

Abstract
Activation of the Akt/mammalian target of rapamycin (mTOR) pathway has been shown to be associated with resistance to endocrine therapy in estrogen receptor alpha (ERα)‐positive breast cancer patients. Utmost importance is attached to strategies aimed at overcoming treatment resistance. In this context, this work aimed to investigate whether, in breast cancer cells, the use of an mTOR inhibitor would be sufficient to reverse the resistance acquired after exposure to endocrine therapy. The ERα‐positive human breast adenocarcinoma derived‐MCF‐7 cells used in this study have acquired both cross‐resistance to hydroxy‐tamoxifen (OH‐Tam) and to fulvestrant and strong activation of the Akt/mTOR pathway. Cell proliferation tests in control cells demonstrated that the mTOR inhibitor rapamycin enhanced cell sensitivity to endocrine therapy when combined to OH‐Tam or to fulvestrant. In resistant cells, rapamycin used alone greatly inhibited cell proliferation and reversed resistance to endocrine therapy by blocking the agonist‐like activity of OH‐Tam on cell proliferation and bypassing fulvestrant resistance. Reversion of resistance by rapamycin was associated with increased ERα protein expression levels and modification of the balance of phospho‐ser167 ERα/total ERα ratio. Pangenomic DNA array experiments demonstrated that the cotreatment of resistant cells with fulvestrant and rapamycin allowed the restoration of 40% of the fulvestrant gene‐expression signature. Taken together, data presented herein strongly support the idea that mTOR inhibitor might be one of the promising therapeutic approaches for patients with ERα‐positive endocrine therapy‐resistant breast cancers. (Cancer Sci 2008; 99: 1992–2003)
Abbreviations:
- mTOR
mammalian target of rapamycin
- ERα
estrogen receptor alpha
- OH‐Tam
hydroxy‐tamoxifen
- ER+
ERα‐positive
- SERD
selective estrogen receptor down‐regulator
- SERM
selective estrogen receptor modulator
- Tam
tamoxifen
- PI3K
phosphatidylinositol 3‐kinase
- Akt/PKB
protein kinase B
- E2
17β‐estradiol
- BrdU
5‐bromodeoxyuridine
- GCOS
GeneChip Operating Software
- RMA
Robust Multi‐array Average
- GO
Gene Ontology
- RTQ‐PCR
Real‐Time Quantitative PCR
Seventy per cent of tumors of breast cancer patients are positive for estrogen receptor alpha (ERα) at diagnosis and are therefore suitable for endocrine therapy, which aims to block the mitogenic action of estrogens on breast cancer cells. However, acquired resistance is a major problem limiting the clinical benefit of endocrine therapy. Approximately 60% of patients with ERα‐positive (ER+) breast cancer who initially responded to the selective estrogen receptor modulator (SERM) tamoxifen (Tam) will develop acquired resistance.( 1 ) Recent clinical trials have also showed that, in patients with advanced breast cancer progressing on prior endocrine therapy with Tam or an aromatase inhibitor, the selective estrogen receptor down‐regulator (SERD) fulvestrant (ICI 182, 780), while yielding clinical benefit, is also prone to the development of resistance.( 2 , 3 )
The signaling pathway composed of phosphatidylinositol 3‐kinase (PI3K), protein kinase B (Akt/PKB) and mammalian target of rapamycin (mTOR) is crucial for cell growth and survival (for review( 4 )). Evidence suggests both the involvement of the PI3K/Akt/mTOR survival pathway in the acquisition of endocrine therapy resistance and the presence of complex interactions between ERα and this pathway. Indeed, non‐genomic actions of ERα are known to include interaction with and activation of the signaling kinase PI3K,( 5 ) and 17β‐estradiol (E2) is capable of activating mTOR via the tuberin/rheb pathway.( 6 ) Activation of the Akt pathway in breast tumor samples has been shown to be associated with resistance to endocrine therapy and a worse outcome in breast cancer patients,( 7 , 8 ) and over‐expression in transfected MCF‐7 cells of constitutively active myr‐Akt confers Tam resistance.( 9 ) Combining Tam with an mTOR inhibitor has led to additive/synergistic cytotoxic effects on MCF‐7 cells.( 10 , 11 , 12 ) Inhibition of proliferation and induction of apoptosis are increased when the mTOR inhibitor RAD001 is combined with an aromatase inhibitor in MCF‐7 cells over‐expressing aromatase.( 13 ) Finally, two studies have shown that mTOR inhibitors are able to restore Tam sensitivity( 14 ) and modify response to fulvestrant in transfected MCF‐7 cells with constitutively active myr‐Akt.( 15 ) However, they have failed to provide evidence of an association between reversion of fulvestrant resistance by RAD001 and enhancement of fulvestrant‐induced apoptosis.( 15 )
No study has ever investigated the impact of an mTOR inhibitor on breast cancer cells that have acquired resistance after exposure to endocrine therapy (a model that mimics the clinical situation of resistance development in Tam‐treated patients). As multifactorial changes leading to the activation of a survival system for cancer cells seem to be involved in the development of endocrine therapy resistance, the aim of the present study was to investigate whether the use of an mTOR inhibitor could be sufficient to reverse endocrine therapy resistance in breast cancer cells having acquired endocrine therapy resistance and activation of the endogenous Akt/mTOR pathway. To address this issue, we tested, at both the proliferation and gene‐expression levels, the impact of the mTOR inhibitor rapamycin on MCF‐7‐derived breast cancer cells (MVLN cells)( 16 ) and on resistant (R)‐MVLN cells (selected after 6‐month exposure of MVLN cells to 200 nM OH‐Tam)( 17 ) that have developed cross‐resistance to OH‐Tam and fulvestrant.
Materials and Methods
Cell culture. MVLN and R‐MVLN cells were grown for 1–2 passages as previously described,( 16 ) then purged for 4 days in Dulbecco's Modified Eagle Medium without phenol red and supplemented with 3% steroid‐depleted, dextran‐coated and charcoal‐treated fetal calf serum (DCC medium).
Cell proliferation analysis. The cells purged in DCC medium for 4 days were plated at 12 000 cells per well on a 96‐well tissue culture plate in DCC medium. One day later, the cells were treated for 1, 3, 5 or 8 days with vehicle (0.02% methanol), 1 nM E2 (Sigma, St Louis, MO, USA), 200 nM OH‐Tam (Sigma), 100 nM fulvestrant (Tocris, Ellisville, MO, USA), 2 nM or 20 nM rapamycin (Ozyme, Saint Quentin en Yvelines, France) or combined treatments. Proliferating cells were analyzed using the Cell Proliferation enzyme‐linked immunosorbent assay (ELISA), 5‐bromodeoxyuridine (BrdU) (colorimetric) Kit (Roche, Meylan, France). Briefly, the cells were labeled for 24 h with BrdU and the labeled nuclei were identified using a specific anti‐BrdU antibody according to the manufacturer's recommendations.
Western blot. Cell extract preparations and Western blot analysis were performed as previously described.( 18 ) ERα and insulin‐like growth factor 1 receptor alpha (IGF1Rα) antibodies were from Santa Cruz Biotechnology (CA, USA); phospho‐ser167 ERα, phospho‐ser473 Akt, total Akt, phospho‐ser136 Bad, phospho‐ser21/9 GSK3α/β, phospho‐thr389 p70S6K and total p70S6K antibodies were from Cell Signaling (Beverly, MA, USA); α‐Tubulin antibody was from Sigma. Blots were quantified using the VisionWorksLS Image Acquisition and Analysis Software.
RNA extraction. Total RNA from cell culture was prepared using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's recommendations and subsequently quantified on a Nanodrop ND1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). RNA integrity was checked using the BioAnalyzer 2100 (Agilent Technologies, Palo Alto, CA, USA).
Human genome U133 Plus 2.0 Affymetrix DNA chip experiment and analysis. Two independent cell culture replicates for each experimental condition (i.e. untreated cells or cells treated for 4 days with 100 nM fulvestrant, 20 nM rapamycin or a combination of both compounds) were used to generate total RNA. Complex probes were produced from these RNA, then hybridized to each array according to the manufacturer's recommendations (Affymetrix, Santa Clara, CA, USA) using ProfilXpert (Lyon, France). Scanned images of microarray chips were analyzed using GCOS (GeneChip Operating Software; Affymetrix) with the default settings. Raw data were normalized using the GC‐RMA algorithm, a modification of the RMA (robust multi‐array average)( 19 ) algorithm that takes into account that the non‐specific affinity of a probe is related to its sequence.( 20 ) We used the implementation of this algorithm available in the GC‐RMA Bioconductor package.( 21 ) Expression values were expressed as the ratio of the treated cell line gene value to that obtained with the untreated cell line (the ratio was called fold change, or FC). Two independent cell culture replicates were used for each pharmacological condition; four FC were obtained in total. Only genes with 4 out of 4 or 3 out of 4 FC values superior to the cut‐off chosen (>1.7 or <–1.7) were considered as differentially expressed. The choice of the FC cut‐off value was based on our previous experience,( 18 , 22 ) and on the consideration that, by exploring independent cell culture replicates, we were working under stringent conditions for data analysis (biological variability). Fatigo was used to analyze gene lists by using Gene Ontology (GO).( 23 ) The heatmaps were computed by using R software.
