

Abstract
Aberrant methylation of promoter CpG islands is known to be a major inactivation mechanism of tumor‐suppressor and tumor‐related genes. In order to identify novel hypermethylated genes in early stage lung adenocarcinoma, we carried out methylated CpG island amplification, modified suppression subtractive hybridization, and methylation‐specific polymerase chain reaction to identify aberrant methylation of CpG islands in the A/J mouse lung adenoma model, which histologically mimics the early stage of human pulmonary adenocarcinoma. Through methylated CpG island amplification, suppression subtractive hybridization, and differential screening, we detected five genes, three of which have human homologs. Two of them showed downregulation of their expression in human lung adenocarcinoma. Of these two genes, we selected sterile α motif domain 14 (SAMD14) and further analyzed its methylation status and expression level by methylation‐specific polymerase chain reaction and quantitative real‐time polymerase chain reaction. Most of the lung adenocarcinoma cell lines showed suppressed expression of SAMD14 together with hypermethylation at the promoter region, although an immortalized bronchial epithelium cell line (PL16B) did not show hypermethylation and did express SAMD14. The expression of SAMD14 in A549 was rescued by treatment with the demethylation agent 5‐aza‐2′‐deoxycytidine. These data indicate that hypermethylation of the SAMD14 gene promoter region is associated with silencing of its expression. Hypermethylation at the CpG site of the SAMD14 promoter region was detected frequently in early invasive adenocarcinoma (8/24, 33.3%) but not in in situ adenocarcinoma (0/7, 0%) or normal lung tissue (0/31, 0%). Hypermethylation of the SAMD14 gene is a specific event in pulmonary adenocarcinogenesis and malignant progression. (Cancer Sci 2008; 99: 2177–2184)
Lung cancer is one of the most common malignancies, and is the leading cause of cancer‐related death in the world, including Japan.( 1 ) Adenocarcinoma is the predominant histological subtype of lung carcinoma in many countries, and its incidence is increasing.( 2 , 3 ) As in other types of cancer, survival of patients with lung adenocarcinoma depends on the stage at which it is diagnosed, the survival rate being higher if diagnosis is made early. Therefore, the best chance for survival in lung cancer is detection and treatment at an early stage and it is extremely important to find sensitive and specific molecular markers for early detection and patient classification.
Development of cancer typically involves inactivation of a series of tumor‐suppressor genes by point mutations and chromosomal deletions.( 4 ) However, it has recently become clear that epigenetic alterations also play an important role in cancer development. Many genes contain CpG islands in their promoter regions, and aberrant methylation of these regions in cancer leads to transcriptional silencing. Epigenetic changes are heritable through cell division, and result in altered gene activity without any accompanying changes in the DNA sequence.( 5 , 6 ) The best‐characterized and most easily quantifiable epigenetic alteration in cancer is a change in the pattern of DNA methylation.( 7 ) Carcinogenesis is also accompanied by changes in the methylation of genomic DNA.( 8 ) These changes include aberrant local hypermethylation of promoters of various tumor‐suppressor genes, which results in their silencing, and whole‐genome hypermethylation accompanied by activation of oncogenes, retrotransponsons, and genomic instability.( 9 ) Aberrant methylation of CpG islands within the promoter region of tumor‐suppressor genes, which leads to epigenetic silencing of these genes, is a frequent and key event in both early stage and late‐stage pathogenesis of lung tumors.( 10 , 11 , 12 )
Small lung adenocarcinomas (2 cm in diameter or less) can be divided into two major groups, each of which is further subdivisible into three types.( 13 ) One of these two major groups is the replacement growth type, which replaces the pulmonary alveolar structures as it grows. The other is the non‐replacement growth type, which destroys the original alveolar framework during growth. The three subtypes in the former group include localized bronchioloalveolar carcinoma without active fibroblastic proliferation (types A and B) (in situ adenocarcinoma) and localized bronchioloalveolar carcinoma with active fibroblastic proliferation (type C; early invasive carcinoma). These three replacement‐type lung adenocarcinomas are thought to be early stage tumors.
