

Abstract
Recently, it has been found that inappropriate expression of microRNAs (miRNAs) is strongly associated with carcinogenesis. In this study, we demonstrated that the expression of miRNAs (miRs) ‐143 and ‐145, the levels of which were previously shown to be reduced in colon cancers and various kinds of established cancer cell lines, was also decreased in most of the B‐cell malignancies examined, including chronic lymphocytic leukemias (CLL), B‐cell lymphomas, Epstein‐Barr virus (EBV)‐transformed B‐cell lines, and Burkitt lymphoma cell lines. All samples from 13 CLL patients and eight of nine B‐cell lymphoma ones tested exhibited an extremely low expression of miRs‐143 and ‐145. The expression levels of miRs‐143 and ‐145 were consistently low in human Burkitt lymphoma cell lines and were inversely associated with the cell proliferation observed in the EBV‐transformed B‐cell lines. Moreover, the introduction of either precursor or mature miR‐143 and ‐145 into Raji cells resulted in a significant growth inhibition that occurred in a dose‐dependent manner and the target gene of miRNA‐143 was determined to be ERK5, as previously reported in human colon cancer DLD‐1 cells. Taken together, these findings suggest that miRs‐143 and ‐145 may be useful as biomarkers that differentiate B‐cell malignant cells from normal cells and contribute to carcinogenesis in B‐cell malignancies by a newly defined mechanism. (Cancer Sci 2007; 98: 1914–1920)
Abbreviations:
- 5‐Aza
5‐Aza‐2′‐deoxycytidine
- C/EBP
CCAAT/enhancer binding protein
- CLL
chronic lymphocytic leukemia
- Ct
threshold cycle
- DLBCL
diffuse large B cell lymphoma
- DNMT‐1
DNA methyltransferase
- EBV
Epstein‐Barr virus
- EDTA
ethylenediaminetetraacetic acid
- ERK
extracellular signal regulated kinase
- FACS
fluorescence‐activated cell sorting
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- GAPDH
glyceraldehyde phosphate dehydrogenase
- HRP
horseradish peroxidase
- MALT
extranodal marginal zone B‐cell lymphoma of mucosa‐assciated lymphoid tissue
- MAPK
mitogen‐activated protein kinase
- miRNAs
microRNAs
- PBL
peripheral blood lymphocytes
- PBS
phosphate‐buffered saline
- PCR
polymerase chain reaction
- PLSD
protected least significant difference
- PVDF
polyvinylidene fluoride
- RISC
RNA‐induced silencing complex
- RT
reverse transcription
- SDS
sodium dodecyl sulfate
- TSA
Tricostatin A
- UTR
untranslated region.
MicroRNAs are endogenous ~22‐nt non‐coding RNAs that regulate gene expression by inhibiting the translation of mRNAs in a sequence‐specific manner.( 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 ) With more than 400 already identified, the human genome may contain up to 1000 miRNAs 8 (http://microrna.sanger.ac.uk/). Up to one‐third of human mRNAs are predicted to be miRNA targets.( 9 ) Each miRNA can target more than 200 transcripts directly or indirectly,( 10 , 11 ) whereas more than one miRNA can converge on a single mRNA target.( 9 , 12 , 13 , 14 , 15 ) Therefore, the potential regulatory circuitry afforded by miRNA is enormous. These findings support the notion that alterations of miRNAs copy number and their regulatory genes highly prevalent in cancer because of genomic aberrations is closely associated with carcinogenesis.
Recent increasing evidence shows that the expression of miRNA genes is deregulated in human cancer.( 16 , 17 , 18 , 19 ) Specific over‐ or underexpression has been shown to correlate with particular tumor types.( 20 , 21 , 22 , 23 , 24 ) miRNA overexpression can result in downregulation of tumor suppressor genes, whereas their underexpression can lead to oncogene upregulation.( 16 , 17 , 18 , 19 ) For example, let‐7, downregulated in lung cancer,( 25 , 26 , 27 ) suppresses Ras,( 26 ) miR‐15 and miR‐16, deleted or downregulated in CLL,( 28 ) suppress BCL2,( 29 ) miR‐17‐5p and miR‐20a control the balance of cell death and proliferation driven by the proto‐oncogene c‐Myc.( 30 ) Clear evidence indicates that miRNA polycistron miR‐17‐92 serves as an oncogene in lymphoma,( 23 ) and lung cancer,( 31 ) and that miR‐372 and miR‐373 are novel oncogenes in testicular germ cell tumors that act by numbing the p53 pathway.( 32 ) Thus, miRNA expression profiles may predict the outcome of disease.( 25 , 27 , 33 )
First, by differential hybridization using a DNA microarray for miRNAs and then by semi‐quantitative RT‐PCR analysis between tumor and non‐tumor tissues, we previously found that miRs‐143 and ‐145, whose genes are located within 1.8 kb of each other in the chromosome 5q32 region, were downregulated in colon cancer,( 34 ) as also reported by Michael et al.( 35 ) Furthermore, all of the various kinds of human cancer cell lines tested exhibited an extremely low‐expression of miRs‐143 and ‐145, whereas the normal tissues in which they originate showed a good expression of both.( 34 )
In the present study, we investigated the expression of miRs‐143 and ‐145 in hematopoietic malignancies, and found that the expression levels of both miRs were significantly decreased in B‐cell malignancies, thus suggesting that they are good markers for B‐cell malignancies, especially for CLL in combination with miRs‐15 and ‐16.( 28 ) Furthermore, the expression levels of miRs‐143 and ‐145 were consistently low in Burkitt lymphoma cell lines and were inversely related to the growth of EBV‐transformed cell lines. The transfection experiment of Raji cells with either precursor miR‐143 or ‐145 demonstrated that both miRs negatively contributed to the cell growth.
