

Abstract
Recently, dendritic cells (DC) transfected with tumor RNA have been used as a cancer vaccine. The efficacy of a cancer vaccine using DC transfected tumor RNA was examined. Of particular interest was whether a vaccine using DC transfected with recrudescent tumor RNA is effective for the treatment of a regrowing tumor after prior immunotherapy. In addition, the usefulness of co‐transfection of granulocyte macrophage colony‐stimulating factor (GM‐CSF) mRNA to augment the DC vaccine was examined. CT26 tumor‐bearing mice were immunized by s.c. injection with DC transfected with CT26 mRNA (DC‐CT26). The cytotoxic activity against CT26 in mice immunized with DC‐CT26 was significantly higher than that in the control group (P < 0.001) and was augmented by GM‐CSF mRNA co‐transfection (P < 0.05), resulting in remarkable therapeutic efficacy in CT26 s.c. tumor models. Cytotoxic T lymphocytes induced by the vaccination using DC transfected with mRNA from the recrudescent tumor showed a potent cytotoxicity against the recrudescent CT26 tumor cells, which was significantly higher than the cytotoxicity induced by the vaccination using DC‐CT26 (P < 0.05). In addition, in a recrudescent tumor model, this vaccination suppressed the regrowing s.c. tumors, and was augmented by GM‐CSF mRNA co‐transfection (P < 0.05). These results suggested that vaccination therapy using DC simultaneously transfected with whole tumor RNA and GM‐CSF mRNA could generate therapeutic immune responses even against recrudescent tumor after prior vaccination. (Cancer Sci 2008; 99: 407–413)
Immunotherapy against cancer has been extensively studied. It has recently been reported that immunotherapy using antigen‐loaded dendritic cells (DC) (sipuleucel‐T) has provided a survival advantage against asymptomatic hormone‐refractory metastatic prostate carcinoma patients for the first time.( 1 ) However, most immunotherapy using single‐antigen‐loaded DC is insufficient because of heterogeneity of tumors. Another approach, utilizing total tumor proteins or tumor associated antigen (TAA) genes as a source of antigen have been developed; for example, a vaccine using DC loaded with tumor lysates,( 2 ) proteins( 3 ) or DNA.( 4 , 5 ) Recently, RNA has also been used for tumor vaccination.( 6 , 7 , 8 ) Boczkowski et al. reported the first functional data that DC transfected with mRNA‐encoding specific tumor antigens or total tumor RNA derived from tumor cells elicited potent specific cytotoxic T lymphocytes (CTL) responses against tumors.( 6 ) The strategy of using RNA has several potential advantages. First, RNA can be effectively amplified from a very small number of cells; therefore, unlike tumor‐extract vaccines, a small amount of tumor tissue is sufficient to prepare the material for vaccination.( 7 ) Second, the tumor antigen need not be identified and the presence of multiple tumor antigens reduces the risk of antigen‐negative escape mutants. Furthermore, it is not necessary to elucidate the molecular nature of the putative tumor antigens. Third, unlike DNA‐based vaccines, there is little danger of incorporation of RNA sequences into the host genome. On the other hand, there are several drawbacks to the use of RNA for vaccination. RNA is unstable, and it seems to be difficult to maintain its quality for clinical application; and, moreover, there is only a low level of protein expression caused by RNA transfection. However, in spite of this, several clinical trials of cancer vaccines using DC transfected with tumor derived RNA showed clinical feasibility and safety in the treatment of patients and also showed that obvious antitumor immune responses were induced.( 9 , 10 , 11 )
Another remarkable advantage of RNA‐loaded DC‐based vaccines is the ability to deal with the recrudescent tumor, even in the same patient, because RNA can be freshly prepared from the recurrent tumor via biopsy. It is very important to establish the method of enhancing the antitumor effect of tumor‐RNA‐loaded DC‐based vaccines, considering future clinical application. The functions of DC are affected by several immunostimulatory cytokines within the local tissue environment.( 12 ) In particular, granulocyte macrophage colony‐stimulating factor (GM‐CSF) is a potent stimulator of DC.( 13 ) Several investigators have reported that GM‐CSF gene‐transduced tumor vaccine results in efficient tumor suppression and survival benefit in mouse models.( 14 , 15 ) Furthermore, DC adenovirally transduced with the whole TAA gene and GM‐CSF gene strengthened T‐cell responses, especially their migratory capacity for draining the lymph node by CC chemokine receptor 7 (CCR7) expression.( 16 ) Therefore, it is possible that GM‐CSF is a good candidate for the enhancement of this vaccine therapy. This study was designed to investigate whether immunotherapy using DC transfected with tumor RNA is useful for the treatment of s.c. tumors. Of particular interest was to examine whether DC transfected with recrudescent tumor RNA are effective for the treatment of a regrowing tumor after vaccination using DC transfected with primary tumor RNA. Furthermore, the augmentation effect of co‐transfection of DC with GM‐CSF mRNA on this vaccine strategy was investigated.
Materials and Methods
Mice and cell lines. Female 6–8‐week‐old BALB/c (H‐2d) mice were purchased from Japan SLC (Hamamatsu, Japan). All of the mice were maintained under specific pathogen‐free conditions and used in accordance with institutional guidelines. The CT 26 (H‐2d), an undifferentiated low‐immunogenic murine colorectal adenocarcinoma cell line, were grown in Dulbecco's modified Eagle's medium (DMEM; Nissui Pharmaceutical, Tokyo, Japan) supplemented with 10% fetal bovine serum (FBS) in a humidified atmosphere of 5% CO2 at 37°C. The Meth‐A (H‐2d), a sarcoma cell line (RIKEN BioResource Center, Ibaraki, Japan), were grown in RPMI‐1640 medium (Nissui) supplemented with 10% FBS.