The goal of the present study was to assess the potential of a molecular marker‐based approach for the early detection of human lung cancer. Although it would be most useful to study abnormal methylation in the early stages of human adenocarcinoma, it is very difficult to obtain fresh high‐quality material from such tumors. In order to screen for abnormal gene methylation in adenocarcinoma, we used the A/J mouse model of adenoma induced by 4‐(methylnitrosamino)‐1‐(3‐pyridyl)‐1‐butanone (NNK).( 14 ) There are several techniques for identifying genes that are differentially methylated between normal and tumor tissues, such as methylated CpG island amplification (MCA)‐suppressive subtractive hybridization (SSH), MCA‐representational difference analysis, and methylation‐specific microarray. In the present study, we chose MCA‐SSH to identify a set of genes that were expressed differentially in A/J mouse lung adenoma and normal lung tissue. We identified a previously uncharacterized gene, sterile α motif domain 14 (SAMD14), and analyzed the methylation status of its promoter region and its expression level in lung cancer cell lines and in the early stages of human pulmonary adenocarcinoma.
Materials and Methods
Animal and chemicals. A/J mice were purchased at 5 weeks of age from Japan SLC (Shizuoka, Japan), and were housed five per cage at 23 ± 2°C, 55 ± 5% relative humidity and 14‐h light/10‐h dark cycle. They were maintained on an MF (Oriental Yeast Company, Tokyo, Japan) diet with water ad libitum throughout the study. After a 2‐week acclimation, at 7 weeks of age each mouse was given a single intraperitoneal dose of 4 mg NNK in saline.( 14 ) All of the mice were killed at 52 weeks after the carcinogen treatment. The lungs were excised immediately and embedded in Tissue‐Tek OCT Compound (Sakura Finetek Japan, Tokyo, Japan), frozen in dry ice–acetone, and stored at –80°C until analysis.
Tissues and histological classification. A/J mouse lung adenoma and normal tissue, obtained as described above, were used for subtractive analysis of hypermethylated CpG islands (Fig. 1a).
Figure 1.
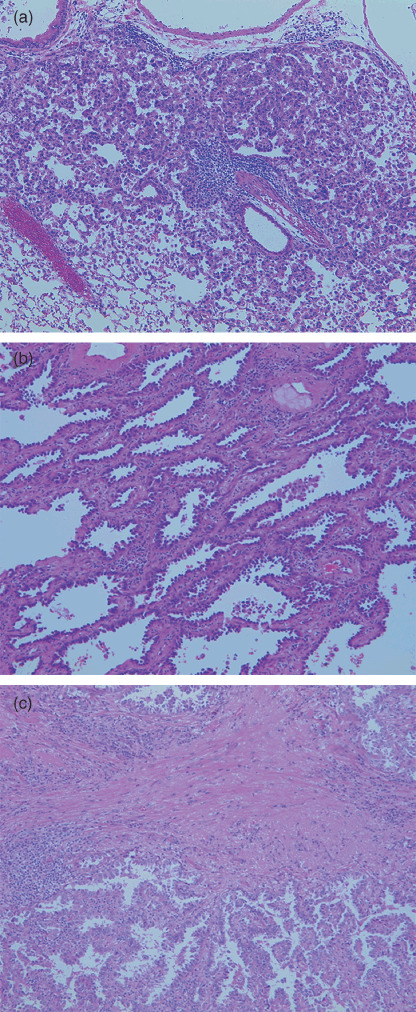
(a) Histology of A/J mouse adenoma used for the methylated CpG island amplification–suppression subtractive hybridization procedure. (b) Human lung adenocarcinoma (in situ adenocarcinoma, type A). (c) Human lung adenocarcinoma (early invasive adenocarcinoma, type C).
Thirty‐one paired samples of small‐sized lung adenocarcinoma, less than 2 cm in diameter, and adjacent non‐cancerous tissue were obtained from a bank of surgically resected material at the University Hospital of Tsukuba (Ibaraki, Japan). All patients had given informed consent for specimen collection. The samples were from 13 male and 18 female patients (average age, 62.8 years; women, 62.7 years; men, 62.9 years). All of the cases were diagnosed histologically according to the World Health Organization classification,( 15 ) and the Noguchi classification for small‐sized adenocarcinomas (Fig. 1b,c).( 13 ) A small amount of each specimen was embedded directly in Tissue‐Tek OCT Compound and frozen immediately in dry ice–acetone. The specimens were then stored at –80°C until analysis.