Materials and Methods
Patients and tissue preparation. All human blood and lymph node samples were obtained from patients who had undergone collection for diagnosis at Nagoya University Hospital and its collaborating hospitals in Nagoya, Aichi Prefecture. Human tonsils were also obtained by tonsilectomy. Informed consent in writing was obtained from each patient. Collection and distribution of the samples were approved by the appropriate Institution Review Board. The patients comprised 13 cases of chronic lymphocytic leukemia (CLL) and nine cases of B‐cell lymphoma (8DLBCL, 1MALT). Pathological review and FACS analysis showed B‐cell non‐Hodgkin lymphoma. PBL from three healthy donors were used as normal controls. Fresh lymphoma biopsy specimens and tonsils obtained by operation were gently minced over a wire mesh screen to obtain a cell suspension, which was then centrifuged over Ficoll‐Hypaque (Amersham Biosciences AB, Uppsala, Sweden). CD19+ B‐cells from the tonsils were used as control cells. Purity was assessed, and isolation carried out by FACS. All samples were prepared by gravity centrifugation through Ficoll‐Hypaque and then frozen in liquid nitrogen until the experiments could be carried out. Such thawed specimens were used for the extraction of total RNA.
Cell culture, viability and treatment with 5‐Aza‐2′‐deoxycytidine or tricostatin A. Human Burkitt cell lines Raji, Daudi, P3, and KHM‐10B; and human EBV‐transformed cells IC, L11, L22, and L25 were grown in RPMI‐1640 medium supplemented with 10% (v/v) heat‐inactivated FBS (Sigma, St. Louis, MO, USA) and 2 mM L‐glutamine under an atmosphere of 95% air and 5% CO2 at 37°C.
The number of viable cells was determined by use of the trypan‐blue dye exclusion test. 5‐Aza (Sigma) was used for demethylation of DNA and histone. TSA (Sigma), a histone deacetylase inhibitor, was also used to examine the effect of acetylation of histone on miR‐143 and ‐145 expression. The cells were treated with these agents for 18 h at various concentrations.
Quantitative RT‐PCR and genomic PCR. Total RNA was isolated from the cells by the phenol/guanidium thiocyanate method with DNase I treatment. To determine the expression of miRNAs by semi‐quantitative RT‐PCR, we measured their levels by using a mirVana™ qRT‐PCR miRNA Detection Kit (Ambion, Austin, TX, USA) and mirVana qRT‐PCR Primer set (Ambion). Briefly, after reverse transcription of 50 ng of total RNA, cDNA was generated. The PCR reaction consisted of 22 cycles (95°C for 15 s, 60°C for 30 s) after an initial denaturation step (95°C for 3 min). The cycle number was initially determined by quantitative PCR. The PCR primer pairs for miRs‐143, ‐145, and ‐15a were obtained commercially from Ambion. The PCR products obtained by using such primer pairs were confirmed to be from loci of miRs‐143 and ‐145 by DNA sequencing. U6 was used as a control and was determined in each case. In addition, in order to examine the expression level in detail we also carried out TaqMan® MicroRNA Assays using real‐time PCR.( 16 ) The Ct is defined as the fractional cycle number at which the fluorescence passes a fixed threshold. miR‐143 and miR145 concentration in each type of cell were measured and were normalized to U6, which was used as an internal control. To determine the level of ERK5 mRNA we prepared cDNA from the total RNA samples by using a PCR purification kit (Qiagen, Hilden, Germany) and used them for PCR (Takara, Ohtsu, Japan). The primers for ERK5 were as follows: ERK5‐sense‐211, 5′‐CCTTCGATGTGACCTTTGAC‐3′; and ERK5‐antisense‐1418, 5′‐TGACACCATTGATCTGACCC‐3′. To examine the presence of the genomic loci of miRs‐143 and ‐145, we extracted DNA from the cell lines tested and used it for PCR (Takara). The primers for genomic loci of miRs‐143 and ‐145 were as follow: 5q32‐sense, 5′‐TTGGTCCTGGGTGCTCAAAT‐3′; and 5q32‐antisense, 5′‐AGGAACTCCCAAGCTCAAGT‐3′. The primers amplified the DNA fragment including both loci at 5q32. The genomic locus of GAPDH was used as an internal control. The PCR reaction consisted of 30 cycles (94°C for 30 s, 57.5°C for 1 min, 72°C for 1 min) after an initial denaturation step (95°C for 1 min). The PCR products were analyzed by electrophoresis on 2% agarose gels.