Validation of array data by Real‐Time Quantitative polymerase chain reaction (RTQ‐PCR). RTQ‐PCR measurements were performed as described previously( 24 , 25 ) using a LightCycler (Roche) or an ABI Prism 7900 Sequence Detection System (Perkin‐Elmer Applied Biosystems, Foster City, CA, USA) with the corresponding SYBR Green Kit, according to the manufacturer's recommendations. Differentially expressed genes were considered for FC > 2 or FC < –2.
Results
The MCF‐7‐derived R‐MVLN cells are cross‐resistant to OH‐Tam and to fulvestrant. The ER+ R‐MVLN cells emerged from selection under 200 nM OH‐Tam treatment (6 months) of the MCF‐7‐derived MVLN cells,( 16 , 17 ) and the exact phenotype developed by the R‐MVLN cells is still not fully characterized. The functionality of the ERα and the basal transcription machinery in MVLN and R‐MVLN cells was assessed by verifying that E2 treatment was able to stimulate cell proliferation in the BrdU assay in both cell lines (Fig. 1a). The amplitude of E2‐stimulation of cell proliferation was significantly greater in R‐MVLN cells than in MVLN cells. The phenotype of OH‐Tam resistance developed by R‐MVLN cells was characterized by the loss of the cytostatic activity of this molecule, such as it is detectable in MVLN cells, to the benefit of a strong stimulation of the R‐MVLN cell proliferation (agonist‐like effect) (Fig. 1b). These data (E2 and OH‐Tam responses) are in accordance with what we previously observed with two other OH‐Tam‐resistant MVLN‐derived cellular clones.( 25 ) Concerning the pharmacological response to fulvestrant, MVLN cells were shown to be sensitive to the cytostatic activity of this coumpound, as expected (Fig. 1c). Strikingly, while selected under OH‐Tam only, R‐MVLN cells have also developed resistance to fulvestrant, characterized by a total insensitivity to this molecule (Fig. 1c). As fulvestrant is known to bind to ERα and to induce the degradation of the receptor via the proteasome,( 26 , 27 ) the loss of fulvestrant‐induced ERα degradation in the R‐MVLN cells would be an obvious explanation for the development of the resistance to this molecule. As shown in Figure 1d, ERα expression was lower in R‐MVLN cells than in MVLN cells, and fulvestrant treatment induced a decrease in ERα protein expression in both cell lines. This suggests that other unknown mechanisms are involved in the development of resistance to this SERD. Taken together, these data show that the MVLN cell line was, as expected, sensitive to the cytostatic effect of both OH‐Tam and fulvestrant, whereas R‐MVLN cells developed under OH‐Tam selection a cross‐resistance to both OH‐Tam and fulvestrant, and thus represent an interesting cellular model to identify strategies to bypass cross‐resistance to endocrine therapy.
Figure 1.
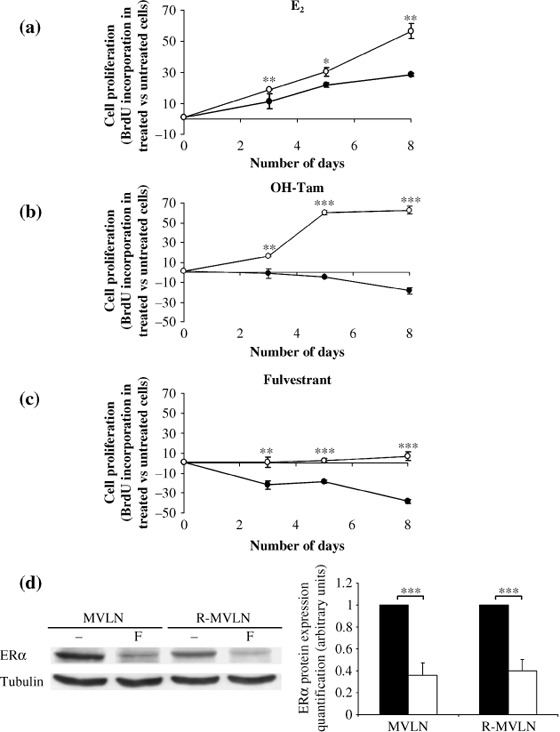
Growth kinetics of MCF‐7‐derived breast cancer (MVLN) and resistant (R)‐MVLN cells under different pharmacological conditions. Proliferative response (5‐bromodeoxyuridine [BrdU] labeling) of MVLN ( ) and R‐MVLN (
) and R‐MVLN ( ) cells treated for 3, 5 or 8 days with (a) 1 nM E2, (b) 200 nM hydroxy‐tamoxifen (OH‐Tam) or (c) 100 nM fulvestrant. Results are means ± SD from three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 versus the corresponding MVLN treatment according to Student's t‐test. (d) Fulvestrant induces estrogen receptor alpha (ERα) degradation in both MVLN and R‐MVLN cells. Western blot analysis of total ERα in the absence (–) or presence of 100 nM fulvestrant (F). Expression of α‐tubulin was measured as an invariant control. The Western blot shown is from one representative experiment that was reproduced at least three times in independent experiments and cell lysates. Histograms represent means ± SD of protein expression levels in untreated‐ (black bars) and 100 nM fulvestrant‐treated (white bars) MVLN and R‐MVLN cells (***P < 0.001 according to Student's t‐test).
) cells treated for 3, 5 or 8 days with (a) 1 nM E2, (b) 200 nM hydroxy‐tamoxifen (OH‐Tam) or (c) 100 nM fulvestrant. Results are means ± SD from three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 versus the corresponding MVLN treatment according to Student's t‐test. (d) Fulvestrant induces estrogen receptor alpha (ERα) degradation in both MVLN and R‐MVLN cells. Western blot analysis of total ERα in the absence (–) or presence of 100 nM fulvestrant (F). Expression of α‐tubulin was measured as an invariant control. The Western blot shown is from one representative experiment that was reproduced at least three times in independent experiments and cell lysates. Histograms represent means ± SD of protein expression levels in untreated‐ (black bars) and 100 nM fulvestrant‐treated (white bars) MVLN and R‐MVLN cells (***P < 0.001 according to Student's t‐test).
The Akt/mTOR signaling pathway is activated in the R‐MVLN cell line. The compiled data suggest that activation of the Akt pathway is associated with endocrine therapy resistance, estrogen deprivation and worse outcome in breast cancer patients.( 7 , 8 , 9 ) The data presented in Figure 2 demonstrates an activation of Akt in the R‐MVLN cells (3.7‐fold increase in the phospho‐ser473 Akt/total Akt ratio, Fig. 2a) paired with an increase in the phosphorylation level of three Akt‐targets: (i) the ser167 located in the ligand‐independent transactivation AF‐1 domain of the ERα protein (2.5‐fold increase in the phospho‐ser167 ERα/total ERα ratio, Fig. 2b); (ii) Bad (1.9‐fold increase in phospho‐ser136 Bad, Fig. 2c), and (iii) GSK3 (2.3‐fold increase in the GSK3 phosphorylation status, Fig. 2d). The PI3K/Akt pathway is normally regulated by upstream receptor tyrosine kinases, especially IGF1R.( 28 ) We found that the expression of the IGF1Rα protein was weakly but significantly greater in R‐MVLN cells than in MVLN cells (Fig. 2e). The downstream Akt target mTOR kinase is known to induce the phosphorylation of several targets, especially the thr389 residue of p70S6K.( 4 ) Figure 2f shows that the phosphorylation status of p70S6K was increased (by 2.7‐fold) in R‐MVLN cells compared with MVLN cells. Taken together, these data suggest an activation of the whole IGF1R/PI3K/Akt/mTOR signaling pathway in the OH‐Tam‐ and fulvestrant‐resistant R‐MVLN cells.
Figure 2.