Pulsing of Meth‐A with the immunodominant peptide, AH‐1. Peptide AH‐1, nonamer SPSYVYHQF, derived from the envelope protein (gp70) of an endogenous ecotropic murine leukemia virus (MuLV) has been proved to be an immunodominant peptide.( 17 ) It was synthesized by Takara to a purity of more than 90% as determined by high‐performance liquid chromatography (HPLC) and acid analysis.( 18 ) Meth‐A (2 × 106) was cultured in complete medium containing 100 µg/mL of AH‐1 at 37°C for 3 h. The cells were then washed twice in phosphate‐buffered saline (PBS) and used as Meth‐A pulsed with AH‐1 (Meth‐A/AH‐1) for the experiments.
Generation of DC in vitro from bone marrow cells. RPMI‐1640 medium supplemented with 10% FBS, 2 mM L‐glutamine, 50 mM 2‐mercaptoethanol (Invitrogen), 100 U/mL penicillin and 100 µg/mL streptomycin (complete medium), was used to generate DC and induce CTL. DC were generated from murine bone marrow precursors.( 19 ) Briefly, murine bone marrow cells were cultured in complete medium that contained 200 U/mL recombinant murine (rm) GM‐CSF for 10 days. On day 10, non‐adherent cells were collected, and then placed at 2 × 106/well (Falcon3003) in 10 mL complete medium with 10 µg/mL lipopolysaccharide (LPS, Escherichia coli 0127: B8; Sigma, St. Louis, MO, USA) and 100 U/mL of rmGM‐CSF. The cells were incubated for 24 h, and non‐adherent cells were collected by gentle pipetting and were used for further studies. These DC preparations were stained with fluorescein isothiocyanate (FITC)‐conjugated monoclonal antibodies (mAb; BD Pharmingen, San Diego, CA, USA), against murine cell surface molecules, and were examined by flow cytometry.
Isolation of mRNA from tumor cell lines and tumors. Total RNA was isolated from tissue culture cells and implanted tumors using a commercial kit (Isogen; Nippon Gene, Japan) as per the manufacturer's protocols. Next, poly (A)+ RNA (mRNA) was isolated from the total RNA using a Poly (A)+ Isolation Kit from Total RNA (Nippon Gene) as per the manufacturer's protocols.
In vitro transcription of RNA. The plasmid pGEM4Z/mGM‐CSF was constructed by ligating an EcoRI and BamHI fragment from the pBluescript SKII+ mGM‐CSF (RIKEN) into the pGEM4Z (Promega, Madison, WI, USA). In vitro synthesis of mRNA was carried out using 2 µg linearized pGEM4Z/mGM‐CSF as template for in vitro transcription with the T7 promoter per the manufacturer's protocols (mMESSAGE mMACHINE Kit; Ambion, Austin, TX, USA) to generate 5′mGppG‐capped in vitro‐transcribed (IVT) mRNA. DNase‐treated mMESSAGE mMACHINE reaction was polyadenylated by a Poly (A) Tailing Kit (Ambion). pQBI‐T7‐GFP (Qbiogene, Irvine, CA, USA) was linearized with ClaI, followed by in vitro transcription. Following DNase‐treatment, the reaction was polyadenylated. The quality of all RNA preparations was determined by electrophoresis on agarose gels stained with ethidium bromide.
Transfection of DC with RNA. DC were transfected with mRNA using a Nucleofector device (Amaxa, Cologu, Germany) according to the manufacturer's protocol. DC were washed twice with PBS, and harvested and 1 × 106 DC were resuspended in 100 µL Mouse Macrophage Nucleofecto Solution (Amaxa). After the addition of mRNA (2–20 µg mRNA), the cells were electroporated using Amaxa program U‐01. Afterwards, the cells were cultured for 6 h in complete medium containing LPS (10 µg/mL) in 6‐well plates (Falcon). The transfection efficiency was monitored using green fluorescent protein (GFP) mRNA. DC were transfected with various amount of GFP mRNA, and cultured for 24 h. Next, the GFP expression was evaluated by flow cytometry. After the optimization of RNA electroporation, CT26 mRNA was electroporated into DC at 2 µg per sample. GM‐CSF mRNA was also electroporated into DC at 2–20 µg/sample. These DC were used for the experiments (DC‐CT26: DC transfected with CT26 mRNA; DC‐GM‐CSF: DC transfected with GM‐CSF mRNA; DC‐CT26/GM‐CSF: DC transfected with CT26 mRNA and GM‐CSF mRNA; DC‐CT26R: DC transfected with CT26 recrudescent tumor mRNA; DC‐GFP: DC transfected with GFP mRNA).
Flow cytometric analysis. The phenotypical analysis of DC preparations was performed using a FACS Calibur (Becton Dickinson, Mountain View, CA, USA) with Cell Quest software. DC (2 × 105) were incubated with specific antibodies in PBS for 30 min at 4 µg and rinsed twice. The cells were analyzed for CD86 and CCR7 expressions. The following antibodies were used for flow cytometry: FITC‐conjugated antimouse CD86 mAb (GL1) (BD Pharmingen) and goat antimouse CCR7 pAb (ImmunoDetect). FITC‐conjugated antigoat immunoglobulin (Ig)G (Dako) was used as a secondary antibody for the unconjugated Ab at 4 µg for 30 min. The expression level of GFP in DC‐GFP was also evaluated 24 h after transfection using flow cytometry.