Transfection of Raji cells with precursor or mature miR‐143 and ‐145 miRNAs. Raji cells were seeded in six‐well plates at a concentration of 1–2 × 105/well on the day before the transfection. The miRs‐143 and ‐145 precursors (20–100 nM/mL; Ambion) and mature miRNAs (20–60 nM/mL) were used for the transfection of the cells, which was achieved by using cationic liposomes (i.e. TransIT‐TKO) (Mirus Bio Company, Madison, WI, USA) according to the manufacturer's lipofection protocol. The transfection efficiency was evaluated by the transfection of the cells with a duplex siRNA‐FITC (Dharmacon, Lafayette, CO, USA). Non‐specific control miRNA (NS, 57% GC content; Ambion) was used as a control for non‐specific effects. The sequences of mature miRNA‐143 (miRNA‐143 m) and ‐145 (miRNA‐145 m) were as follows: UGAGAUGAAGCACUGUAGCUCA and GUCCAGUUUUCCCAGGAAUCCCUU, respectively. The effects manifested by the introduction of the precursor or mature miRNAs into the cells were assayed at 36 h after the transfection. At the same time, semi‐quantitative RT‐PCR was carried out on the cells transfected with the precursors.
Western blotting. The cells were homogenized in chilled lysis buffer comprising 10 mM Tris‐HCl (pH 7.4), 1% NP‐40, 0.1% deoxycholic acid, 0.1% SDS, 150 mM NaCl, 1 mM EDTA, and 1% protease inhibitor cocktail (Sigma) and stood for 30 min on ice. After centrifugation at 16 000 g for 20 min at 4°C, the supernatants were collected as protein samples. Protein contents were measured with a DC protein assay kit (Biorad, Hercules, CA, USA). 10 µg of lysate protein for western blotting of ERK5 and c‐myc was separated by SDS‐PAGE using a 10% polyacrylamide gel and electroblotted onto a PVDF membrane (DuPont, Boston, MA, USA). After blockage of non‐specific binding sites for 1 h with 5% non‐fat milk in PBS containing 0.1% Tween 20, the membrane was incubated overnight at 4°C with antihuman ERK5 antibody (Cell Signaling Tec. Inc., Beverly, MA, USA), antihuman c‐myc antibody (Santa Cruz, Santa Cruz, CA, USA) or with DNMT‐1 (Santa Cruz). The membranes were then washed three times with PBS containing 0.1% Tween 20, incubated further with HRP‐conjugated sheep antimouse or donkey antirabbit Ig antibody (Amersham Biosciences, Piscataway, NJ, USA) at room temperature, and then washed three times with PBS containing 0.1% Tween 20. The immunoblots were visualized by use of an enhanced chemiluminescence detection kit (New England Biolabs, Beverly, MA, USA).
Statistics. Differences were statistically evaluated by one‐way anova followed by Fisher's PLSD. A P‐value of less than 0.05 was considered to be statistically significant.
Results
Expression levels of miRs‐143 and ‐145 were significantly decreased in B‐cell malignancies. We examined the expression of miRs‐143 and ‐145 in the samples from patients with B‐cell malignancies by conducting semi‐quantitative RT‐PCR and TaqMan assays using real‐time PCR. Representative bands of expression obtained by the former and the mean values of TaqMan assays from two independent experiments using the latter are presented in Fig. 1. There was no patient with any abnormality of chromosome 5q32, where miRs‐143 and ‐145 are colocalized within a 1.8‐kb distance from each other. In TaqMan assays, Ct values of the samples detected by TaqMan probes corresponding to U6, miR‐143 and miR‐145 are shown in Table 1. Notably, all of the cases of CLL showed an extremely low‐expression (Fig. 1a). Among the B‐cell lymphomas, all of the samples except Bl‐18 exhibited a low level (Fig. 1b). Recent reports by Calin et al.( 28 ) demonstrated that the expression levels of mir‐15a and ‐16 from 13q13.4 were decreased in CLLs with an incidence of approximately 68%. However, the incidence of reduced miR‐15a expression in our Japanese CLLs was not so high (approximately 54%, Fig. 1a).
Figure 1.