Cell lines. We used the lung cancer cell lines A549,( 16 ) Calu‐3,( 17 ) PC‐14 (Riken Cell Bank, Tsukuba, Japan), RERF‐LC‐KJ,( 18 ) and NCI‐H23.( 19 ) PL16T was derived from atypical adenomatous hyperplasia (AAH) and PL16B was derived from the resected end of the normal bronchus of the same patient.( 20 ) Most cell lines were propagated in RPMI‐1640 (Invitrogen, Carlsbad, CA, USA) and 10% fetal calf serum, except A549 and Calu‐3, which were cultured in Dulbecco's modified Eagle Medium: Nutrient Mixture F‐12 (DMEM/F‐12) and mininum essential media alpha (MEM‐alpha) (Invitrogen) containing 10% fetal calf serum.
Methylated CpG island amplification of A/J mouse lung adenoma tissue and normal lung tissue. MCA was carried out as described by Toyota et al.( 21 ) We used two frozen cryostat sections (10 µm) of A/J mouse normal lung and adenoma tissue. Genomic DNA was extracted by digestion with proteinase K, followed by phenol–chloroform extraction. Five micrograms of DNA extracted from A/J mouse lung adenoma tissue or its normal counterpart was digested with 100 U SmaI for 16 h (all restriction enzymes were from New England Biolabs, Beverly, MA, USA). The DNA was then digested with 20 U XmaI for 16 h. Purified DNA (0.5 µg) was ligated to RXMA12/24 (RXMA12, 5′‐CCGGGTCGGTGA‐3′; RXMA24, 5′‐AGCACTCTCCAGCCTCTCACCGAC‐3′) or RMCA12/24 (RMCA12, 5′‐CCGGGCAGAAAG‐3′; RMCA24, 5′‐CCACCGCCATCCGAGCCTTTCTGC‐3′) adaptors with T4 ligase (New England Biolabs) and amplified by polymerase chain reaction (PCR).
Modified suppression subtractive hybridization( 22, 23 ) and differential screening. SSH was carried out on MCA amplicons between A/J mouse lung adenoma tissue and counterpart tissue by using a PCR‐Select cDNA subtraction kit (BD Bioscience Clontech, Palo Alto, CA, USA) with some modifications.( 23 ) Briefly, MCA amplicons were digested with the restriction enzyme SmaI to remove the RMCA 24‐mer adaptor or the RXMA 24‐mer adaptor and purified with CHROMA SPIN Columns (Clontech Laboratories, Mountain View, CA, USA). SSH was carried out according to the manufacturer's instructions, and the subtractive hybridization products were amplified by PCR. First, diluted tester DNA (MCA amplicon of A/J mouse lung adenoma tissue) was ligated to adaptor with T4 DNA ligase (both from the Clontech PCR‐Select cDNA Subtraction Kit) and subtraction was carried out. After the subtraction analysis, the forward subtracted MCA amplicon of A/J mouse adenoma tissue was inserted into the T/A cloning vector pCR 2.1 (Invitrogen) and the plasmids were transformed into One Shot DH5α‐T1R E. coli (Invitrogen). Individual transformants carrying subtracted DNA fragments were isolated from white colonies on 5‐bromo‐4‐chloro‐3‐indolyl‐β‐D‐galactopyranoside agar plates.
A total of 930 clones were picked randomly. Their insert DNA was amplified by PCR, blotted onto nylon membranes (BM Equipment, Tokyo, Japan), and hybridized with the forward‐ and reverse‐subtraction probes that had been radiolabeled with [32P]dCTP by random priming. Dot blot hybridization was carried out according to the manufacturer's protocol (PCR‐Select Differential screening kit; BD Bioscience Clontech). Visualization was achieved by exposure to Kodak BioMax XAR films (Eastman Kodak, Rochester, NY, USA).
Sequencing and homology search. One hundred and seven clones were selected by differential screening for sequence analysis using the BigDye Terminator v3.1 Cycle Sequencing ready reaction kit and an ABI PRISM 310 Genetic Analyzer (both from Applied Biosystems Japan, Tokyo, Japan). Sequence homologies were identified from the National Center for Biotechnology Information (NCBI) resource with the basic local alignment search tool (BLAST).