Assays for GM‐CSF secretion. DC‐GM‐CSF were seeded at a concentration of 1 × 106 cells/well in a total volume of 1 mL complete medium in a 48‐well plate (Falcon) and cultured for 24 h, and then supernatants were harvested, and the GM‐CSF content was measured in duplicate using a mouse GM‐CSF ELISA kit (Endogen, Woburn, MA, USA).
Induction of tumor‐specific CTL and cytotoxic assay. To determine whether the administration of DC‐CT26 would induce CT26‐specific CTL and CTL activity would be enhanced by co‐transfection of GM‐CSF mRNA, BALB/c mice were immunized three times every 5 days by s.c. injections of 1 × 106 DC‐CT26, DC‐CT26/GM‐CSF or DC‐GFP. The spleens were removed 14 days after the last immunization, and then the in vivo‐primed spleen cells were co‐cultured (4 × 106/mL) with irradiated (10 000 rads) CT26 cells (4 × 105/mL) in complete medium containing rmIL‐2 50 IU/mL (BD Pharmingen). After 5 days of co‐culture, the in vivo‐restimulated spleen cells were assayed in a 4‐h 51Cr release assay as described previously.( 20 , 21 )
Experimental design of DC vaccine therapy for s.c. tumor model. To assess whether a preexisting s.c. tumor could be suppressed after immunization with DC transfected with mRNA derived from tumor, BALB/c mice (6–8 weeks old) were injected s.c. in the right flank with a lethal dose of 5 × 105 CT26 cells. Five days later, tumor‐bearing mice were injected three times every 5 days s.c. in the opposite flank with 1 × 106 DC transfected with mRNA. These mice were randomly divided into the following four groups (n = 7/group): group 1, treated with PBS; group 2, treated with DC‐GFP; group 3, treated with DC‐CT26; and group 4, treated with DC‐CT26/GM‐CSF. The size of the tumor was evaluated every 5 days by measuring the diameters with calipers and the volume was calculated using the following formula: (short diameter)2 × long diameter × 0.52. The animals were killed when the tumor reached a volume set before therapy according to the local animal committee requirements or when they became moribund before the expected tumor volume.
CT26 recrudescent tumor cell. Regrowing CT26 tumors after two vaccinations of DC‐CT26 is defined as recrudescent tumors. BALB/c mice were inoculated s.c. in the right flank with 5 × 105 CT26. Five days later, tumor‐bearing mice were treated twice every 5 days with 1 × 106 DC‐CT26. Three days after the last treatment, animals were sacrificed and the tumor in the right flank was removed. The tumor tissues were minced with opposing scalpels and cultured in complete medium for 3 days. Thereafter, the cells were used as CT26 recrudescent tumors (CT26R).
Induction of recrudescent tumor‐specific CTL and cytotoxicity assay. To determine if immunization of DC‐CT26R could induce CTL showing cytotoxicity against CT26 tumor, BALB/c mice were immunized three times with DC‐CT26, DC‐CT26R or DC‐GFP. Spleen cells were isolated 14 days after last immunization and restimulated in vitro for 5 days. Meth‐A, Meth‐A/AH‐1, CT26 and CT26R were used as target cells to determine the cytotoxic activity of effector cells. The results are shown as the means ± SD (n = 4/group).
Experimental design of DC vaccine therapy for recrudescent tumor model by revaccination. To determine whether the revaccination of DC‐CT26R could induce an antitumor effect on recrudescent tumor after vaccination of DC‐CT26 and whether co‐transfection of GM‐CSF mRNA could augment the antitumor effect of the revaccination with DC‐CT26R, BALB/c mice were inoculated s.c. in the right flank with 5 × 105 CT26 and in the left flank with 5 × 104 CT26. Five days later, tumor‐bearing mice (n = 5/group) were treated twice every 5 days with 1 × 106 DC‐CT26. Three days after the last treatment, animals were placed in a supine position after induction of anesthesia and the tumor in the right flank was removed and mRNA was extracted. Two days after tumor resection, they were randomly divided into the following four groups (n = 5/group): group 1, treated with PBS; group 2, treated with DC‐CT26; group 3, treated with DC‐CT26R (DC transfected with mRNA from the right flank tumor); and group 4, treated with DC‐CT26R/GM‐CSF and by vaccinations. The volume of the tumor in the left flank was evaluated every 5 days.
Statistical analysis. Quantitative results are expressed as the mean ± SD. A statistical analysis was performed by anova and Fisher's test using Statview 5.0 software (Abacus Concepts, Berkeley, CA, USA). P < 0.05 was considered statistically significant.
Results
mRNA transfection of DC by electroporation. The electroporation was first optimized by monitoring the transfection efficiency and DC viability using GFP mRNA. GFP expression was detected at all tested doses (2–20 µg mRNA), and increased in a dose‐dependent manner. The use of 20 µg mRNA resulted in the highest GFP expression (54.5%). On the other hand, the viability of transfected DC was similar to that of non‐transfected DC at any tested dose. Transgene expression in DC was detected first at 6 h after transfection and peaked at 24–48 h (data not shown).