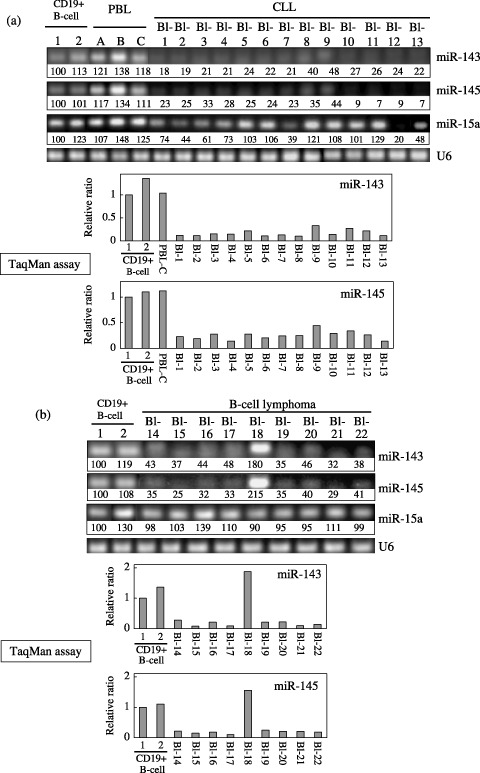
Expression of microRNAs (miRNAs)‐143 and ‐145 in human B‐cell malignancies examined by quantitative reverse transcription‐polymerase chain reaction (RT‐PCR). Blood samples or lymph nodes (Bl) from patients were obtained on admission or after relapse of the disease. Thirteen cases of chronic lymphocytic leukemia (CLL) (a) and nine cases of B‐cell lymphoma (b) were examined. Peripheral blood lymphocytes (PBL) from individuals a, b, and c and CD19+ B‐cells from the tonsils (1 and 2) were used as controls. In CLLs, the expression of miR‐15a( 28 ) was also examined by semi‐quantitative RT‐PCR. U6 was used as an internal standard. The intensity of the bands for miR‐143, ‐145 and ‐15a expression was determined by densitometry and the value is given under each band. The levels of CD19+ B‐cells for CLL and B‐cell lymphoma were designated as 100 in semi‐quantitative RT‐PCR and as 1 in quantitative RT‐PCR by TaqMan assays using a real‐time PCR. The results of real‐time PCR are expressed as the mean values of two independent experiments.
Table 1.
The Ct values of U6, miR‐143, and miR‐145 in real‐time PCR using the TaqMan probes.
| Standard Curve | Ct | |
|---|---|---|
| Mean ± SD | CV (%) | |
| U6 | 27.1 ± 0.12 | 0.4 |
| miR‐143 | 31.0 ± 0.12 | 0.4 |
| miR‐145 | 28.1 ± 0.08 | 0.3 |
Ct, cycle threshold; CV, coefficient variation; miR, miRNA; SD, standard deviation.
Relationship between the expression levels of miRs‐143 and ‐145 and cell growth. In order to clarify the relationship between the expression levels of miRs‐143 and 145 and cell growth in B‐cell lines, we examined the expression levels of EBV‐transformed B‐cell lines from healthy donors (IC, L11, L22, and L25) and established Burkitt cell lines (Raji, Daudi, P32, and KHM‐10B; Fig. 2a). It should be noted that the expression levels were inversely related to the cell growth of EBV‐transformed cells from the data of semi‐quantitative RT‐PCR (Ambion) (Fig. 2a,b; expression level, L25 < L22 = L11 < IC; growth, IC < L22 < L11 < L25). Furthermore, all of the Burkitt cell lines, in which cells have genetic aberrations including c‐Myc, showed a fairly low level of the expression of both miRNAs like the L25 cells (Fig. 2a), which showed the highest cell proliferation among the EBV‐transformed B‐cell lines (Fig. 2b). The data in Fig. 2a,b clearly indicate an inverse relationship between the expression levels of miR‐143 and ‐145 and cell growth.
Figure 2.
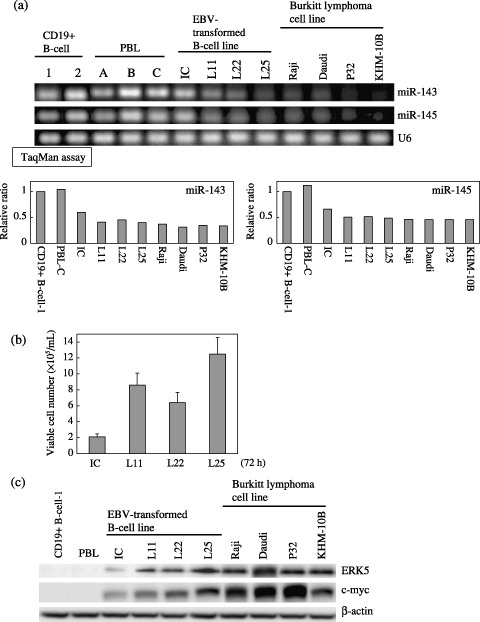
Expression of microRNAs (miRNAs)‐143 and ‐145 and cell growth in B‐cell cultured cell lines. (a) Evaluation of expression of miRNAs‐143 and ‐145 in human Epstein‐Barr virus (EBV)‐transformed B‐cell lines and Burkitt lymphoma cell lines by use of semi‐quantitative reverse transcription‐polymerase chain reaction (RT‐PCR) and TaqMan MicroRNA assays using real‐time PCR. U6 was used as an internal standard. CD19+ B‐cell‐1 was designated as 1 in quantitative RT‐PCR by TaqMan miRNA assays using real‐time PCR. The results of real‐time PCR were expressed as the mean values of two independent experiments. (b) Steady‐state cell growth of EBV‐transformed B‐cell lines at 72 h after seeding at the concentration of 1 × 105/mL (c) Western blot analysis of ERK5 and c‐myc in control and B‐cell lines in the same samples as in (b), and in human Burkitt lymphoma cell lines. β‐actin was used as an internal control.