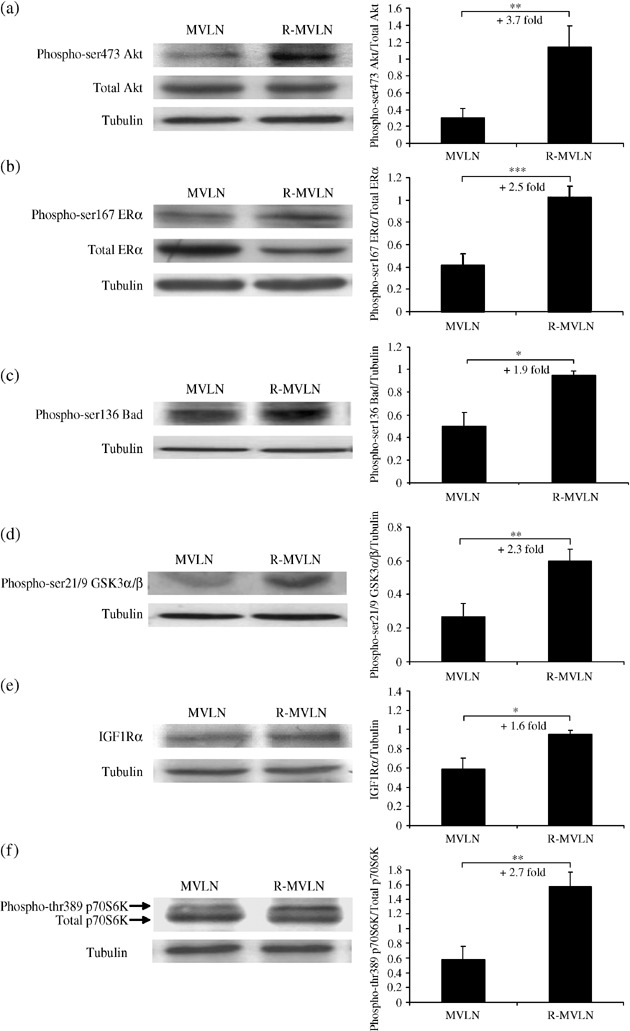
Activated Akt/mammalian target of rapamycin (mTOR) signaling pathway is present in resistant MCF‐7‐derived breast cancer (R‐MVLN) cells. Western blot analysis of (a) phospho‐ser473 Akt and total Akt, (b) phospho‐ser167 estrogen receptor alpha (ERα) and total ERα, (c) phospho‐ser136 Bad, (d) phospho‐ser21/9 GSK3α/β, (e) IGF1Rα and (f) phospho‐thr389 p70S6 K and total p70S6 K as described in the Materials and methods section using specific antibodies. Expression of α‐tubulin was measured as an invariant control. The Western blots shown are from one representative experiment that was reproduced at least three times in independent experiments and cell lysates. Histograms represent means ± SD of protein expression levels in independent experiments (*P < 0.05, **P < 0.01 and ***P < 0.001 according to Student's t‐test).
Impact of the mTOR inhibitor rapamycin on the phosphorylation level of p70S6K, Akt and GSK3 proteins. Treatment of MVLN and R‐MVLN cells by 20 nM rapamycin led, in both cell lines, to the total abrogation of the phosphorylation of the mTOR‐target thr389 p70S6K protein (Fig. 3a,d). This suggests that rapamycin was able to block, as expected, the mTOR pathway. As several studies have observed that rapamycin exposure is associated with a feedback regulation of the Akt pathway,( 29 , 30 ) we investigated the phosphorylation status of Akt and of the Akt‐target GSK3 in rapamycin‐exposed MVLN and R‐MVLN cells. No significant variation in the phospho‐ser473 Akt/total Akt ratio or in the GSK3 phosphorylation status was observed in rapamycin‐treated MVLN cells (Fig. 3b–d). In contrast, in R‐MVLN cells, rapamycin exposure induced a weak but significant decrease in the phospho‐ser473 Akt/total Akt ratio (P < 0.01) associated with a decrease in the phosphorylation level of GSK3 (P < 0.001) (Fig. 3b–d). Thus, under rapamycin exposure, a feedback regulation seems to occur upstream of mTOR in R‐MVLN cells but not in MVLN cells, leading to a decreased Akt activation.
Figure 3.
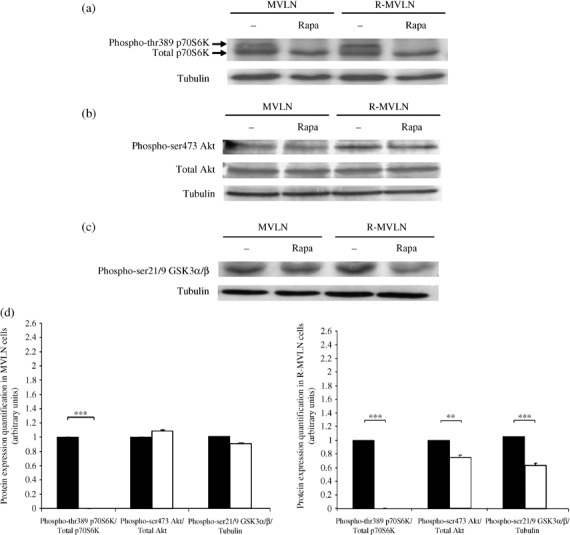
Consequences of 4‐day rapamycin exposure on p70S6K, Akt and GSK3 protein phosphorylation. Western blot analysis of (a) phospho‐thr389 p70S6 K and total p70S6 K, (b) phospho‐ser473 Akt and total Akt, (c) phospho‐ser21/9 GSK3α/β in the absence (–) or presence of 20 nM rapamycin (Rapa). α‐Tubulin protein level was measured to verify equal loading. The Western blots shown are from one representative experiment that was reproduced at least three times in independent experiments and cell lysates. (d) Histograms represent means ± SD of protein expression levels in untreated‐ (black bars) and 20 nM rapamycin‐treated (white bars) MVLN and R‐MVLN cells. **P < 0.01 and ***P < 0.001 versus the corresponding untreated cells according to Student's t‐test.
Rapamycin affects ERα protein expression levels and the balance of phospho‐ser167 ERα/total ERα in R‐MVLN cells. As shown in Figure 4, rapamycin exposure had no impact on ERα protein expression in MVLN cells but was associated with a two‐fold increase in ERα protein expression in R‐MVLN cells (Fig. 4a,b,d). By RTQ‐PCR experiments, we did not find under the same experimental conditions any variation in ESR1/ERα mRNA levels (data not shown), suggesting that the up‐regulation of ERα protein expression in rapamycin‐treated R‐MVLN cells occurs at a post‐transcriptional level. As phosphorylation of the ERα AF‐1 domain is one of the possible mechanisms involved in the regulation of the agonist‐like activity of Tam,( 31 ) we investigated the phospho‐ser167 ERα/total ERα ratio in the presence of the mTOR inhibitor. In rapamycin‐treated MVLN cells, no significant variation in the phospho‐ser167 ERα/total ERα ratio was observed (Fig. 4c,d). In contrast, in R‐MVLN cells rapamycin exposure induced a decrease in the phospho‐ser167 ERα/total ERα ratio (P < 0.001) (Fig. 4c,d). This observation was probably the consequence of both the increase in total ERα expression (Fig. 4b) and the decrease in Akt activation in rapamycin‐treated R‐MVLN cells (Fig. 3b). Taken together, these data suggest that under rapamycin exposure, a feedback regulation seems to occur upstream of mTOR in R‐MVLN cells but not in MVLN cells, leading to a decreased Akt activation associated with a decreased proportion of the phospho‐ser167 ERα population among the total population of ERα. As total ERα expression is increased in rapamycin‐treated R‐MVLN cells, these data suggest that, in R‐MVLN resistant cells, mTOR inhibition affects the balance of phospho‐ser167 ERα/total ERα to the benefit of the non‐phosphorylated form of the receptor.
Figure 4.
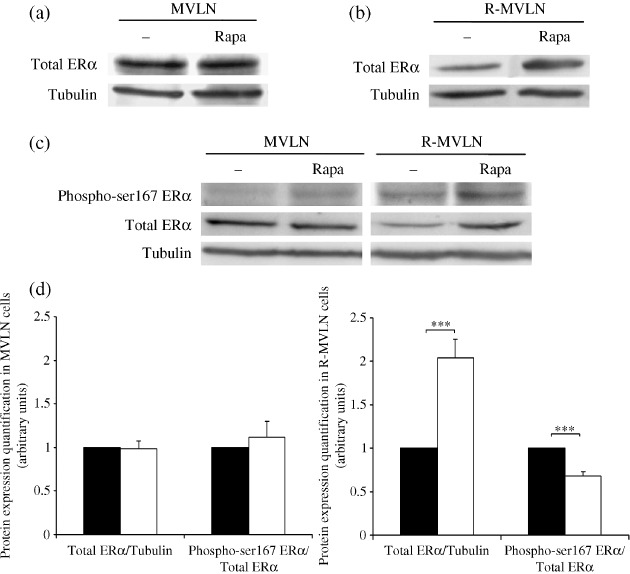
Rapamycin affects the estrogen receptor alpha (ERα) protein expression levels and the phospho‐ser167 ERα/total ERα ratio in the resistant MCF‐7‐derived breast cancer (R‐MVLN) cells. Western blot analysis of total ERα in (a) MVLN and (b) R‐MVLN cells and of (c) phospho‐ser167 ERα and total ERα in the absence (–) or presence of 20 nM rapamycin (Rapa). α‐Tubulin protein level was measured to verify equal loading. The Western blots shown are from one representative experiment that was reproduced at least three times in independent experiments and cell lysates. (d) Histograms represent means ± SD of protein expression levels in untreated‐ (black bars) and 20 nM rapamycin‐treated (white bars) MVLN and R‐MVLN cells. ***P < 0.001 versus the corresponding untreated cells according to Student's t‐test.