Real‐time quantitative reverse transcription–polymerase chain reaction gene expression analysis in A/J mouse tissue. Total RNA was isolated with Trizol (Invitrogen) according to the manufacturer's instructions, and samples were stored at –70°C until use. The quality of RNA was determined with an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). A 1‐µg sample of RNA was used for cDNA synthesis with TaKaRa ExscriptR RT reagent Kit (Perfect Real Time; Takara Bio, Otsu, Shiga, Japan). Expression of target genes was evaluated by real‐time reverse transcription–PCR based on the SYBR Green I method and a Gene Amp 7300 real‐time PCR system (Applied Biosystems Japan). The PCR primers were purchased from Takara Bio and the primer sequences used are shown in Table 1. Numbers of target cDNA molecules were normalized to numbers of glyceraldehyde‐3‐phosphate dehydrogenase cDNA molecules as a control.
Table 1.
Primer sets used for quantitative reverse transcription–polymerase chain reaction
| GenBank accession no. | Gene name | Primer (5′‐3′) |
|---|---|---|
| NM_146025 | Mus‐Samd14 | Sense: 5′‐GCTCCGAGAACCTGTGGATGA‐3′ Antisense: 5′‐AGCCCTGGCTTTGTGTAAGCTG‐3′ |
| NM_016705 | Mus‐Kif21a | Sense: 5′‐TGACTCCCAGCAGGAGCAGA‐3′ Antisense: 5′‐CGTCAAATCCGGTTCCCATC‐3′ |
| NM_147776 | Mus‐Vwa1 | Sense: 5′‐GATGCTGTTCTGGACTGCGTTC‐3′ Antisense: 5′‐CATAGTGTGACACGCTGGCTGA‐3′ |
| NM_174920 | Hs‐SAMD14 | Sense: 5′‐AGACGCTGAAGATGACCGATG‐3′ Antisense: 5′‐ACGGGTTCTCGGAGCTTTG‐3′ |
| NM_017641 | Hs‐KIF21A | Sense: 5′‐CGAGAGTCCAGGCCTTACCAAC‐3′ Antisense: 5′‐GCCACTTCATGCGAGCTGTC‐3′ |
HomoloGene search and quantitative real‐time polymerase chain reaction expression check for homologous genes in human tissue. Human homolog genes were identified using the HomoloGene search in the NCBI. The SAMD14 and KIF21A genes were examined for their expression levels in human lung adenocarcinoma cell lines and resected materials, including tumor tissue and adjacent normal tissue, by quantitative real‐time polymerase chain reaction (qRT‐PCR). The primer sequences used are shown in Table 1.
Bisulfite sequencing (BS) and methylation‐specific polymerase chain reaction (MSP) of the SAMD14 promoter region. DNA from human lung adenocarcinoma tissue and its counterpart normal tissue was subjected to bisulfite treatment. One microgram of DNA was denatured with NaOH and modified with sodium bisulfite as reported previously.( 24 , 25 ) CpG islands in the promoter regions of SAMD14 were identified using CpG Island Searcher.( 26 ) CpG islands were defined by the following criteria: CG > 55%, observed CpG/expected CpG > 0.65, and length > 500 bp. For bisulfite sequencing, modified genomic DNA of five pairs of samples was subjected to PCR with the following bisulfite sequencing primer set: BS (sense), 5′‐TTTTGTTAGTTGGGTTTA TGG‐3′; and BS (antisense), 5′‐TACAACTACTTCTCTACCCRC‐3′. The region from –454 to –147 was amplified and five clones were sequenced from each of the specimens. For MSP, the primers for the methylated reaction were: MSP‐SAMD14 (sense), 5′‐AGGGGTGTTTTTTTTC‐3′; and MSP‐SAMD14 (antisense), 5′‐ACGATAAACAAAACCCCCG‐3′. The primers for the unmethylated reaction were: USP‐SAMD14 (sense), 5′‐GTTAGGG GTGTGTTTTTTTTTTT‐3′; and USP‐SAMD14 (antisense), 5′‐AACAATAAACAAAACCCCCACC‐3′. The PCR amplification of modified DNA samples consisted of: one cycle of 94°C for 10 min; 35 cycles of 94°C for 30 s, 64°C for 45 s, and 72°C for 30 s; and one cycle of 72°C for 7 min. Methyl Primer Express Software v. 1.0 (Applied Biosystems Japan) was used to design the biased MSP, unmethylated‐specific PCR (USP), and BS primers.
