardiovasc Medical 2008; 5: 637–48. [DOI] [PubMed] [Google Scholar]
- 13. Krizhanovsky V, Yon M, Dickins RA et al . Senescence of activated stellate cells limits liver fibrosis. Cell 2008; 134: 657–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigensis. Curr Opin Cell Biol 2008; 20: 150–5. [DOI] [PubMed] [Google Scholar]
- 15. Deng Y, Chan SS, Chang S. Telomere dysfunction and tumour suppression: the senescence connection. Nat Rev Cancer 2008; 8: 450–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature 1990; 345: 458–60. [DOI] [PubMed] [Google Scholar]
- 17. de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 2005; 19: 2100–10. [DOI] [PubMed] [Google Scholar]
- 18. Bodnar AG, Ouellette M, Frolkis M et al . Extension of life‐span by introduction of telomerase into normal human cells. Science 1998; 279: 349–52. [DOI] [PubMed] [Google Scholar]
- 19. Blasco MA, Lee HW, Hande MP et al . Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 1997; 91: 25–34. [DOI] [PubMed] [Google Scholar]
- 20. Chang S, Multani AS, Cabrera NG et al . Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet 2004; 36: 877–82. [DOI] [PubMed] [Google Scholar]
- 21. Choudhury AR, Ju Z, Djojosubroto MW et al . Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat Genet 2007; 39: 99–105. [DOI] [PubMed] [Google Scholar]
- 22. Tomás‐Loba A, Flores I, Fernández‐Marcos PJ et al . Telomerase reverse transcriptase delays aging in cancer‐resistant mice. Cell 2008; 135: 609–22. [DOI] [PubMed] [Google Scholar]
- 23. Neumann AA, Reddel RR. Telomere maintenance and cancer – look, no telomerase. Nat Rev Cancer 2002; 2: 879–84. [DOI] [PubMed] [Google Scholar]
- 24. Shay JW, Wright WE. Hallmarks of telomeres in ageing research. J Pathol 2007; 211: 114–23. [DOI] [PubMed] [Google Scholar]
- 25. Sherr CJ, Depinho RA. Cellular senescence: mitotic clock or culture shock? Cell 2000; 102: 407–10. [DOI] [PubMed] [Google Scholar]
- 26. Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 2003; 5: 741–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lloyd AC. Limits to lifespan. Nat Cell Biol 2002; 4: E25–7. [DOI] [PubMed] [Google Scholar]
- 28. Ince TA, Richardson AL, Bell GW et al . Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell 2007; 12: 160–70. [DOI] [PubMed] [Google Scholar]
- 29. Packer L, Fuehr K. Low oxygen concentration extends the lifespan of cultured human diploid cells. Nature 1977; 267: 423–5. [DOI] [PubMed] [Google Scholar]
- 30. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell cenescence associated with accumulation of p53 and p16INK4a . Cell 1997; 88: 593–602. [DOI] [PubMed] [Google Scholar]
- 31. von Zglinicki T, Saretzki G, Ladhoff J, d’Adda di Fagagna F, Jackson SP. Human cell senescence as a DNA damage response. Mech Ageing Dev 2005; 126: 111–17. [DOI] [PubMed] [Google Scholar]
- 32. Lee AC, Fenster BE, Ito H et al . Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem 1999; 274: 7936–40. [DOI] [PubMed] [Google Scholar]
- 33. Hara E, Tsurui H, Shinozaki A, Nakada S, Oda K. Cooperative effect of antisense‐Rb and antisense‐p53 oligomers on the extension of life span in human diploid fibroblasts, TIG‐1. Biochem Biophys Res Commun 1991; 179: 528–34. [DOI] [PubMed] [Google Scholar]
- 34. Shay JW, Pereira‐Smith OM, Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res 1991; 196: 33–9. [DOI] [PubMed] [Google Scholar]
- 35. Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer 2002; 2: 910–17. [DOI] [PubMed] [Google Scholar]
- 36. Sharpless NE, DePinho RA. p53: good cop/bad cop. Cell 2002; 110: 9–12. [DOI] [PubMed] [Google Scholar]
- 37. Stevaux O, Dyson NJ. A revised picture of the E2F transcriptional network and RB function. Curr Opin Cell Biol 2002; 14: 684–91. [DOI] [PubMed] [Google Scholar]
- 38. Maehara K, Yamakoshi K, Ohtani N et al . Reduction of total E2F/DP activity induces senescence‐like cell cycle arrest in cancer cells lacking functional pRB and p53. J Cell Biol 2005; 168: 553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell‐cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993; 366: 704–7. [DOI] [PubMed] [Google Scholar]
- 40. Hunter T. Braking the cycle. Cell 1993; 75: 839–41. [DOI] [PubMed] [Google Scholar]
- 41. Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Regulation of p16CDKN2 expression and its implication for cell immortalization and senescence. Mol Cell Biol 1996; 16: 859–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Noda A, Ning Y, Venable SF, Pereira‐Smith OM, Smith JR. Cloning of senescent cell‐derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res 1994; 211: 90–8. [DOI] [PubMed] [Google Scholar]