OH‐Tam and fulvestrant cytostatic activities were enhanced by rapamycin in control MVLN cells. With the aim to assess the effect of rapamycin on the proliferation of control MVLN cells, we performed a BrdU test (assay measuring the proportion of cells that entered S phase) with either vehicle, rapamycin, OH‐Tam, fulvestrant, or rapamycin combined with OH‐Tam or fulvestrant (Fig. 5a). The effect of rapamycin on MVLN and R‐MVLN cells was investigated after prolonged exposure (4 days) to fulfill BrdU assay conditions in order to detect any impact of the mTOR inhibitor on the pharmacological response to OH‐Tam or to fulvestrant. In our experimental conditions, rapamycin alone had a weak inhibitory effect on MVLN cell proliferation (similar to that of OH‐Tam alone), but was able to greatly enhance the sensitivity of these cells to the cytostatic activity of OH‐Tam (Fig. 5a, 412% increase of cell proliferation inhibition, with 2 nM and 20 nM rapamycin, respectively, P < 0.01 and P < 0.001). When combined with fulvestrant, rapamycin exposure also enhanced the cytostatic action of this molecule (Fig. 5a, 33% increase of cell proliferation inhibition, P < 0.05). No difference was observed between the two rapamycin concentrations tested (2 nM and 20 nM).
Figure 5.
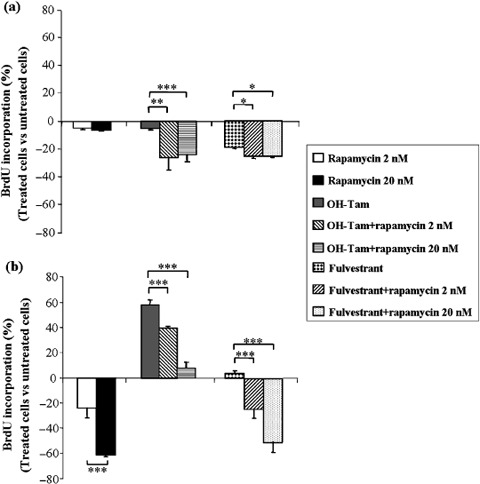
Impact of rapamycin treatment on the pharmacological response to hydroxy‐tamoxifen (OH‐Tam) and to fulvestrant using 5‐bromodeoxyuridine (BrdU) cell proliferation assay. For each experimental condition, the proliferation response was assessed by BrdU labeling (treated cells versus untreated cells) as described by the supplier. (a) MCF‐7‐derived breast cancer (MVLN) and (b) resistant (R)‐MVLN cells were cultured for 4 days in steroid‐depleted, dextran‐coated and charcoal‐treated fetal calf serum (DCC medium) in the presence of 2 nM rapamycin, 20 nM rapamycin, 200 nM OH‐Tam, 200 nM OH‐Tam combined with 2 nM or 20 nM rapamycin, 100 nM fulvestrant, and 100 nM fulvestrant combined with 2 nM or 20 nM rapamycin. Results are means ± SD from three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 according to Student's t‐test.
Rapamycin blocks the stimulating action of OH‐Tam on cell proliferation and reverses fulvestrant resistance in the resistant R‐MVLN cells. In contrast to what was observed in MVLN cells, rapamycin exposure of R‐MVLN cells induced a strong inhibition of cell proliferation (Fig. 5b, 24% and 60% inhibition of cell proliferation with 2 nM and 20 nM rapamycin, respectively, P < 0.001). Furthermore, the agonist‐like activity of OH‐Tam on cell proliferation developed by the resistant R‐MVLN cells was totally impaired in the presence of rapamycin, in a dose‐dependent manner (Fig. 5b, P < 0.001). Finally, the most striking data was that the resistance of R‐MVLN cells to fulvestrant was totally bypassed in the presence of rapamycin (Fig. 5b, 22% and 51% inhibition of cell proliferation with, respectively, 2 nM and 20 nM rapamycin, P < 0.001). Taken together, these data demonstrated first a great sensitivity of endocrine therapy‐resistant R‐MVLN cells to the antiproliferative activity of rapamycin, and second, that mTOR inhibitor was capable of reverting resistance to both OH‐Tam and fulvestrant.
Specific gene‐expression profiles are associated with the restoration of the fulvestrant signature in rapamycin‐treated R‐MVLN cells. To identify the molecular mechanisms involved in the reversion of the endocrine therapy resistance in R‐MVLN cells under rapamycin exposure, we investigated gene‐expression profiles using the pangenomic Affymetrix Human Genome U133 Plus 2.0 Array. We designed our gene‐expression experiment to decipher whether the reversion of the fulvestrant resistance observed in resistant R‐MVLN cells, when both fulvestrant and rapamycin were combined (Fig. 5b), was effectively due to the restoration of the pharmacological response to fulvestrant, or was simply the consequence of an antiproliferative action of rapamycin per se.
The sensitivity of MVLN cells to fulvestrant is illustrated in Figure 6a by the identification of a great number of variations in gene expression (n = 1241), whereas the resistance developed by R‐MVLN cells is highlighted at the gene‐expression level, as only 234 gene‐expression deregulations were observed under fulvestrant exposure. Only 10.9% (136 genes) of the MVLN ± fulvestrant signature was still present in the R‐MVLN ± fulvestrant gene‐expression profile. Exposure to combined rapamycin and fulvestrant in R‐MVLN cells generated a dramatically different gene‐expression pattern (n = 2095 deregulations of expression), and the gray region of the Venn diagram presented in Figure 6a (genes uncommon to the R‐MVLN ± fulvestrant signature) reflected the reversion of fulvestrant resistance by rapamycin. Strikingly, 498 genes of this signature (set 1) were similarly deregulated in fulvestrant‐treated MVLN cells, demonstrating that 40% (498/1241) of the gene‐expression profile associated with sensitivity to fulvestrant was restored in fulvestrant‐resistant R‐MVLN cells in the presence of an mTOR inhibitor.
Figure 6.
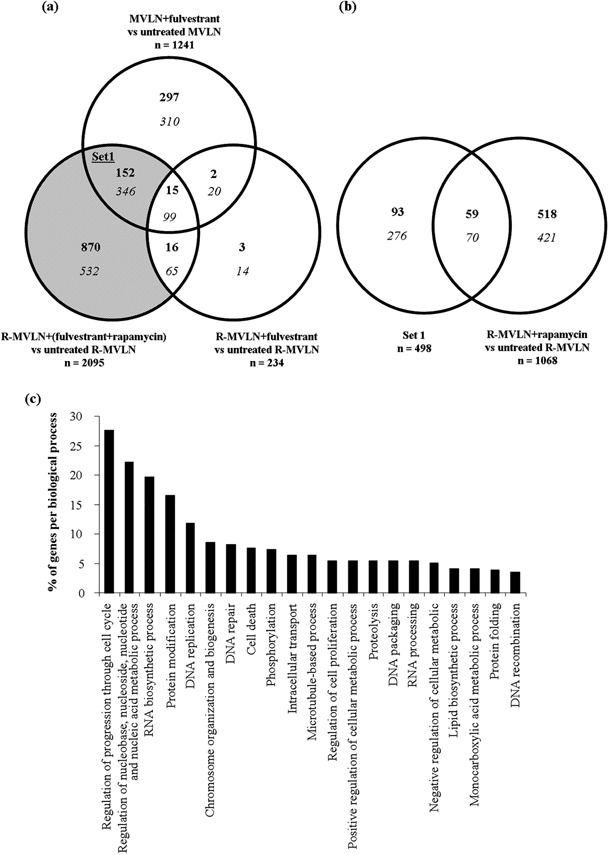
Reversion of the fulvestrant resistance in resistant MCF‐7‐derived breast cancer (R‐MVLN) cells is associated with specific gene‐expression profiles. (a) Venn diagram showing the distribution of the differentially regulated genes identified in MVLN cells treated with fulvestrant versus untreated cells, R‐MVLN cells treated with fulvestrant versus untreated cells and R‐MVLN cells treated with fulvestrant + rapamycin versus untreated cells. Differentially regulated genes were those whose expression levels were up‐regulated (bold characters) if fold change (FC) > 1.7 or down‐regulated (italic characters) if FC < –1.7. The gray region represents the genes associated with the reversion of fulvestrant resistance by rapamycin in R‐MVLN cells. Set 1 is the set of genes commonly deregulated in MVLN ± fulvestrant and R‐MVLN ± (fulvestrant+rapamycin) but not in R‐MVLN ± fulvestrant. (b) Venn diagram showing the commonly differentially regulated genes between set 1 and R‐MVLN ± rapamycin. (c) Illustration of the most representative gene ontology (GO) biological processes of genes belonging to set 1. Each gene can be a member of more than one GO category.