In an assay for GM‐CSF secretion, DC transfected with GM‐CSF mRNA at various doses produced GM‐CSF in a dose‐dependent manner. In contrast, non‐transfected DC produced very low amounts of GM‐CSF. The use of 20 µg GM‐CSF mRNA resulted in the highest GM‐CSF production (4072.6 ± 454.6 pg/1 × 106 cells/mL, Table 1).
Table 1.
Granulocyte macrophage colony‐stimulating factor (GM‐CSF) production of dendritic cells (DC) transfected with GM‐CSF mRNA at various volumes (µg/1 × 106 cells)
| GM‐CSF mRNA (µg) | GM‐CSF (pg) |
|---|---|
| 0 | 94.9 ± 21.2 |
| 1 | 269.1 ± 29.3 |
| 2 | 491.9 ± 32.5 |
| 5 | 1883.8 ± 44.4 |
| 10 | 2599.6 ± 81.7 |
| 15 | 3596.2 ± 154.4 |
| 20 | 4072.6 ± 454.6 |
Expression of CD86 and CCR7 on DC transfected with tumor mRNA and GM‐CSF mRNA. Approximately 90% of the immature DC showed high levels of CD11c, more than 99% DC showed high levels of CD80, major histocompatibility complex (MHC) class I and MHC class II, and approximately 30% showed low levels of CD40 and CD86. DC stimulated with LPS (LPS‐DC) showed a higher expression of CD86 and CCR7 than immature DC. There was no difference in the expression of CD86 on LPS‐DC, DC‐CT26 and DC‐CT26/GM‐CSF. The expression of CCR7 on DC‐CT26/GM‐CSF was markedly higher than that on DC‐CT26, thus suggesting that it was markedly upregulated by GM‐CSF mRNA co‐transfection (P < 0.001, Fig. 1).
Figure 1.
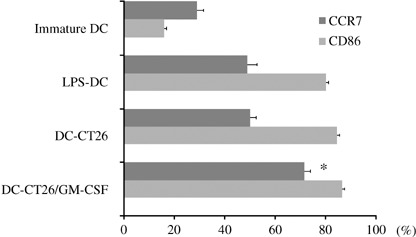
Expressions of CD86 and CCR7 on dendritic cells (DC) incubated with medium alone (immature DC), lipopolysaccharide (LPS)‐DC, DC‐CT26 and DC‐CT26/granulocyte colony‐stimulating factor (GM‐CSF). The percentages of positively stained cells are shown as the mean ± SD (n = 3/group). The expressions of CD86 and CCR7 on DC were analyzed by flow cytometry. *Significantly different from DC‐CT26 and LPS‐DC (P < 0.001).
Cytotoxic activity in spleen cells in mice immunized with DC transfected with tumor mRNA. The cytotoxic activity against CT26 in spleen cells of mice immunized with DC‐CT26 was significantly higher than that of mice immunized with DC‐GFP or PBS (P < 0.001; Fig. 2a). On the other hand, the cytotoxic activity against Meth‐A was low in all groups (Fig. 2b). The cytotoxic activity against CT26 in the spleen cells of mice immunized with DC‐CT26 was enhanced by co‐transfection with GM‐CSF mRNA (P < 0.05; Fig. 2a). The cytotoxic activity against CT26 induced by immunization with DC‐CT26 was almost completely blocked by an anti‐CD8 monoclonal antibody or anti‐MHC class I antibody, while this activity was not blocked by an anti‐CD4 antibody at all, thus suggesting that an MHC class I‐restricted CT26‐specific CD8+ CTL immunized response was induced by the immunization with DC‐CT26 (data not shown).
Figure 2.
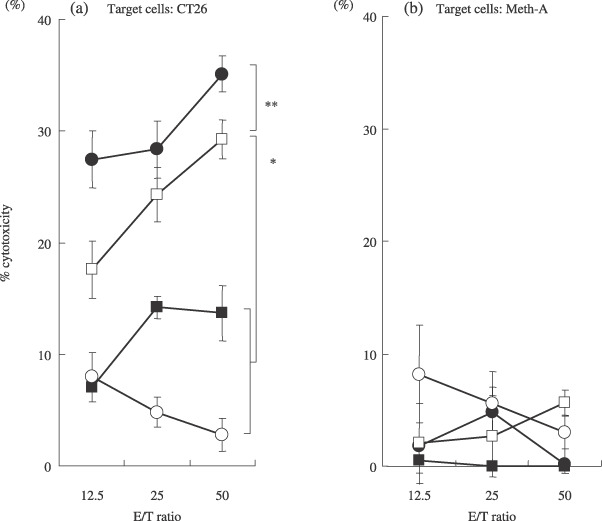
BALB/c mice (n = 4/group) received three vaccinations of DC‐CT26/GM‐CSF ( ), DC‐CT26 (
), DC‐CT26 ( ), DC‐green fluorescent protein (GFP) (
), DC‐green fluorescent protein (GFP) ( ) or phosphate‐buffered saline (PBS) (
) or phosphate‐buffered saline (PBS) ( ). Fourteen days after immunization, spleen cells were harvested in complete medium, and incubated with irradiated CT26. The cytotoxic activity was then examined using a 51Cr‐release assay. CT26 and Meth‐A were used as targets to determine the cytotoxic activity of effector cells. Results are shown as the means ± SD (n = 4/group). *Significantly different from DC‐GFP or PBS (P < 0.001; Fig. 2a). **Significantly different from DC‐CT26 RNA (P < 0.05).