Transfection of RAJI cells with precursor or mature miR‐143 or ‐145, respectively, causes growth inhibition. In order to examine the suppressive function of miR‐143 and ‐145 with respect to cell growth and to examine which enzymatic modification during miRNA biogenesis is perturbed, we transfected low‐expressant Raji cells with precursor miR‐143 (miR‐143p) or ‐145 (miR‐145p), in which transfection resulted in a significant growth inhibition that occurred in a dose‐dependent manner (Fig. 3a). Semi‐quantitative RT‐PCR using the primers for miR‐143 or ‐145 demonstrated a significant increase in the levels of miR‐143 and 145 in the Raji cells transfected with the respective precursors compared with their levels in the control cells (Fig. 3a). In order to further confirm that miR‐143 or ‐145 has a growth inhibition, we carried out a transfection experiment using mature miR‐143 (miR‐143 m) or ‐145 (miR‐145 m) (Fig. 3b). In both experiments, a dose‐dependent growth inhibition by the introduction of mature miR‐143 or ‐145, which results were very similar to those obtained when the transfection was carried out with precursor miR‐143 or ‐145. These findings indicate that miR‐143 and ‐145 negatively contribute to cell growth, because the compensation of miR‐143 or ‐145 by the transfection induced growth inhibition and that the perturbation of processing of miR‐143 or ‐145 in the nucleus including transcription and microprocessor of Drosha and DG8 could cause the cells to reduce their expression of both.
Figure 3.
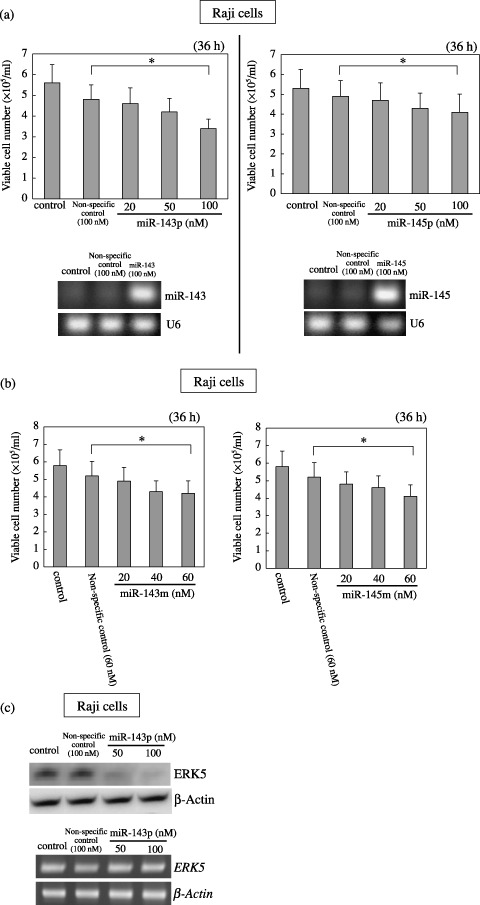
Effect of transfection of human Burkitt lymphoma Raji cells with either precursor (a) or mature type (b) of microRNAs (miRNAs)‐143 and ‐145, and ERK5 expression in the cells transfected with the precursor miR‐143 (c). (a, b) Number of viable transfected or control cells at 36 h after transfection is shown. Data are presented as the mean ± standard deviation (SD) of three different experiments, each carried out in duplicate. Levels of miRNAs‐143 and ‐145 in Raji cells at 36 h after the transfection of the cells with miR‐143 or ‐145 precursor miRNAs at 100 nM are shown in (a). U6 was used as an internal standard. The difference between the non‐specific control and 100 (a) or 60 (b) nM treatment was significant (*P < 0.01). (c) Expression levels of ERK5 protein and the mRNA at 36 h after the transfection of Raji cells with miR‐143 precursor miRNAs, as evaluated by western blot analysis (upper panel) and by quantitative RT‐PCR (lower panel), respectively. β‐actin was used as an internal standard. Control cells were incubated in medium containing transfection reagent alone.
Genomic status and epigenetic change in miR‐143 and ‐145 loci on 5q32. In order to examine the chromosomal aberrations of miRs‐143 and ‐145 loci on 5q32, we carried out genomic PCR on the B‐cell lines by using a primer pair covering both loci.( 34 ) As shown in Fig. 4a, more than one allele was confirmed to be present in all EBV‐transformed and Burkitt lymphoma cell lines, as in the placenta, used as the positive control. Furthermore, the treatment with 2–10 µM 5‐Aza, which completely reduced the level of DNMT‐1, did not upregulate the expression of miRNAs‐143 and ‐145 at all in L25 cells (Fig. 4b); nor did that with TSA. Thus, it appears that some genomic aberration or epigenetic change did not cause the low expression of miR‐143 and ‐145 in the cells.
Figure 4.