We then sought to differentiate in set 1 the gene deregulations resulting from the action of rapamycin from those resulting from the combination of rapamycin and fulvestrant. Cross‐analyzing set 1 with the gene‐expression signature obtained in R‐MVLN cells treated by rapamycin alone indicated that only 26% (129/498) of set 1 genes were the consequence of the intrinsic action of rapamycin per se (Fig. 6b). These data indicate that for 74% of the gene‐expression variations identified in set 1, the restoration of the fulvestrant gene‐expression signature was effectively the consequence of the combination of fulvestrant and rapamycin. A similar result was obtained when we considered the totality of the genes reflecting the reversion of fulvestrant resistance by rapamycin (gray region of the Venn diagram presented in Fig. 6a), as the deregulation of 60% of these genes was shown to be effectively the consequence of the combination of fulvestrant and rapamycin and not the consequence of rapamycin alone (data not shown).
Finally, 342 of the 498 genes belonging to set 1 could be classified using Gene Ontology (Fatigo). The biological functions most represented were (Fig. 6c): regulation of progression through cell cycle (27.6%), regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolic process (22.2%), RNA biosynthetic process (19.7%), protein modification (16.5%), DNA replication (11.8%), chromosome organization and biogenesis (8.6%), DNA repair (8.3%) and cell death (7.6%). The amplitude of the gene‐expression variations detected in the MVLN ± fulvestrant condition, absent in the R‐MVLN ± fulvestrant condition, and restored in the R‐MVLN ± (fulvestrant + rapamycin) condition is illustrated in Figure 7 for these most‐represented biological processes. In accordance with the results shown in Figure 6b, exposure of R‐MVLN cells to rapamycin did not alter the expression of most of the genes (Fig. 7). Finally, the array data were validated by RTQ‐PCR for 25 genes (Table 1). Taken together, the current results demonstrated that mTOR inhibition was not only capable of reversing fulvestrant resistance at the cell proliferation level (Fig. 5) but also of restoring, at the gene‐expression level, 40% of the fulvestrant signature in R‐MVLN cells (6, 7, Table 1).
Figure 7.
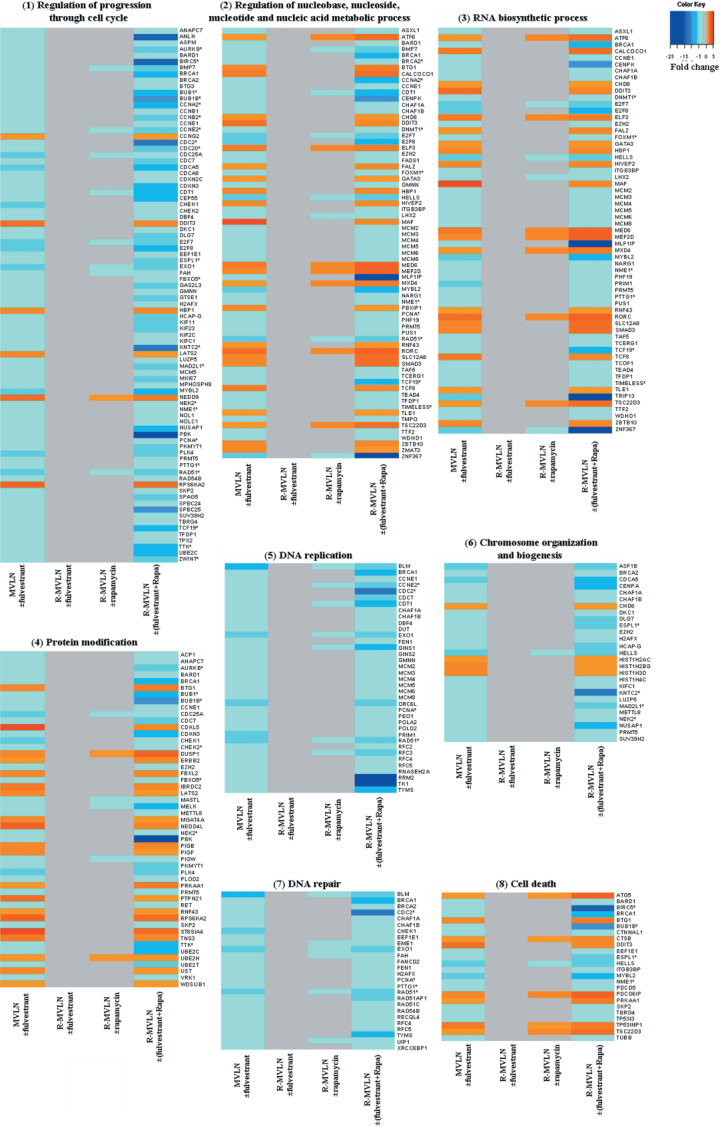
Gene‐expression variations measured by Affymetrix arrays in the eight most representative biological processes in gene ontology (GO) of set 1. Each row represents a gene and each column represents a different pharmacological condition: MCF‐7‐derived breast cancer (MVLN) ± fulvestrant; R‐MVLN ± fulvestrant; R‐MVLN ± rapamycin; R‐MVLN ± (fulvestrant+rapamycin). The results are expressed as the mean of fold change (FC) values; a color scale is indicated at the top. Genes highlighted in red correspond to up‐regulated genes (FC > 1.7). Genes highlighted in blue correspond to down‐regulated genes (FC < –1.7). Invariant gene‐expressions are represented in gray. Genes with asterisks are those whose expression variations were validated by real‐time quantitative polymerase chain reaction (RTQ‐PCR).
Table 1.
Validation by RTQ‐PCR of the gene expression variations (fold change (FC)) measured by Affymetrix array. Biological processes were: (1) regulation of progression through cell cycle, (2) regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolic process, (3) RNA biosynthetic process, (4) protein modification, (5) DNA replication, (6) chromosome organization and biogenesis, (7) DNA repair, (8) cell death, (9) phosphorylation, (10) microtubule‐based process, (11) intracellular transport, (12) regulation of cell proliferation, (13) proteolysis and (14) regulation of cell size. Bold characters represent expression variations greater than the cut‐offs used as described in the Materials and methods section
| Gene symbol | Description | Biological process | FC array | FC RTQ‐PCR | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MVLN ± fulvestrant | R‐MVLN ± fulvestrant | R‐MVLN ± rapamycin | R‐MVLN (± fulvestrant + rapamycin) | MVLN ± fulvestrant | R‐MVLN ± fulvestrant | R‐MVLN ± rapamycin | R‐MVLN (± fulvestrant + rapamycin) | |||
| AURKB | aurora kinase B | 1, 4, 9 | –3.8 | 1.1 | –1.4 | –5.3 | –3.2 | –1.1 | –1.1 | –5.7 |
| BIRC5 | baculoviral IAP repeat‐containing 5 (survivin) | 1, 8, 11 | –3.1 | –1.2 | –1.6 | –14.3 | –3.0 | 1.0 | –1.2 | –7.0 |
| BUB1 | BUB1 budding uninhibited by benzimidazoles 1 homolog | 1, 4, 9 | –2.7 | –1.2 | –1.6 | –6.9 | –2.4 | 1.1 | –1.1 | –3.7 |
| BUB1B | BUB1 budding uninhibited by benzimidazoles 1 homolog beta | 1, 4, 8, 9, 11 | –3.4 | –1.4 | –1.5 | –10.5 | –2.2 | 1.1 | 1.1 | –6.4 |
| CCNA2 | cyclin A2 | 1, 2 | –3.1 | –1.3 | –1.6 | –7.9 | –3.2 | 1.0 | –1.1 | –12.3 |
| CCNB2 | cyclin B2 | 1 | –2.3 | 1.1 | –1.1 | –5.3 | –3.6 | 1.8 | –1.8 | –6.2 |
| CCNE2 | cyclin E2 | 1, 5 | –3.6 | –1.6 | –2.4 | –6.4 | –4.3 | –1.5 | –3.4 | –4.5 |
| CDC2 | cell division cycle 2, G1 to S and G2 to M | 1, 5, 7 | –2.5 | –1.0 | –1.7 | –10.8 | –2.1 | –1.0 | –1.5 | –7.6 |
| CDC20 | CDC20 cell division cycle 20 homolog | 1, 13 | –3.6 | –1.1 | 1.1 | –4.9 | –2.3 | 1.1 | 1.1 | –4.8 |
| DNMT1 | DNA (cytosine‐5‐)‐ methyltransferase 1 | 2, 3 | –1.9 | –1.3 | –1.3 | –1.9 | –6.7 | –1.5 | –1.4 | –3.5 |
| ESPL1 | extra spindle poles like 1 | 1, 6, 8, 11, 13 | –2.5 | –1.1 | –1.6 | –4.2 | –2.3 | 1.4 | –1.0 | –5.0 |
| FBXO5 | F‐box only protein 5 | 1, 4 | –2.7 | –1.4 | –1.6 | –2.9 | –2.2 | –1.1 | –1.3 | –2.9 |
| FOXM1 | forkhead box M1 | 2, 3 | –2.0 | –1.2 | –1.4 | –3.0 | –2.0 | –1.2 | –1.1 | –4.8 |
| IL17RB | interleukin 17 receptor B | 14 | –2.5 | –1.4 | –1.4 | –2.1 | –2.2 | –1.8 | –1.2 | –7.5 |
| KNTC2 | kinetochore associated 2 | 1, 6, 11 | –2.6 | 1.1 | –1.6 | –10.6 | –2.7 | 1.1 | 1.0 | –6.8 |
| MAD2L1 | MAD2 mitotic arrest deficient‐like 1 | 1, 6 | –3.9 | –1.5 | –1.5 | –9.3 | –2.8 | –1.2 | –1.6 | –3.5 |
| NEK2 | NIMA (never in mitosis gene a)‐related kinase 2 | 1, 4, 6, 9, 13 | –3.5 | –1.4 | –1.6 | –5.3 | –2.0 | 1.2 | –1.0 | –5.2 |
| NME1 | non‐metastatic cells 1, protein (NM23A) expressed in | 1, 2, 3, 8, 12 | –2.3 | –1.5 | –1.5 | –3.0 | –2.2 | –1.6 | –1.6 | –3.3 |
| PCNA | proliferating cell nuclear antigen | 1, 2, 5, 7, 10 | –2.8 | –1.2 | –1.6 | –3.0 | –13.0 | –1.6 | –1.9 | –3.9 |
| PTTG1 | pituitary tumor‐transforming 1 | 1, 3, 7 | –2.1 | 1.4 | 1.6 | –3.6 | –2.1 | 1.4 | 1.9 | –2.7 |
| RAD51 | RAD51 homolog | 1, 2, 5, 7 | –5.3 | –1.6 | –2.7 | –6.4 | –3.2 | –1.3 | –3.1 | –5.0 |
| TCF19 | transcription factor 19 (SC1) | 1, 2, 3 | –4.0 | –1.4 | –1.6 | –7.8 | –3.1 | –1.1 | –1.3 | –6.0 |