Using these primers, lung adenocarcinoma cell lines, including A549, Calu‐3, RERF‐LC‐KJ, NCI‐H23, PL16B, and PL16T, and 31 cases of early stage lung adenocarcinoma were examined for their methylation status at the promoter region of the SAMD14 gene with the MSP method.
5‐Aza‐2′‐deoxycytidine treatment. The lung cancer cell line A549 was treated with culture medium containing a demethylating agent, 5‐aza‐2′‐deoxycytidine (5‐Aza; Sigma‐Aldrich Japan, Tokyo, Japan). 5‐Aza was dissolved in dimethyl sulfoxide (DMSO). A549 cells (1 × 106 cells/100 mm dish) were incubated in culture medium with 5‐Aza (final concentration 2 or 10 µmol/L) and without 5‐Aza (DMSO final concentration 2 or 10 µmol/L) for 2 days. The culture medium was changed every day. After cell harvesting, RNA was extracted for qRT‐PCR.
Statistical analysis. Associations of SAMD14 and KIF21A gene expression were evaluated with the two‐sided Student's t‐test and Mann–Whitney U‐test. Associations between SAMD14 promoter methylation status in human adenocarcinoma and its counterpart normal lung tissue were evaluated with Fisher's exact test. A P‐value of <0.05 was taken to signify statistical significance.
Animal welfare. Animal experiments were carried out in a humane manner after receiving approval from the Institutional Animal Experiment Committee of the University of Tsukuba. All studies were carried out in accordance with the Regulation for Animal Experiments in our university and the Fundamental Guideline for Proper Conduct of Animal Experiments and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Results
Identification of methylated genes in A/J mouse adenoma by methylated CpG island amplification–suppressive subtractive hybridization differential screening. To identify aberrantly methylated genes that were also differentially expressed in A/J mouse lung adenoma tissue and its counterpart normal tissue, we carried out MCA‐SSH genome scanning. This approach first involves isolation of CpG‐rich SmaI‐digested fragments and generation of a methylation‐specific library by the MCA method reported previously by Toyota et al.( 21 ) Then, using A/J mouse adenoma tissue as the tester and its counterpart normal tissue as the driver, we carried out SSH to exclude sequences common to the tester and driver populations. The amplified subtracted DNA fragments (A/J mouse adenoma minus normal lung) were then subcloned. After transformation, 930 white colonies were selected randomly and cultured. By T/A cloning and differential screening, 107 clones from a total of 930 clones were selected for sequence analysis and BLAST searching. However, most of the sequences did not match with real genes, and only five differentially methylated genes were identified, namely LOC674120, Kif21a, Samd14, EG436235, and Vwa1. Detailed information regarding these five genes can be found in Table 2 and the supplementary material.
Table 2.
Genes identified in the DNA fragments isolated by methylated CpG island amplification–suppressive subtractive hybridization
| Official symbol Mus musculus | Gene name | Official symbol Homo sapiens |
|---|---|---|
| LOC674120 | Similar to Ig heavy chain V region MC101 precursor | – |
| Kif21a | Kinesin family member 21A | KIF21A |
| Samd14 | Sterile α motif domain containing 14 | SAMD14 |
| EG436235 | Similar to regulator of G‐protein signaling 3 | – |
| Vwa1 | Von Willebrand factor A domain containing 1 | VWA1 |
Quantitative expression analysis of genes in A/J mouse lung adenoma tissue and normal tissue, and of homologous genes in human pulmonary adenocarcinoma tissue and normal tissue. We used Entrez Gene at the NCBI (http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene) to browse HomoloGene for human homologs of the five genes identified. Human homologs of three of the genes were identified, namely KIF21A, SAMD14, and VWA1 (Table 2). To assess differences in the expression levels of the genes selected by MCA‐SSH differential screening, we carried out qRT‐PCR assays using seven pairs of A/J mouse lung adenoma and non‐tumorous tissues. As shown in Figure 2, qRT‐PCR confirmed that the genes Samd14 and Kif21A were expressed at significantly lower levels in A/J mouse lung adenoma tissues than in paired non‐tumorous tissues. Consequently, the two human genes SAMD14 and KIF21A were selected for further analysis of their expression levels in human pulmonary adenocarcinoma and counterpart normal lung. We carried out qRT‐PCR on total RNA extracted from 18 lung adenocarcinoma tissues and nine counterpart normal lung tissues. As shown in Figure 3, the expression levels of the SAMD14 and KIF21A genes were significantly lower in human pulmonary adenocarcinoma tissue than in counterpart normal lung tissue.