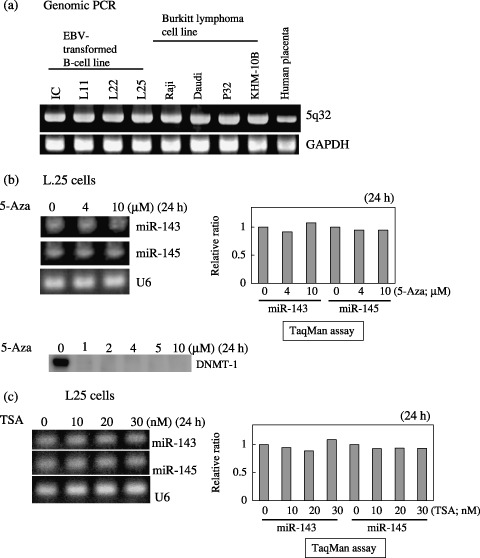
Confirmation of the presence of the genomic loci of microRNAs (miRNAs)‐143 and ‐145 at chromosome 5q32 by genomic polymerase chain reaction (PCR) (a) and expression of the miRNAs in Epstein‐Barr virus (EBV)‐transformed L25 cells after 24‐h treatment with 5‐Aza‐2′‐deoxycytidine (5‐Aza, b) or tricostatin A (TSA, c). (a) The primers amplified the DNA fragment including the loci of miRs‐143 and ‐145. Placental DNA was used as a positive control. The genomic locus of glyceraldehyde phosphate dehydrogenase (GAPDH) was used as an internal control. (b, c) The expression of the miRNAs in L25 cells after the treatment with either agent at the indicated concentrations is shown. The value for the control cells is designated as 1 in quantitative reverse transcription (RT)‐PCR by TaqMan miRNA assays using a real‐time PCR. The results of real‐time PCR are expressed as the mean values of two independent experiments. U6 was used as an internal standard. Results of Western blot analysis of DNA methyltransferase (DNMT)‐1 are also given in (b).
MicroRNA‐143 targets the ERK‐5 mRNAs. Previously, we determined that one of the target genes of miR‐143 was ERK5 in the human colon cancer cell line DLD‐1.( 34 ) ERK5 is a recently characterized MAPK, which is most similar to the well‐studied ERK1/2 subfamily, but uses distinct mechanisms to elicit responses. The physiological importance of this signaling cascade is underscored by the early embryonic death caused by the targeted deletion of the erk5 or the mek5 genes in mice.( 36 ) In Fig. 2c, the level of ERK5 expression in Burkitt lymphoma cell lines was approximately several times higher than that in the other B‐cells including CD19+ B‐cells and EBV transformed B‐cell lines except L25, where the pattern was very similar to that of c‐myc. We clearly demonstrated that the expression levels between miR‐143 and ERK5 were inversely correlated in regard to cell growth in EBV‐transformed cell lines (Fig. 2). Based on these results, ERK‐5 was shown to be a growth‐related MAPK even in B‐cells. In order to further confirm that the target mRNA of miR‐143 is ERK5, we examined the expression of ERK5 and the mRNA in the RAJI cells transfected with precursor miR‐143 (Fig. 3). Expectedly the protein expression levels were significantly decreased in a dose‐dependent manner, whereas the mRNA levels were almost unchanged (Fig. 3c). Thus, these findings and the previous results in colon cancer( 34 ) altogether indicate that miR‐143 could target the ERK5 gene even in B‐cells.
Discussion
In the current study, we demonstrated that the expression levels of miRs‐143 and ‐145 were significantly reduced in the B‐cell malignancies tested and further that the expression levels were inversely related to the cell growth in the EBV‐transformed B‐cell lines, which have no genomic aberrations. The introduction of miR‐143 or ‐145 precursors or mature types into the Raji cells led to a significant growth inhibition. Such data suggest that miRs‐143 and ‐145 could negatively contribute to cell growth by targeting growth‐related genes.
The increased expression of miR‐143 by the transfection with precursor miR‐143 into Raji cells lowered the protein expression levels of ERK5, whereas the mRNA level of ERK5 remained unchanged, which suggests that ERK5 is one of the target genes of miR‐143, as shown in previous reports by us,( 34 ) and Esau et al.( 37 )
Recent studies indicate that proteins involved in miRNA biogenesis, including Drosha and double‐stranded‐RNA‐binding protein DGCR8, Dicer 1, Argonaute 2, and RISC, may also participate in the complex interactions that regulate miRNA expression, together with additional mechanisms that regulate miRNA at the epigenetic, transcriptional level. Since the transfection of Raji cells with the precursor miR‐143 or ‐145 exhibited growth inhibition, transcription and/or processing by Drosha and DGCR8 microprocessor in the nucleus was possibly perturbed, which is true in the case of human colon cancer DLD‐1 cells.( 34 ) Furthermore, we did not obtain any data that indicated an inappropriate expression caused by genomic aberration or epigenetic change such as methylated DNA and histone. Recently, miR‐223 was shown to be upregulated by the retinoic acid‐induced replacement of NFI‐A with C/EBP α, resulting in promotion of human granulocyte differentiation.( 38 ) As miR‐223 repressed NFI‐A translation, the upregulation of miR‐223 by C/EBP α and granulopoiesis was further accelerated through positive feedback. Therefore, the machinery involved in the transcription step of miRs‐143 and ‐145, whose primary miRNAs are most likely identical, because their genomic loci are located within a 1.8‐kb span, should be clarified for achieving a fully comprehensive view of the processes operating in carcinogenesis.