| TIMELESS | timeless homolog | 2, 3 | –2.3 | –1.2 | –1.4 | –2.7 | –2.0 | –1.1 | –1.2 | –2.6 |
| TTK | TTK protein kinase | 1, 4, 9, 11, 12 | –2.4 | –1.4 | –1.6 | –7.4 | –2.1 | –1.0 | –1.1 | –3.6 |
| ZWINT | ZW10 interactor | 1, 11 | –2.9 | –1.2 | –1.7 | –6.0 | –2.5 | –1.3 | –1.7 | –3.6 |
RTQ‐PCR, real‐time quantitative polymerase chain reaction; MVLN, MCF‐7‐derived breast cancer cells; R‐MVLN, resistant MCF‐7‐derived breast cancer cells.
Discussion
Resistance to endocrine therapy is one of the main challenges in the treatment of ER+ breast cancer and the use of signal transduction inhibitors represents one of the most promising therapeutic approaches in this indication. Rapamycin and its analogs (temsirolimus, everolimus and AP23573) are currently being tested in clinical trials as novel‐target anticancer agents,( 32 , 33 ) and a recent study suggests that over 50% of breast cancer patients could be potential candidates for treatment by mTOR inhibition.( 34 ) The present work is, to our knowledge, the first report of an extensive investigation of the impact of an mTOR inhibitor on ER+ breast cancer cells having acquired resistance after exposure to endocrine therapy. The R‐MVLN cells used in this study are an interesting model because: (i) they mimic the clinical situation of resistance development in Tam‐treated patients; (ii) they have acquired cross‐resistance to OH‐Tam and to fulvestrant (a phenotype rarely observed under OH‐Tam selection); and (iii) they have acquired strong activation of their endogenous Akt/mTOR pathway.
Prolonged exposure (4 days) to rapamycin was performed for two reasons: (i) to fulfill BrdU assay conditions required to detect any impact of the mTOR inhibitor on the pharmacological response to OH‐Tam or fulvestrant; and (ii) to mimic the long‐term effects induced by the clinical use of mTOR inhibitors. By cell proliferation assay, the current study showed that rapamycin‐induced inhibition of cell proliferation was greater in R‐MVLN cells (60% inhibition) than in the MVLN cells (7% inhibition), in accordance with the, respectively, high and low levels of Akt/mTOR pathway activation in each cell line. More interestingly, mTOR inhibition led to an enhanced cytostatic activity of both OH‐Tam and fulvestrant in control MVLN cells. In accordance with our data, previous studies have observed an increased Tam sensitivity of mTOR inhibitor‐treated MCF‐7 cells.( 10 , 11 , 12 ) Finally, in the resistant R‐MVLN cells, rapamycin was able to reverse acquired resistance to endocrine therapy by abolishing the agonist‐like activity of OH‐Tam on cell proliferation and by bypassing fulvestrant resistance. Taken together, these data showed that: (i) even in ER+ breast cancer cells having a low level of activation of the Akt/mTOR pathway, rapamycin is able to enhance endocrine therapy efficiency; (ii) mTOR inhibition by itself is sufficient to strongly inhibit the proliferation and reverse the OH‐Tam and fulvestrant acquired cross‐resistance of breast cancer cells possessing an activated Akt/mTOR pathway.
As the PI3K/Akt/mTOR pathway is an important survival pathway for cancer cells, another important issue is the determination of the impact of rapamycin upstream of mTOR. Initial studies have reported down‐regulation of phospho‐ser473 Akt in response to mTOR knock‐down in both Drosophila and mammalian cells.( 35 ) Subsequent reports have found that mTOR inhibition induced Akt phosphorylation and activation through PI3K‐dependent pathways,( 29 , 30 , 36 , 37 ) illustrating the complexity of this regulation. However, one can observe that after prolonged exposure to rapamycin (2–3 days), the level of activated Akt returned to basal level in MCF‐7 breast cancer cells.( 30 ) Consistent with these data, the MVLN cells treated for 4 days with rapamycin did not display any variation in phospho‐ser473 Akt/total Akt ratio. However, in the resistant R‐MVLN cells, rapamycin treatment was associated with decreased Akt activation. These data suggest the existence of regulation mechanisms between mTOR inhibition and the upstream Akt pathway in the R‐MVLN cells, and that these complex feedback regulations could vary with the cellular context.
Rapamycin is known to have antiproliferative effects via inhibition of the G1 to S transition,( 38 ) but the exact mechanism involved has not been totally elucidated.( 39 , 40 , 41 ) To our knowledge, no study has ever investigated the impact of mTOR inhibition on the gene‐expression profile of breast cancer cells. In the present study, we sought to determine whether the cytostatic activity observed with the combination of rapamycin and fulvestrant was the consequence of the intrinsic antiproliferative action of rapamycin or was due to the combination of the two drugs. Our gene‐expression profiling study clearly demonstrated that the reversion of fulvestrant resistance by rapamycin in R‐MVLN cells was a direct consequence of the combination with fulvestrant, and was associated with the restoration of 40% of the fulvestrant gene‐expression signature observed in control MVLN cells. With particular focus on the biological processes associated with this restoration of the fulvestrant signature by rapamycin (Fig. 6c), we found that the ‘regulation of progression through the cell cycle’ was, not surprisingly, the biological process most represented. However, unexpected biological functions such as ‘RNA biosynthetic process’ were also identified, even though mTOR is mainly known as a ‘master regulator’ of the translational machinery (for review( 42 )). Yet, it has been observed that TOR could also control transcription, both in yeast and in mammals (for review( 43 )). Since 19.7% of the genes selected in the present study belong to the ‘RNA biosynthetic process’, future work is needed to decipher the importance of this function in rapamycin activity and/or reversion of endocrine therapy resistance. Finally, several genes selected in the present study have previously been found to be associated with endocrine therapy resistance and/or poor prognosis in ER + breast cancer cells: IL17RB;( 44 ) PTTG1;( 45 ) GATA3;( 46 ) CHD6;( 47 ) ORC6L, PRC1, CENPA, MCM6, RFC4, CCNE2;( 48 ) TIMELESS;( 49 ) BIRC5;( 50 ) BTG3, CTSB and TTK. ( 51 ) Taken together, our results have allowed the identification of genes that might represent key actors both in the development of endocrine therapy resistance and in the strategies aimed at reverting such resistance.