). Fourteen days after immunization, spleen cells were harvested in complete medium, and incubated with irradiated CT26. The cytotoxic activity was then examined using a 51Cr‐release assay. CT26 and Meth‐A were used as targets to determine the cytotoxic activity of effector cells. Results are shown as the means ± SD (n = 4/group). *Significantly different from DC‐GFP or PBS (P < 0.001; Fig. 2a). **Significantly different from DC‐CT26 RNA (P < 0.05).
Therapeutic efficacy of DC transfected with tumor mRNA in s.c. tumor models. The therapeutic efficacy of vaccination therapy was assessed using DC transfected with tumor mRNA in vivo. In addition, the ability of co‐transfection of DC with GM‐CSF mRNA to augment the therapeutic efficacy of this vaccination was also assessed. Vaccination using DC‐CT26 showed remarkable therapeutic efficacy compared with vaccination using DC‐GFP (P < 0.001) or PBS (P < 0.0001) in this preexisting tumor model. In particular, the vaccination of DC‐CT26/G‐CSF elicited a more potent efficacy than that of DC‐CT26 (P < 0.01; Fig. 3), suggesting that the antitumor effect on CT26 was obviously induced by the vaccination of DC‐CT26, and it was significantly enhanced by co‐transfection with GM‐CSF mRNA. Interestingly, a tumor‐free mouse was observed only in the DC‐CT26/G‐CSF vaccination group (one of seven).
Figure 3.
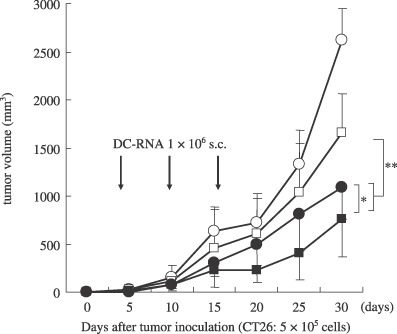
Inhibition of CT26 tumor growth in mice immunized with DC‐RNA in the tumor model. BALB/c mice were inoculated s.c. in the right flank with 5 × 105 CT26 cells. Five days later, tumor‐bearing mice (n = 7 mice/group) were treated with s.c. injection in the opposite flank with 1 × 106 DC‐CT26/GM‐CSF (), DC‐CT26 (), DC‐GFP () or PBS (). The results are presented as the mean tumor volumes ± SD of mice that developed tumors in each group. *Significantly different from DC‐CT26 (P < 0.01). **Significantly different from DC‐GFP (P < 0.001).
Cytotoxic activity in spleen cells in mice immunized with DC transfected with recrudescent tumor RNA. The cytotoxic activity against Meth‐A/AH‐1 in spleen cells in mice immunized with DC‐CT26 was similar to that in the mice immunized with DC‐CT26R, although the cytotoxic activity against Meth‐A was not recognized in both mice (Fig. 4a,b). The cytotoxic activity against CT26 in spleen cells of mice immunized with DC‐CT26 was higher than that in mice immunized with DC‐CT26R, but the difference was not significant (P = 0.554; Fig. 4c). On the other hand, the cytotoxic activity against CT26R in spleen cells in mice immunized with DC‐CT26R was significantly higher than that of mice immunized with DC‐CT26 (P < 0.05; Fig. 4d). These results suggested that the recrudescent tumor evaded the immune surveillance after the prior vaccine by modifying its antigeneity.
Figure 4.
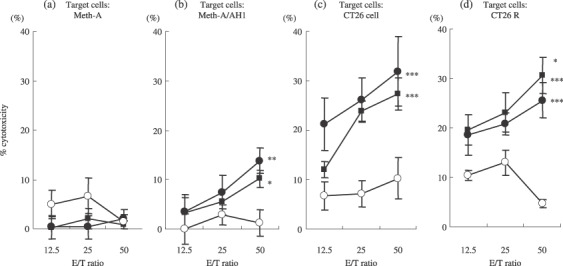
Efficacy of DC‐RNA vaccination in priming CTL. BALB/c mice were immunized three times by the s.c. injection of DC‐CT26 (), DC‐CT26R () or DC‐GFP () every 5 days. Spleen cells were isolated 14 days after immunization and restimulated in vitro for 5 days with irradiated CT26 cells in DC‐CT26 group and with CT26 recrudescent tumor in DC‐CT26R group. Meth‐A and Meth‐A/AH1, CT26 recrudescent tumor and CT26 cell were used as targets to determine the cytotoxic activity of effector cells. Results are shown as the means ± SD (n = 4/group). (b) *Significantly different from DC‐GFP (P < 0.05). **Significantly different from DC‐GFP (P < 0.01). (c) ***Significantly different from DC‐GFP (P < 0.001). (d) *Significantly different from DC‐CT26 (P < 0.05). ***Significantly different from DC‐GFP (P < 0.001).
Therapeutic efficacy of DC‐RNA in s.c. recrudescent tumor model. The therapeutic efficacy of vaccination therapy using DC transfected with mRNA from recrudescent CT26 tumor for the treatment of recrudescent tumor was examined after two vaccinations using DC transfected with mRNA from the primary tumor. The revaccination that used DC‐CT26R elicited a more potent therapeutic efficacy compared with a vaccination that used DC‐CT26 (P < 0.01). Furthermore, the co‐transfection of DC with GM‐CSF mRNA augmented the efficacy of this revaccination against CT26 recrudescent tumor (P < 0.05; Fig. 5). These results suggested that the revaccination using DC transfected with mRNA from recrudescent tumor is a favorable strategy for the treatment of recrudescent tumors, and co‐transfection with GM‐CSF mRNA is useful for the augmentation of this revaccination.