Figure 2.
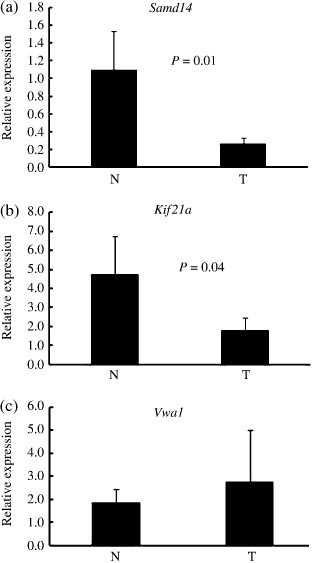
Quantitative real‐time reverse transcription–polymerase chain reaction analysis of genes: (a) Samd14, (b) Kif21a, and (c) Vwa1 in A/J mouse lung adenoma tissue (T) and its adjacent normal tissue (N). All of the genes selected by methylated CpG island amplification, modified suppression subtractive hybridization, and differential screening. (N, seven cases; T, seven cases). Bars show means ± SD. *Difference was statistically significant (P < 0.05).
Figure 3.
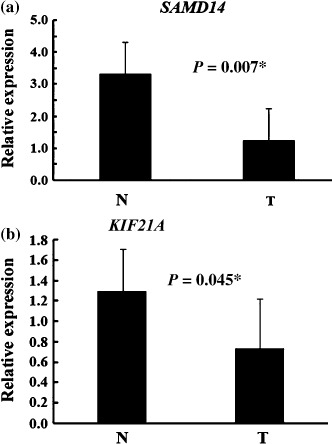
Quantitative real‐time reverse transcription–polymerase chain reaction analysis of the (a) SAMD14 and (b) KIF21A genes in small‐sized human lung adenocarcinomas (T) and human normal lung tissue (N). All measurements are shown relative to the expression level of the glyceraldehyde 3‐phosphate dehydrogenase gene. Bars show means ± SD. *Difference was statistically significant (P < 0.05). (N, 18 cases; T, nine cases).
Methylation status of the SAMD14 promoter region and downregulation of its expression in lung cancer cell lines. On the basis of the qRT‐PCR results, we investigated the methylation status of the promoter regions of the SAMD14 and KIF21A genes, located upstream of the transcription start site, by bisulfite sequencing and MSP. The promoter regions of these two genes were identified by use of WWW Promoter Scan (http://www‐bimas.cit.nih.gov/molbio/proscan/), and CpG islands in the promoter regions of these two genes were identified using CpG Island Searcher.( 26 ) In this experiment, the designed primer for bisulfite sequencing of the KIF21A gene did not yield PCR products. For KIF21A, we also designed 10 sets of MSP primers, but none of them worked. Therefore, in the present report we focused on the SAMD14 gene.
The promoter site of SAMD14 has a CG content of over 68%. For bisulfite sequencing, modified genomic DNA of five pairs of samples was subjected to PCR using bisulfite sequencing primer sets designed to amplify the region of interest. Then, PCR products were subcloned and sequenced. We examined five clones for each sample. The sequencing results showed that most clones of lung adenocarcinoma tissues were methylated but that no clones showed methylation in normal lung tissue (Fig. 4). From the results of bisulfite sequencing, we defined primer sets targeting the sequence around the most frequently methylated sites and carried out MSP using these primer sets.