Following the demonstration that rapamycin exposure of R‐MVLN cells led both to reversion of endocrine therapy resistance at the proliferation level and to the reversion of part of the fulvestrant gene‐expression signature, the precise mechanisms by which mTOR inhibition leads to such events remained to be elucidated. However, the current study supports the existence of a close cross‐talk between the ERα and the mTOR pathways as a decrease in the proportion of the phospho‐ser167 ERα population to the benefit of an increase in the total amount of ERα was observed in rapamycin‐treated R‐MVLN cells. Complementary experiments demonstrated that the phosphorylation level of another serine residue (ser118), also located in the ligand‐independent transactivation AF‐1 domain of the ERα protein, was not affected by rapamycin treatment, whereas total ERα protein expression was still increased (data not shown). This would suggest, in rapamycin‐treated R‐MVLN cells, a shift of the balance between ligand‐independent/ligand‐dependent activity of ERα toward the ligand‐dependent activity which could represent part of the mechanisms involved in the reversion of OH‐Tam resistance and/or of fulvestrant resistance. ERα‐mediated activity is known to occur through either direct genomic activity of the receptor (estrogen response element (ERE)‐driven transcriptional activity), indirect genomic activity (indirect regulation of transcription by interference with other transcription factors such as the AP‐1 complex, NF‐κB or Sp1) or ERα‐non‐genomic activity.( 52 ) One study analyzed global gene expression in Tam‐resistant MCF‐7 cells and fulvestrant‐resistant MCF‐7 cells and showed that although Tam resistance preferentially alter expression of E2‐responsive genes, fulvestrant resistance is characterized by a strong remodelling of gene expression.( 53 ) This could explain why it was not possible to support the molecular observation of a shift in the total/phosphorylated ERα balance on well‐characterized ERE‐regulated genes such as TFF1/pS2 and PGR using the DNA array data obtained with fulvestrant ± rapamycin‐treated R‐MVLN cells (data not shown). However, one can not exclude that the modification in the total/phosphorylated ERα balance could represent part of the mechanisms mainly involved in the reversion of OH‐Tam resistance and not in the reversion of fulvestrant resistance in the R‐MVLN cells. Other rapamycin‐induced complex mechanisms are also certainly involved and future investigation is thus needed.
In conclusion, the present study has demonstrated that mTOR inhibition is sufficient to reverse endocrine therapy resistance in a breast cancer cell model that has acquired both endocrine therapy resistance and activation of an endogenous Akt/mTOR pathway under OH‐Tam exposure. This supports the idea that mTOR inhibitors might become very helpful for the clinical management of ER+ breast cancer, both to enhance the efficiency of endocrine therapies and to bypass or reverse endocrine therapy resistance. Thus, the association of an mTOR inhibitor with first‐line endocrine therapy, or with second‐line endocrine therapy in patients developing resistance to the first‐line treatment, would enhance treatment efficiency. This is consistent with data from a recent clinical phase II study testing a combination of mTOR inhibitors with the aromatase inhibitor letrozole and reporting a better progression‐free survival in the combination arm than in the letrozole alone arm.( 54 ) Another important question to be addressed in future studies is the impact of mTOR inhibitor treatment on de novo endocrine therapy resistance. Taken together, our observations support the idea that mTOR inhibitors represent one of the most promising therapeutic approaches for patients with ER+ breast cancers.
Declaration of interest
All co‐authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research report.
Acknowledgments
We thank M.D. Reynaud for editing the manuscript. S.E. Ghayad was supported by a fellowship from the Ministère de l’Education Nationale et de l’Enseignement Supérieur (France). This work was supported by grants from the Ligue Nationale Contre le Cancer (Comité de l’Ardèche, Comité de la Saône et Loire, Comité de Savoie) and the ARC (Association pour la Recherche sur le Cancer).
References
- 1. Veronesi U, Boyle P, Goldhirsch A, Orecchia R, Viale G. Breast cancer. Lancet 2005; 365 (9472): 1727–41. [DOI] [PubMed] [Google Scholar]
- 2. Osborne CK, Pippen J, Jones SE et al . Double‐blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: results of a North American trial. J Clin Oncol 2002; 20 (16): 3386–95. [DOI] [PubMed] [Google Scholar]
- 3. Perey L, Paridaens R, Hawle H et al . Clinical benefit of fulvestrant in postmenopausal women with advanced breast cancer and primary or acquired resistance to aromatase inhibitors: final results of phase II Swiss Group for Clinical Cancer Research Trial (SAKK 21/00). Ann Oncol 2007; 18 (1): 64–9. [DOI] [PubMed] [Google Scholar]
- 4. Huang S, Houghton PJ. Targeting mTOR signaling for cancer therapy. Curr Opin Pharmacol 2003; 3 (4): 371–7. [DOI] [PubMed] [Google Scholar]
- 5. Sun M, Paciga JE, Feldman RI et al . Phosphatidylinositol‐3‐OH Kinase (PI3K)/AKT2, activated in breast cancer, regulates and is induced by estrogen receptor alpha (ERalpha) via interaction between ERalpha and PI3K. Cancer Res 2001; 61 (16): 5985–91. [PubMed] [Google Scholar]
- 6. Yu J, Henske EP. Estrogen‐induced activation of mammalian target of rapamycin is mediated via Tuberin and the small GTPase Ras Homologue Enriched in Brain. Cancer Res 2006; 66 (19): 9461–6. [DOI] [PubMed] [Google Scholar]
- 7. Perez‐Tenorio G, Stal O. Activation of AKT/PKB in breast cancer predicts a worse outcome among endocrine treated patients. Br J Cancer 2002; 86 (4): 540–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tokunaga E, Kimura Y, Oki E et al . Akt is frequently activated in HER2/neu‐positive breast cancers and associated with poor prognosis among hormone‐treated patients. Int J Cancer 2006; 118 (2): 284–9. [DOI] [PubMed] [Google Scholar]
- 9. Campbell RA, Bhat‐Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3‐kinase/AKT‐mediated activation of estrogen receptor alpha: a new model for anti‐estrogen resistance. J Biol Chem 2001; 276 (13): 9817–24. [DOI] [PubMed] [Google Scholar]
- 10. Chang SB, Miron P, Miron A, Iglehart JD. Rapamycin inhibits proliferation of estrogen‐receptor‐positive breast cancer cells. J Surg Res 2007; 138 (1): 37–44. [DOI] [PubMed] [Google Scholar]
- 11. Sadler TM, Gavriil M, Annable T, Frost P, Greenberger LM, Zhang Y. Combination therapy for treating breast cancer using antiestrogen, ERA‐923, and the mammalian target of rapamycin inhibitor, temsirolimus. Endocr Relat Cancer 2006; 13 (3): 863–73. [DOI] [PubMed] [Google Scholar]
- 12. Treeck O, Wackwitz B, Haus U, Ortmann O. Effects of a combined treatment with mTOR inhibitor RAD001 and tamoxifen in vitro on growth and apoptosis of human cancer cells. Gynecol Oncol 2006; 102 (2): 292–9. [DOI] [PubMed] [Google Scholar]
- 13. Boulay A, Rudloff J, Ye J et al . Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin Cancer Res 2005; 11 (14): 5319–28. [DOI] [PubMed] [Google Scholar]
- 14. DeGraffenried LA, Friedrichs WE, Russell DH et al . Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt Activity. Clin Cancer Res 2004; 10 (23): 8059–67. [DOI] [PubMed] [Google Scholar]
- 15. Beeram M, Tan QT, Tekmal RR, Russell D, Middleton A, DeGraffenried LA. Akt‐induced endocrine therapy resistance is reversed by inhibition of mTOR signaling. Ann Oncol 2007; 18 (8): 1323–8. [DOI] [PubMed] [Google Scholar]
- 16. Demirpence E, Duchesne MJ, Badia E, Gagne D, Pons M. MVLN cells: a bioluminescent MCF‐7‐derived cell line to study the modulation of estrogenic activity. J Steroid Biochem Mol Biol 1993; 46 (3): 355–64. [DOI] [PubMed] [Google Scholar]
- 17. Badia E, Duchesne MJ, Semlali A et al . Long‐term hydroxytamoxifen treatment of an MCF‐7‐derived breast cancer cell line irreversibly inhibits the expression of estrogenic genes through chromatin remodeling. Cancer Res 2000; 60 (15): 4130–8. [PubMed] [Google Scholar]