Figure 5.
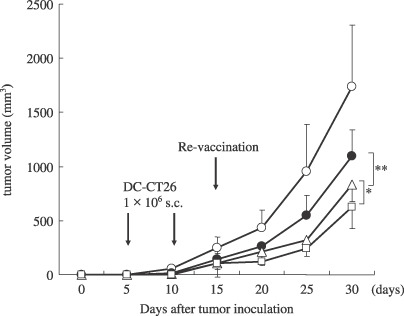
Therapeutic efficacy of genetically modified DC in s.c. tumor models. BALB/c mice were inoculated 5 × 105 CT26 cells in the right flank and 5 × 104 CT26 cells in the left flank. Five days later, tumor‐bearing mice (n = 5 group) were treated with s.c. injection in the other site with 1 × 106 DC‐CT26 twice 5 days apart. Three days after the second treatment, the tumors in the left flank were resected after induction of anesthesia and RNA was extracted from the tumor. Two days after, DC transfected with mRNA from the right flank tumor were inoculated as revaccinations. The results are presented as the mean tumor volumes in each group. *Significantly different from DC‐CT26R (, P < 0.05). **Significantly different from DC‐CT26 (P < 0.01).
Discussion
The present study demonstrated that vaccination therapy using DC transfected with tumor RNA could induce an antitumor immune response as Boczkowski et al. have reported.( 6 ) The vaccination using DC transfected with CT26 mRNA elicited tumor‐specific CTL, because they selectively lyzed CT26 tumor cells and AH‐1 pulsed Meth‐A. However, the cytotoxicity of these CTL was not sufficient to achieve an in vivo antitumor effect against CT26 s.c. tumors. Therefore, a mechanism for the augmentation of the antitumor effect is necessary. GM‐CSF mRNA was employed for the enhancement of this cancer vaccine therapy. GM‐CSF is a potent stimulator of DC and GM‐CSF gene transduction enhances the capacity of DC to induce a primary immune response.( 13 , 22 ) GM‐CSF plays an important role in the maturation, function and migration capacity of DC and mediates homing to lymphoid organs in response to the cognate ligands CCL19 and CCL21.( 23 ) In addition, it has recently been reported that GM‐CSF mRNA encapsulated in cationic liposomes enhances immunological responses induced by the vaccination with tumor associated antigens.( 24 ) Our previous studies demonstrated that a vaccination using DC co‐transduced with the TAA gene and the GM‐CSF gene elicited potent therapeutic immunity in s.c. tumors in mouse models,( 16 , 25 ) and also showed that the co‐transduction of the GM‐CSF gene enhanced the migratory capacity of DC for draining the lymph node by upregulation of CCR7 expression.( 16 ) In addition, GM‐CSF has other important functions for DC. We also showed that the lifespan of DC transfected with the GM‐CSF gene was prolonged, and GM‐CSF might protect DC from apoptosis induced by tumor‐derived transforming growth factor (TGF)‐β‐1 in the regional lymph nodes.( 26 )
In the present study, DC were simultaneously transfected with CT26 mRNA and the GM‐CSF mRNA, and it was confirmed that GM‐CSF was really secreted by transfected DC. These findings showed that specific CTL activity against CT26 in the spleen cells from mice immunized with DC transfected with CT26 was enhanced by co‐transfection of DC with the GM‐CSF mRNA. In addition, the antitumor effect of the vaccination was enhanced by co‐transfection with GM‐CSF mRNA in CT26 tumor models. However, this augmenting efficiency by co‐transfection of GM‐CSF mRNA was not as potent as that of adenoviral GM‐CSF gene transduction shown in a previous study.( 16 ) The reason for this is because the amount of GM‐CSF produced by DC transfected with GM‐CSF mRNA was reported to be only approximately 1% of the optimal dose produced by DC adenovirally transduced with the GM‐CSF gene in the previous study.( 16 )
One of the major advantages in using mRNA for TAA gene transfection into DC is to deal with residual tumors or recrudescent tumors. The critical obstacle of cancer immunotherapy is that tumors evade immune surveillance via several mechanisms, such as loss or downregulation of human leukocyte antigen (HLA) class I,( 27 ) antigen alternation or loss of tumor antigens,( 28 ) influence of immunosuppressive cytokines,( 29 ) and suppressor T cells.( 30 ) It is well documented that tumor antigen expression is heterogeneous, even within the same tumor. Decreased antigen expression has also been found in residual tumors after peptide vaccination.( 31 , 32 ) In the clinical studies of peptide vaccine for melanoma, the expression of tumor antigen has been found to obviously decrease in residual tumors after peptide vaccination.( 33 ) In the animal experiments, antigenic drift has been reported to cause tumor evasion.( 34 ) Tumor escape variants probably emerge after treatment with increasingly effective immunotherapy.( 35 ) It seems likely that, as T‐cell‐based tumor immunotherapy becomes stronger, escape mechanisms such as antigen loss are likely to become more prominent. Tumors can alter their antigen expression qualitatively through the mutation of antigenic epitopes. In fact, in the present study, CTL induced by the vaccination using DC‐transfected mRNA from the recrudescent tumor showed a potent cytotoxicity against the recrudescent tumor cells, which was significantly higher than the cytotoxicity induced by the vaccination using DC transfected with the primary tumor. These data were consistent with the therapeutic effect observed in s.c. tumor models. We speculated that the CT26 tumor altered the antigenicity after the vaccination using DC transfected with the primary CT26 tumor, and then a subpopulation of CT26 tumor cells survived and evaded immune surveillance, and therefore the vaccination using DC transfected with the recrudescent CT26 tumor was more effective than the vaccination using DC transfected with the primary CT26 tumor. However, further experiments comparing the gene expression profiles between CT26 and CT26R are necessary to assess the difference between CT26 and CT26R in terms of antigenicity.