- 43. McConnell BB, Gregory FJ, Stott FJ, Hara E, Peters G. Induced expression of p16INK4a inhibits both CDK4‐ and CDK2‐associated kinase activity by reassortment of cyclin–CDK–inhibitor complexes. Mol Cell Biol 1999; 19: 1981–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mitra J, Dai CY, Somasundaram K et al . Induction of p21WAF1/CIP1 and inhibition of Cdk2 mediated by the tumor suppressor p16INK4a . Mol Cell Biol 1999; 19: 3916–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin‐dependent kinase inhibitor p16INK4a in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci USA 1996; 93: 13 742–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zambetti GP. The p53 mutation ‘gradient effect’ and its clinical implications. J Cell Physiol 2007; 213: 370–3. [DOI] [PubMed] [Google Scholar]
- 47. Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta 1998; 1378: F115–77. [DOI] [PubMed] [Google Scholar]
- 48. Sage J, Miller AL, Pérez‐Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re‐entry. Nature 2003; 424: 223–8. [DOI] [PubMed] [Google Scholar]
- 49. Dirac AM, Bernards R. Reversal of senescence in mouse fibroblasts through lentiviral suppression of p53. J Biol Chem 2003; 278: 11 731–4. [DOI] [PubMed] [Google Scholar]
- 50. Beauséjour CM, Krtolica A, Galimi F et al . Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003; 22: 4212–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dai CY, Enders GH. p16INK4a can initiate an autonomous senescence program. Oncogene 2000; 19: 1613–22. [DOI] [PubMed] [Google Scholar]
- 52. Takahashi A, Ohtani N, Yamakoshi K et al . Mitogenic signalling and the p16INK4a–Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol 2006; 8: 1291–7. [DOI] [PubMed] [Google Scholar]
- 53. Wheaton K, Riabowol K. Protein kinase C delta blocks immediate‐ early gene expression in senescent cells by inactivating serum response factor. Mol Cell Biol 2004; 24: 7298–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang X, Yu K, Hao Y et al . LATS1 tumour suppressor affects cytokinesis by inhibiting LIMK1. Nat Cell Biol 2004; 6: 609–17. [DOI] [PubMed] [Google Scholar]
- 55. Miyauchi H, Minamino T, Tateno K, Kunieda T, Toko H, Komuro I. Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21‐dependent pathway. EMBO J 2004; 23: 212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nogueira V, Park Y, Chen CC et al . Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 2008; 14: 458–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ramsey MR, Sharpless NE. ROS as a tumour suppressor? Nat Cell Biol 2006; 8: 1213–15. [DOI] [PubMed] [Google Scholar]
- 58. Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA. Deletions of the cyclin‐dependent kinase‐4 inhibitor gene in multiple human cancers. Nature 1994; 368: 753–6. [DOI] [PubMed] [Google Scholar]
- 59. Hussussian CJ, Struewing JP, Goldstein AM et al . Germline p16 mutations in familial melanoma. Nat Genet 1994; 8: 15–21. [DOI] [PubMed] [Google Scholar]
- 60. Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995; 83: 993–1000. [DOI] [PubMed] [Google Scholar]
- 61. Stott FJ, Bates S, James MC et al . The alternative product from the human CDKN2A locus, p14ARF, participates in a regulatory feedback loop with p53 and MDM2. EMBO J 1998; 17: 5001–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gil J, Peters G. Regulation of the INK4b–ARF–INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol 2006; 7: 667–77. [DOI] [PubMed] [Google Scholar]
- 63. Kamijo T, Zindy F, Roussel MF et al . Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF . Cell 1997; 91: 649–59. [DOI] [PubMed] [Google Scholar]
- 64. Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A. Loss of p16INK4a confers susceptibility to metastatic melanoma in mice. Nature 2001; 413: 83–6. [DOI] [PubMed] [Google Scholar]
- 65. Sharpless NE, Bardeesy N, Lee KH et al . Loss of p16INK4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001; 413: 86–91. [DOI] [PubMed] [Google Scholar]
- 66. Sharpless NE, Ramsey MR, Balasubramanian P, Castrillon DH, DePinho RA. Loss of p16INK4a with retention of p19Arf predisposes mice to tumorigenesis. Oncogene 2004; 23: 379–85. 14724566 [Google Scholar]