- 18. Vendrell JA, Magnino F, Danis E et al . Estrogen regulation in human breast cancer cells of new downstream gene targets involved in estrogen metabolism, cell proliferation and cell transformation. J Mol Endocrinol 2004; 32 (2): 397–414. [DOI] [PubMed] [Google Scholar]
- 19. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 2003; 31 (4): e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu Z, Irizarry RA, Gentleman RC, Martinez Murillo F, Spencer F. A Model Based Background Adjustment for Oligonucleotide Expression Arrays. Baltimore: Johns Hopkins University; 2004. [Google Scholar]
- 21. Gentleman RC, Carey VJ, Bates DM et al . Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004; 5 (10): R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cohen P, Bouaboula M, Bellis M et al . Monitoring cellular responses to Listeria monocytogenes with oligonucleotide arrays. J Biol Chem 2000; 275 (15): 11 181–90. [DOI] [PubMed] [Google Scholar]
- 23. Al‐Shahrour F, Diaz‐Uriarte R, Dopazo J. FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics 2004; 20 (4): 578–80. [DOI] [PubMed] [Google Scholar]
- 24. Girault I, Tozlu S, Lidereau R, Bieche I. Expression analysis of DNA methyltransferases 1, 3A, and 3B in sporadic breast carcinomas. Clin Cancer Res 2003; 9 (12): 4415–22. [PubMed] [Google Scholar]
- 25. Vendrell JA, Bieche I, Desmetz C et al . Molecular changes associated with the agonist activity of hydroxy‐tamoxifen and the hyper‐response to estradiol in hydroxy‐tamoxifen‐resistant breast cancer cell lines. Endocr Relat Cancer 2005; 12 (1): 75–92. [DOI] [PubMed] [Google Scholar]
- 26. Fan M, Bigsby RM, Nephew KP. The NEDD8 pathway is required for proteasome‐mediated degradation of human estrogen receptor (ER)‐alpha and essential for the antiproliferative activity of ICI 182 780 in ERalpha‐positive breast cancer cells. Mol Endocrinol 2003; 17 (3): 356–65. [DOI] [PubMed] [Google Scholar]
- 27. Dauvois S, Danielian PS, White R, Parker MG. Antiestrogen ICI 164 384 reduces cellular estrogen receptor content by increasing its turnover. Proc Natl Acad Sci USA 1992; 89 (9): 4037–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oldham S, Hafen E. Insulin/IGF and target of rapamycin signaling: a TOR de force in growth control. Trends Cell Biol 2003; 13 (2): 79–85. [DOI] [PubMed] [Google Scholar]
- 29. Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF‐1R‐dependent mechanism. Oncogene 2007; 26 (13): 1932–1940. [DOI] [PubMed] [Google Scholar]
- 30. O’Reilly KE, Rojo F, She QB et al . mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006; 66 (3): 1500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Glaros S, Atanaskova N, Zhao C, Skafar DF, Reddy KB. Activation function‐1 domain of estrogen receptor regulates the agonistic and antagonistic actions of tamoxifen. Mol Endocrinol 2006; 20 (5): 996–1008. [DOI] [PubMed] [Google Scholar]
- 32. Johnston SR. Targeting downstream effectors of epidermal growth factor receptor/HER2 in breast cancer with either farnesyltransferase inhibitors or mTOR antagonists. Int J Gynecol Cancer 2006; 16 (Suppl 2): 543–8. [DOI] [PubMed] [Google Scholar]
- 33. Morgensztern D, McLeod HL. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer Drugs 2005; 16 (8): 797–803. [DOI] [PubMed] [Google Scholar]
- 34. Noh WC, Kim YH, Kim MS et al . Activation of the mTOR signaling pathway in breast cancer and its correlation with the clinicopathologic variables. Breast Cancer Res Treat 2008; 110 (3): 477–483. [DOI] [PubMed] [Google Scholar]
- 35. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor‐mTOR complex. Science 2005; 307 (5712): 1098–101. [DOI] [PubMed] [Google Scholar]
- 36. Shi Y, Yan H, Frost P, Gera J, Lichtenstein A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up‐regulating the insulin‐like growth factor receptor/insulin receptor substrate‐1/phosphatidylinositol 3‐kinase cascade. Mol Cancer Ther 2005; 4 (10): 1533–40. [DOI] [PubMed] [Google Scholar]
- 37. McDonald PC, Oloumi A, Mills J et al . Rictor and integrin‐linked kinase interact and regulate Akt phosphorylation and cancer cell survival. Cancer Res 2008; 68 (6): 1618–24. [DOI] [PubMed] [Google Scholar]
- 38. Hashemolhosseini S, Nagamine Y, Morley SJ, Desrivieres S, Mercep L, Ferrari S. Rapamycin inhibition of the G1 to S transition is mediated by effects on cyclin D1 mRNA and protein stability. J Biol Chem 1998; 273 (23): 14 424–9. [DOI] [PubMed] [Google Scholar]
- 39. Grewe M, Gansauge F, Schmid RM, Adler G, Seufferlein T. Regulation of cell growth and cyclin D1 expression by the constitutively active FRAP‐p70s6K pathway in human pancreatic cancer cells. Cancer Res 1999; 59 (15): 3581–7. [PubMed] [Google Scholar]
- 40. Law M, Forrester E, Chytil A et al . Rapamycin disrupts cyclin/cyclin‐dependent kinase/p21/proliferating cell nuclear antigen complexes and cyclin D1 reverses rapamycin action by stabilizing these complexes. Cancer Res 2006; 66 (2): 1070–80. [DOI] [PubMed] [Google Scholar]
- 41. Nelsen CJ, Rickheim DG, Tucker MM, Hansen LK, Albrecht JH. Evidence that cyclin D1 mediates both growth and proliferation downstream of TOR in hepatocytes. J Biol Chem 2003; 278 (6): 3656–63. [DOI] [PubMed] [Google Scholar]
- 42. Proud CG. Signalling to translation: how signal transduction pathways control the protein synthetic machinery. Biochem J 2007; 403 (2): 217–34. [DOI] [PubMed] [Google Scholar]
- 43. Mayer C, Grummt I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene 2006; 25 (48): 6384–91. [DOI] [PubMed] [Google Scholar]
- 44. Goetz MP, Suman VJ, Ingle JN et al . A two‐gene expression ratio of homeobox 13 and interleukin‐17B receptor for prediction of recurrence and survival in women receiving adjuvant tamoxifen. Clin Cancer Res 2006; 12 (7 Pt 1): 2080–7. [DOI] [PubMed] [Google Scholar]
- 45. Solbach C, Roller M, Fellbaum C, Nicoletti M, Kaufmann M. PTTG mRNA expression in primary breast cancer: a prognostic marker for lymph node invasion and tumor recurrence. Breast 2004; 13 (1): 80–1. [DOI] [PubMed] [Google Scholar]
- 46. Oh DS, Troester MA, Usary J et al . Estrogen‐regulated genes predict survival in hormone receptor‐positive breast cancers. J Clin Oncol 2006; 24 (11): 1656–64. [DOI] [PubMed] [Google Scholar]
- 47. Jansen MP, Foekens JA, Van Staveren IL et al . Molecular classification of tamoxifen‐resistant breast carcinomas by gene expression profiling. J Clin Oncol 2005; 23 (4): 732–40. [DOI] [PubMed] [Google Scholar]
- 48. Van 't Veer LJ, Dai H, Van De Vijver MJ et al . Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002; 415 (6871): 530–6. [DOI] [PubMed] [Google Scholar]
- 49. Tozlu‐Kara S, Roux V, Andrieu C et al . Oligonucleotide microarray analysis of estrogen receptor‐positive postmenopausal breast carcinomas: identification of HRPAP20 and TIMELESS as outstanding candidate markers to predict the response to tamoxifen. J Mol Endocrinol 2007; 39: 305–18. [DOI] [PubMed] [Google Scholar]
- 50. Yamashita S, Masuda Y, Kurizaki T et al . Survivin expression predicts early recurrence in early‐stage breast cancer. Anticancer Res 2007; 27 (4C): 2803–8. [PubMed] [Google Scholar]
- 51. Becker M, Sommer A, Kratzschmar JR, Seidel H, Pohlenz HD, Fichtner I. Distinct gene expression patterns in a tamoxifen‐sensitive human mammary carcinoma xenograft and its tamoxifen‐resistant subline MaCa 3366/TAM. Mol Cancer Ther 2005; 4 (1): 151–68. [PubMed] [Google Scholar]
- 52. Normanno N, Di Maio M, De Maio E et al . Mechanisms of endocrine resistance and novel therapeutic strategies in breast cancer. Endocr Relat Cancer 2005; 12 (4): 721–47. [DOI] [PubMed] [Google Scholar]
- 53. Fan M, Yan PS, Hartman‐Frey C et al . Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogens tamoxifen and fulvestrant. Cancer Res 2006; 66 (24): 11 954–66. [DOI] [PubMed] [Google Scholar]
- 54. Chollet P, Abrial C, Tacca O et al . Mammalian target of rapamycin inhibitors in combination with letrozole in breast cancer. Clin Breast Cancer 2006; 7 (4): 336–8. [DOI] [PubMed] [Google Scholar]