Considering the clinical application, it is not possible to obtain a large amount of tumor tissue from patients with recurrent tumors. Therefore, a vaccine using DC pulsed with tumor lysate is not feasible in such situations. However, there is no such problem if tumor mRNA is used, because it can be amplified without loss of function.( 7 ) There is another advantage in using mRNA for genetic modification of DC: it is easy to transfect several cytokines at the same time. In the present study, DC were transfected with tumor mRNA together with GM‐CSF mRNA to augment the vaccine efficiency. Moreover, co‐transfection of other cytokine genes such as interleukin (IL)‐12 would be expected to further modify DC function.( 25 ) However, this vaccine strategy may not be expected to reduce a grown tumor, considering reports of previous clinical studies.( 9 , 10 , 11 ) Therefore, this strategy should be used to obtain a maximum benefit. For example, a cancer vaccine using DC transfected with mRNA extracted from surgical specimens may be useful as an individualized adjuvant immunotherapy to prevent metastasis and recurrences after surgery. Once the tumor has recurred, mRNA could be extracted from biopsy specimens of the recurrent tumor and amplified. Then, it could be transfected into DC together with cytokine mRNA such as GM‐CSF and so on. Vaccination using these DC may be useful for further immunotherapy.
In conclusion, the vaccine strategy using DC transfected simultaneously with tumor mRNA and GM‐CSF mRNA resulted in the generation of efficient therapeutic immune responses, even against recrudescent tumors. Therefore, this therapeutic strategy is promising for clinical application as an effective cancer vaccine both in an adjuvant setting after a surgical resection and in the treatment of recurrent tumors.
Acknowledgments
This study was supported by a Grant‐in‐Aid (no. 16390367) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
References
- 1. Small EJ, Schellhammer PF, Higano CS et al . Placebo‐controlled phase III trial of immunologic therapy with sipuleucel‐T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol 2006; 24: 3089–94. [DOI] [PubMed] [Google Scholar]
- 2. Cohen PA, Cohen PJ, Rosenberg SA, Mule JJ. CD4+ T‐cells from mice immunized to syngeneic sarcomas recognize distinct, non‐shared tumor antigens. Cancer Res 1994; 54: 1055–8. [PubMed] [Google Scholar]
- 3. Paglia P, Chiodoni C, Rodolfo M, Colombo MP. Murine dendritic cells loaded in vitro with soluble protein prime cytotoxic T lymphocytes against tumor antigen in vivo . J Exp Med 1996; 183: 317–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Condon C, Watkins SC, Celluzzi CM, Thompson K, Falo LD Jr. DNA‐based immunization by in vivo transfection of dendritic cells. Nat Med 1996; 2: 1122–8. [DOI] [PubMed] [Google Scholar]
- 5. Masafumi M, Tetsuya N, Kazunori Y et al . DNA vaccination of HSP105 leads to tumor rejection of colorectal cancer and melanoma in mice through activation of both CD4+ T cells and CD8+ T cells. Cancer Sci 2005; 96: 695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boczkowski D, Nair SK, Snyder D, Gilboa E. Dendritic cells pulsed with RNA are potent antigen‐presenting cells in vitro and in vivo . J Exp Med 1996; 184: 465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boczkowski D, Nair SK, Nam JH, Lyerly HK, Gilboa E. Induction of tumor Immunity and cytotoxic T lymphocyte responses using dendritic cells transfected with messenger RNA amplified from tumor cells. Cancer Res 2000; 60: 1028–34. [PubMed] [Google Scholar]
- 8. Nair SK, Morse M, Boczkowski D et al . Induction of tumor‐specific cytotoxic T lymphocytes in cancer patients by autologous tumor RNA‐transfected dendritic cells. Ann Surg 2002; 235: 540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hsu AK, Kerr BM, Jones KL, Lock RB, Hart DN, Rice AM. RNA loading of leukemic antigens into cord blood‐derived dendritic cells for immunotherapy. Biol Blood Marrow Transplant 2006; 12: 855–67. [DOI] [PubMed] [Google Scholar]
- 10. Morse MA, Nair SK, Mosca PJ et al . Immunotherapy with autologous, human dendritic cells transfected with carcinoembryonic antigen mRNA. Cancer Invest 2003; 21: 341–9. [DOI] [PubMed] [Google Scholar]
- 11. Caruso DA, Orme LM, Neale AM et al . Results of a phase 1 study utilizing monocyte‐derived dendritic cells pulsed with tumor RNA in children and young adults with brain cancer. Neuro-Oncol 2004; 6: 236–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jonuleit H, Knop J, Enk AH. Cytokines and their effects on maturation, differentiation and migration of dendritic cells. Arch Dermatol Res 1996; 289: 1–8. [DOI] [PubMed] [Google Scholar]