- 67. Krimpenfort P, Ijpenberg A, Song JY et al . p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature 2007; 448: 943–6. [DOI] [PubMed] [Google Scholar]
- 68. Ramsey MR, Krishnamurthy J, Pei XH et al . Expression of p16Ink4a compensates for p18Ink4c loss in cyclin‐dependent kinase 4/6‐dependent tumors and tissues. Cancer Res 2007; 67: 4732–41. [DOI] [PubMed] [Google Scholar]
- 69. Wiedemeyer R, Brennan C, Heffernan TP et al . Feedback circuit among INK4 tumor suppressors constrains human glioblastoma development. Cancer Cell 2008; 13: 355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Huot TJ, Rowe J, Harland M et al . Biallelic mutations in p16INK4a confer resistance to Ras‐ and Ets‐induced senescence in human diploid fibroblasts. Mol Cell Biol 2002; 22: 8135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wei W, Hemmer RM, Sedivy JM. Role of p14ARF in replicative and induced senescence of human fibroblasts. Mol Cell Biol 2001; 21: 6748–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb‐group gene bmi‐1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999; 397: 164–8. [DOI] [PubMed] [Google Scholar]
- 73. Itahana K, Zou Y, Itahana Y et al . Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi‐1. Mol Cell Biol 2003; 23: 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kotake Y, Cao R, Viatour P, Sage J, Zhang Y, Xiong Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4a tumor suppressor gene. Genes Dev 2007; 21: 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bracken AP, Kleine‐Kohlbrecher D, Dietrich N et al . The Polycomb group proteins bind throughout the INK4A‐ARF locus and are disassociated in senescent cells. Genes Dev 2007; 21: 525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008; 132: 363–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Acosta JC, O’Loghlen A, Banito A et al . Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008; 133: 1006–18. [DOI] [PubMed] [Google Scholar]
- 78. Kuilman T, Michaloglou C, Vredeveld LC et al . Oncogene‐induced senescence relayed by an interleukin‐dependent inflammatory network. Cell 2008; 133: 1019–31. [DOI] [PubMed] [Google Scholar]
- 79. Coppé JP, Patil CK, Rodier F et al . Senescence‐associated secretory phenotypes reveal cell‐nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLOS Biol 2008; 6: 2853–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tyner SD, Venkatachalam S, Choi J et al . p53 mutant mice that display early ageing‐associated phenotypes. Nature 2002; 415: 45–53. [DOI] [PubMed] [Google Scholar]
- 81. Krishnamurthy J, Torrice C, Ramsey MR et al . Ink4a/Arf expression is a biomarker of aging. J Clin Invest 2004; 114: 1299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ohtani N, Zebedee Z, Huot TJ et al . Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature 2001; 409: 1067–70. [DOI] [PubMed] [Google Scholar]
- 83. Higuchi T, Bartel FO, Masuya M et al . Thymomegaly, microsplenia, and defective homeostatic proliferation of peripheral lymphocytes in p51‐Ets1 isoform‐specific null mice. Mol Cell Biol 2007; 27: 3353–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fujikawa M, Katagiri T, Tugores A, Nakamura Y, Ishikawa F. ESE‐3, an Ets family transcription factor, is up‐regulated in cellular senescence. Cancer Sci 2007; 98: 1468–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Baker DJ, Perez‐Terzic C, Jin F et al . Opposing roles for p16INK4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol 2008; 10: 825–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ito K, Hirao A, Arai F et al . Regulation of oxidative stress by ATM is required for self‐renewal of haematopoietic stem cells. Nature 2004; 431: 997–1002. [DOI] [PubMed] [Google Scholar]
- 87. Matheu A, Maraver A, Klatt P et al . Delayed ageing through damage protection by the Arf/p53 pathway. Nature 2007; 448: 375–9. [DOI] [PubMed] [Google Scholar]
- 88. Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 2002; 105: 1541–4. [DOI] [PubMed] [Google Scholar]
- 89. Yao H, Yang SR, Edirisinghe I et al . Disruption of p21 attenuates lung inflammation induced by cigarette smoke, LPS, and fMLP in mice. Am J Respir Cell Mol Biol 2008; 39: 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ohtani N, Imamura Y, Yamakoshi K et al . Visualizing the dynamics of p21Waf1/Cip1 cyclin‐dependent kinase inhibitor expression in living animals. Proc Natl Acad Sci USA 2007; 104: 15 034–9. [DOI] [PMC free article] [PubMed] [Google Scholar]