Figure 4.
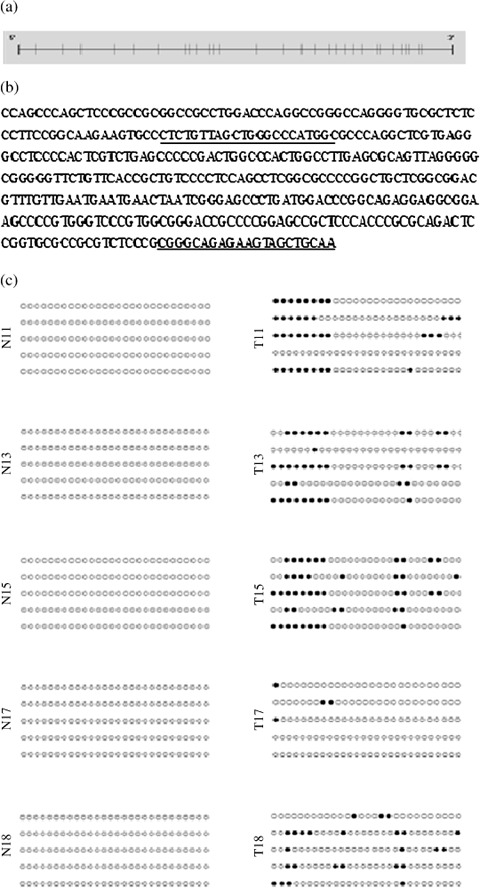
Methylation status of the SAMD14 promoter in lung adenocarcinoma tissues and its adjacent normal tissues. (a) SAMD14 gene promoter. The upright line shows the CpG site. (b) SAMD14 promoter sequence before bisulfate modification. The underlining shows the primer sequence. (c) Methylation status of the SAMD14 promoter region in 5 lung adenocarcinomas (T) and their adjacent normal tissues (N). , Methylation CpG site;
, Methylation CpG site;  , unmethylation site.
, unmethylation site.
To confirm the relationship between methylation and expression of SAMD14, the promoter methylation status of SAMD14 was investigated by MSP and qRT‐PCR in five lung cancer cell lines and two immortalized cell lines originating from atypical adenomatous hyperplasia (PL16T) and normal bronchial epithelium (PL16B). As shown in Figure 5, the expression of SAMD14 was suppressed in four of five adenocarcinoma cell lines that showed hypermethylation of the SAMD14 promoter region. However, the immortalized normal bronchial cell line PL16B showed no methylation of SAMD14 and its mRNA expression of SAMD14 was preserved.
Figure 5.
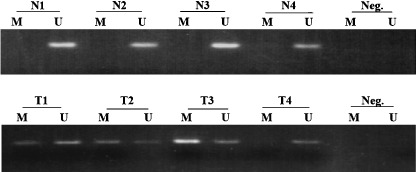
Representative methylation‐specific polymerase chain reaction (MSP) of the SAMD14 promoter region in human pulmonary adenocarcinoma tissues (T) and their counterpart normal tissues (N). The presence of a visible polymerase chain reaction product in lane M indicates the presence of methylation and in lane U indicates a lack of methylation.
Next, to determine whether promoter methylation played a role in SAMD14 expression, we picked up A549, which showed low expression of SAMD14 and hypermethylation of its promoter region (Fig. 6). A549 was treated with the demethylation agent 5‐Aza and the rescue of its SAMD14 expression was examined. As shown in Figure 7, the expression of SAMD14 was rescued by treatment with 5‐Aza, indicating that aberrant DNA methylation is one of the pathways of transcriptional silencing of the SAMD14 gene in the A549 cell line.
Figure 6.
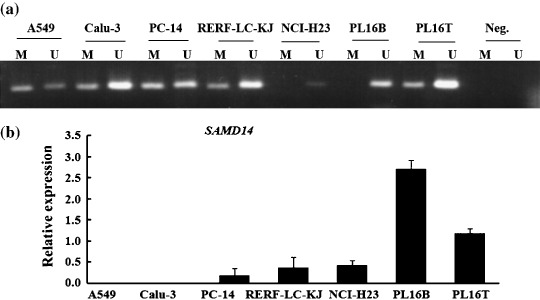
(a) Methylation‐specific polymerase chain reaction (MSP) of the SAMD14 promoter region in lung adenocarcinoma cell lines, the AAH cell line (PL16T), and its adjacent normal cell line (PL16B). (b) Quantitative real‐time reverse transcription–polymerase chain reaction analysis of the SAMD14 gene in lung adenocarcinoma cell lines.