- 13. Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony‐stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor. J Exp Med 1994; 179: 1109–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dranoff G, Jaffee E, Lazenby A et al . Vaccination with irradiated tumor cells engineered to secrete murine granulocyte‐macrophage colony‐stimulating factor stimulates potent, specific, and long‐lasting anti‐tumor immunity. Proc Natl Acad Sci 1993; 90: 3539–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Motoyuki T, Masaharu Y, Toshiro O et al . Inhibition of RL male 1 tumor growth in BALB/c mice by introduction of the RLakt gene coding for antigen recognized by cytotoxic T‐lymphocytes and the GM‐CSF gene by in vivo electroporation. Cancer Sci 2003; 94: 308–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nakamura M, Iwahashi M, Nakamori M et al . Dendritic cells genetically engineered to simultaneously express endogenous tumor antigen and granulocyte macrophage colony‐stimulating factor elicit potent therapeutic antitumor immunity. Clin Cancer Res 2002; 8: 2742–9. [PubMed] [Google Scholar]
- 17. Huang AY, Gulden PH, Woods AS et al . The immunodominant major histocompatibility complex class I‐restricted antigen of a murine colon tumor derives from an endogenous retroviral gene product. Proc Natl Acad Sci 1996; 93: 9730–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakamura M, Iwahashi M, Nakamori M et al . Dendritic cells transduced with tumor‐associated antigen gene elicit potent therapeutic antitumor immunity: comparison with immunodominant peptide‐pulsed DCs. Oncol 2005; 68: 163–70. [DOI] [PubMed] [Google Scholar]
- 19. Lutz MB, Kukutsch N, Ogilvie AL et al . An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Meth 1999; 223: 77–92. [DOI] [PubMed] [Google Scholar]
- 20. Iwahashi M, Tanimura H, Yamaue H et al . Defective autologous mixed lymphocyte reaction (AMLR) and killer activity generated in the AMLR in cancer patients. Int J Cancer 1992; 51: 67–71. [DOI] [PubMed] [Google Scholar]
- 21. Terasawa H, Tanimura H, Nakamori M et al . Antitumor effects of interleukin‐2 gene‐modified fibroblasts in an orthotopic colon cancer model. Jpn J Cancer Res 1999; 90: 1000–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Curiel‐Lewandrowski C, Mahnke K, Labeur M et al . Transfection of immature murine bone marrow‐derived dendrite cells with the granulocyte‐macrophage colony‐stimulating factor gene potently enhances their in vivo antigen‐presenting capacity. Immunol 1999; 163: 174–83. [PubMed] [Google Scholar]
- 23. Randolph GJ. Dendritic cell migration to lymph nodes: cytokines, chemokines, and lipid mediators. Semin Immunol 2001; 13: 267–74. [DOI] [PubMed] [Google Scholar]
- 24. Hess PR, Boczkowski D, Nair SK, Snyder D, Gilboa E. Vaccination with mRNAs encoding tumor‐associated antigens and granulocyte‐macrophage colony‐stimulating factor efficiently primes CTL responses, but is insufficient to overcome tolerance to a model tumor/self antigen. Cancer Immunol Immunother 2006; 55: 672–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ojima T, Iwahashi M, Nakamura M et al . Successful cancer vaccine therapy for carcinoembryonic antigen (CEA)‐expressing colon cancer using genetically modified dendritic cells that express CEA and T helper‐type 1 cytokines in CEA transgenic mice. Int J Cancer 2007; 120: 585–93. [DOI] [PubMed] [Google Scholar]
- 26. Ojima T, Iwahashi M, Nakamura M et al . Benefits of gene transduction of granulocyte macrophage colony‐stimulating factor in cancer vaccine using genetically modified dendritic cells. Int J Oncol 2007; 31: 931–9. [PubMed] [Google Scholar]
- 27. Hicklin DJ, Marincola FM, Ferrone S. HLA class I antigen downregulation in human cancers: T‐cell immunotherapy revives an old story. Mol Med Today 1999; 5: 178–86. [DOI] [PubMed] [Google Scholar]
- 28. Sanchez‐Perez L, Kottke T, Diaz RM et al . Potent selection of antigen loss variants of B16 melanoma following inflammatory killing of melanocytes in vivo . Cancer Res 2005; 65: 2009–17. [DOI] [PubMed] [Google Scholar]
- 29. Girolomoni G, Ricciardi‐Castagnoli P. Dendritic cells hold promise for immunotherapy. Immunol Today 1997; 18: 102–4. [DOI] [PubMed] [Google Scholar]
- 30. Sakaguchi S, Sakaguchi N, Shimizu J et al . Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev 2001; 182: 18–32. [DOI] [PubMed] [Google Scholar]
- 31. Jager E, Ringhoffer M, Karbach J, Arand M, Oesch F, Knuth A. Inverse relationship of melanocyte differentiation antigen expression in melanoma tissues and CD8+ cytotoxic‐T‐cell responses: evidence for immunoselection of antigen‐loss variants in vivo. Int J Cancer 1996; 66: 470–6. [DOI] [PubMed] [Google Scholar]
- 32. Lee KH, Panelli MC, Kim CJ et al . Functional dissociation between local and systemic immune response during anti‐melanoma peptide vaccination. J Immunol 1998; 161: 4183–94. [PubMed] [Google Scholar]
- 33. Riker A, Cormier J, Panelli M et al . Immune selection after antigen‐specific immunotherapy of melanoma. Surgery 1999; 126: 112–20. [PubMed] [Google Scholar]
- 34. Bai XF, Liu J, Li O et al . Antigenic drift as a mechanism for tumor evasion of destruction by cytolytic T lymphocytes. J Clin Invest 2003; 111: 1487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khong HT, Restifo NP. Natural selection of tumor variants in the generation of ‘tumor escape’ phenotypes. Nat Immunol 2002; 3: 999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]