Rescued expression of downregulated or functionally‐deficient miRNAs and/or inhibitors of overexpressed miRNAs may contribute to rebalanced the expression of large gene clusters implicated in oncogenesis, tumor progression, and cell death. One of the target genes for miR‐143 was presently shown to be ERK5 MAPK, which was also shown to be targeted in DLD‐1 cells by us,( 34 ) and in adipocytes by others.( 37 ) Also, we suggest that miR‐145 may target MAP3K3 and MAPK4K4 and that other potential targets for miR‐145, which have oncogenic functions, are MYCN, FOS, YES, and FLI (http://microrna.sanger.ac.uk/) and cell‐cycle promoters such as cyclin D2 and L1. Particularly, the MAPK signal via ERK5 contains c‐myc in the downstream of ERK5.( 39 ) Thus, the reduced expression of both miRNAs directly or indirectly affects cell fate such as growth, survival, and death signals; that is, low‐expression of miR‐143 and ‐145 possibly by perturbed transcription and/or microprocessing, could result in an unbalanced signaling cascade including MAPK, which would lead to a sustained cell proliferation.
To our knowledge, upregulation of mir‐155 from BIC RNA in B‐cell lymphomas,( 40 ) and downregulation of miR‐14 and ‐15, which originated from the deletion of a region of chromosome 13q13.4,( 28 ) have been reported in B‐cell malignancies. Furthermore, the target gene of miR‐15 and ‐16 was shown to be the antiapoptotic gene BCL‐2.( 29 ) Therefore, the genomic deletion of 13q13.4 causes an inappropriate expression of miR‐15 and ‐16 that targets antiapoptotic BCL‐2 mRNAs, which negatively works against cell death. We found that the level of miR‐15a was reduced in seven of 13 CLLs; however, the level of decrease was not as great as in the case of miRNAs‐143 or ‐145. Our results suggested that the incidence of the 13q13.4 deletion in Japanese CLLs may be lower than that in Caucasian CLLs.( 28 ) Recently, the same group also evaluated the miRNA expression profiles of 41 samples of CLL, and found 25 genes (of 161 analyzed) to have a unique miRNA expression signature.( 41 ) However, miR‐143 or ‐145 was not found to be significant with respect to the miRNA expression profile, although it is not clear whether miR‐143 and ‐145 were examined. Previously, we reported that the expression of miRs‐143 and 145 was strongly related to tumorigenesis in colon cancer,( 34 ) because more than 80% of the cases were shown to have low‐expression compared with non‐cancerous tissues. In addition, all of the malignant cell lines tested were shown to be downregulated for these miRs.( 34 )
Thus, miRs‐143 and ‐145 play pivotal roles in the pathogenesis of B‐cell malignancies and could be plausible biomarkers to differentiate B‐cell malignant cells from normal B‐cells, being even better ones than miRs‐15a and ‐16 in Japanese CLLs. Also, we propose miR‐143 or ‐145 as a candidate for the development of RNA medicine against cancer. Further detailed study to decipher the transcription machinery of miR‐143 and ‐145 including their promoter region on 5q32 and their binding sites in the 3′ UTR of ERK5 will be needed for a better understanding of the carcinogenesis involving miRNAs.
Acknowledgments
This work was supported by a grant‐in‐aid for scientific research from the Ministry of Education, Science, Sports, and Culture of Japan. We thank Dr Ogio for giving tonsil as control B‐cells.
References
- 1. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin‐4 encodes small RNAs with antisense complementarity to lin‐14. Cell 1993; 75: 843–54. [DOI] [PubMed] [Google Scholar]
- 2. Lagos‐Quintana M, Rauhut R, Lendeckel W, Tuschl T. Incorporating structure to predict microRNA targets. Science 2001; 294: 853–8. [DOI] [PubMed] [Google Scholar]
- 3. Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001; 294: 858–62. [DOI] [PubMed] [Google Scholar]
- 4. Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science 2001; 294: 862–4. [DOI] [PubMed] [Google Scholar]
- 5. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116: 281–97. [DOI] [PubMed] [Google Scholar]
- 6. Ambros V. The functions of animal microRNAs. Nature 2004; 431: 350–5. [DOI] [PubMed] [Google Scholar]
- 7. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 2004; 5: 522–31. [DOI] [PubMed] [Google Scholar]
- 8. Zamore PD, Haley B. Ribo‐gnome: the big world of small RNAs. Science 2005; 309: 1519–24. [DOI] [PubMed] [Google Scholar]
- 9. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005; 120: 15–20. [DOI] [PubMed] [Google Scholar]
- 10. Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet 2004; 5: 396–400. [DOI] [PubMed] [Google Scholar]
- 11. Lim LP, Lau NC, Garrett‐Engele P et al . Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005; 433: 769–73. [DOI] [PubMed] [Google Scholar]
- 12. Lewis BP, Shih IH, Jones‐Rhoades MW et al . Prediction of mammalian microRNA targets. Cell 2003; 115: 787–98. [DOI] [PubMed] [Google Scholar]
- 13. John B, Enright AJ, Aravin A et al . Human MicroRNA targets. PLoS Biol 2004; 2: e363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kiriakidou M, Nelson PT, Kouranov A et al . A combined computational‐experimental approach predicts human microRNA targets. Genes Dev 2004; 18: 1165–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krek A, Grun D, Poy MN et al . Combinatorial microRNA target predictions. Nat Genet 2005; 37: 495–500. [DOI] [PubMed] [Google Scholar]
- 16. Croce CM, Calin GA. miRNAs, cancer, and stem cell division. Cell 2005; 122: 6–7. [DOI] [PubMed] [Google Scholar]
- 17. Gregory RI, Shiekhattar R. MicroRNA biogenesis and cancer. Cancer Res 2005; 65: 3509–12. [DOI] [PubMed] [Google Scholar]
- 18. Calin GA, Sevignani C, Dumitru CD et al . Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA 2004; 101: 2999–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McManus MT. MicroRNAs and cancer. Semin Cancer Biol 2003; 13: 253–8. [DOI] [PubMed] [Google Scholar]
- 20. Lu J, Getz G, Miska EA et al . MicroRNA expression profiles classify human cancers. Nature 2005; 435: 834–8. [DOI] [PubMed] [Google Scholar]
- 21. Volinia S, Calin GA, Liu C‐G et al . A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA 2006; 103: 2257–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Calin GA, Liu C‐G, Sevignani C et al . A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA 2004; 101: 11755–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. He L, Thomson JM, Hemann MT et al . A microRNA expression signature of human solid tumors defines cancer gene targets. Nature 2005; 435: 828–33. 15944707 [Google Scholar]
- 24. Cummins JM, He Y, Leary RJ et al . The colorectal microRNAome. Proc Natl Acad Sci USA 2006; 103: 3687–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takamizawa J, Konishi H, Yanagisawa K et al . Reduced expression of the let‐7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res 2004; 64: 3753–6. [DOI] [PubMed] [Google Scholar]
- 26. Johnson SM, Grosshans H, Shingara J et al . RAS is regulated by the let‐7 microRNA family. Cell 2005; 120: 635–47. [DOI] [PubMed] [Google Scholar]
- 27. Yanaihara N, Caplen N, Bowman E et al . Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006; 9: 189–98. [DOI] [PubMed] [Google Scholar]
- 28. Calin GA, Dumitru CD, Shimizu M et al . Frequent deletions and down‐regulation of micro‐RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA 2002; 99: 15524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cimmino A, Calin GA, Fabbri M et al . miR‐15 and miR‐16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA 2005; 102: 13944–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. O'Donnell KA, Wentzel EA, Zeller KI et al . c‐Myc‐regulated microRNAs modulate E2F1 expression. Nature 2005; 435: 839–43. [DOI] [PubMed] [Google Scholar]
- 31. Hayashita Y, Osada H, Tatematsu Y et al . A polycistronic microRNA cluster, miR‐17‐92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res 2005; 65: 9628–32. [DOI] [PubMed] [Google Scholar]
- 32. Voorhoeve PM, Le Sage C, Schrier M et al . A genetic screen implicates miRNA‐372 and miRNA‐373 as oncogenes in testicular germ cell tumors. Cell 2006; 124: 1169–81. [DOI] [PubMed] [Google Scholar]
- 33. Calin GA, Ferracin M, Cimmino A et al . A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 2005; 353: 1793–801. [DOI] [PubMed] [Google Scholar]
- 34. Akao Y, Nakagawa Y, Naoe T. MicroRNAs 143 and 145 are possible common onco‐microRNAs in human cancers. Oncol Reports 2006; 16: 845–50. [PubMed] [Google Scholar]
- 35. Michael MZ, O’Connor SM, Van Holst Pellekaan NG et al . Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res 2003; 1: 882–91. [PubMed] [Google Scholar]
- 36. Nishimoto S, Nishida E. MAPK signalling: ERK5 versus ERK1/2. EMBO Rep Rev 2006; 7: 782–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Esau C, Kang X, Peralta E et al . MicroRNA‐143 regulates adipocyte differentiation. J Biol Chem 2004; 279: 52361–5. [DOI] [PubMed] [Google Scholar]
- 38. Fazi F, Rosa A, Fatica A et al . A minicircuitry comprised of microRNA‐223 and transcription factors NFI‐A and C/EBPα regulates human granulopoiesis. Cell 2005; 123: 819–31. [DOI] [PubMed] [Google Scholar]
- 39. English JM, Pearson G, Baer R et al . Identification of substrates and regulators of the mitogen‐activated protein kinase ERK5 using chimeric protein kinases. J Biol Chem 1998; 273: 3854–60. [DOI] [PubMed] [Google Scholar]
- 40. Eis PS, Tam W, Sun L et al . Accumulation of miR‐155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci USA 2005; 102: 3627–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Calin GA, Liu CG, Sevignani C et al . MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci USA 2004; 101: 11755–60. [DOI] [PMC free article] [PubMed] [Google Scholar]