Figure 7.
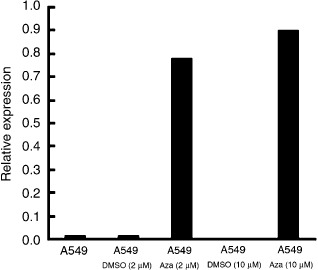
The expression level of the SAMD14 gene in the A549 cell line before and after 5‐aza‐2′‐deoxycytidine (Aza) treatment. DMSO, dimethyl sulfoxide.
Methylation status of the SAMS14 promoter region and its association with clinicopathological features. As indicated in Table 3, hypermethylation of SAMD14 was detected in eight cases of adenocarcinoma (8/31, 25.8%) but not in adjacent normal tissue (0/31, 0%). This result suggests that the promoter region of SAMD14 is specifically hypermethylated in pulmonary adenocarcinoma. The relationship between the clinicopathological features of patients examined and the methylation status of SAMD14 are summarized in Table 4. The promoter region of SAMD14 showed hypermethylation in early invasive pulmonary adenocarcinoma (type C) (8/24, 33.3%) but no methylation in in situ adenocarcinoma (types A and B) (0/7, 0%). Both stage II and III tumors showed hypermethylation (2/2, 100%), but some stage I tumors did not. There were no significant histological differences between early invasive adenocarcinomas with and without hypermethylation of SAMD14.
Table 3.
Frequency of methylation of the SAMD14 promoter region in human pulmonary adenocarcinoma (type C, early invasive adenocarcinoma) and its counterpart normal tissue
| Methylation | N | T | P‐value |
|---|---|---|---|
| M | 0 (0%) | 8 (25.8%) | *0.0023 |
| U | 31 (100%) | 23 (74.2%) | |
| Total | 31 | 31 |
The difference was statistically significant (P < 0.05). M, methylated cytosine; N, human normal lung tissue; T, human small‐sized adenocarcinoma tissue; U, unmethylated cytosine.
Table 4.
SAMD14 gene promoter methylation status and clinicopathological features in patients with early stage pulmonary adenocarcinoma
| Clinicopathological feature | All patients | SAMD14 promoter methylation status | P‐value | |
|---|---|---|---|---|
| No. methylated | No. unmethylated | |||
| All patients | 31 | 8 | 23 | |
| Mean age (years) | 62.8 | 51.1 | 62.9 | |
| Sex | ||||
| Male | 13 | 5 | 8 | |
| Female | 18 | 3 | 15 | 0.13 |
| Pathological stage | ||||
| Stage I | 29 | 6 | 23 | |
| Stage II | 1 | 1 | 0 | |
| Stage III | 1 | 1 | 0 | 0.06 |
| Stage IV | 0 | 0 | 0 | |
| Ly factor | ||||
| Negative | 26 | 6 | 20 | |
| Positive | 5 | 2 | 3 | 0.25 |
| V factor | ||||
| Negative | 24 | 6 | 18 | |
| Positive | 7 | 2 | 5 | 0.75 |
| Lymph node status | ||||
| N0 | 28 | 6 | 18 | |
| N1 and N2 | 7 | 2 | 5 | 0.75 |
| Noguchi classification | ||||
| Type A and B | 7 | 0 | 7 | |
| Type C | 24 | 8 | 16 | 0.09 |
Stage I includes IA and IB, stage II includes IIA and IIB, stage III includes IIIA and IIIB. Ly factor, lymphatic vessel invasion; V factor, vascular vessel invasion.
Discussion
Aberrant methylation of promoter regions in association with gene silencing is one of the major mechanisms responsible for the inactivation of tumor‐suppressor genes, and has been observed in various cancers.( 27 ) We sought to identify biomarkers for lung cancer and to find potential key genes involved in the molecular mechanism of pulmonary adenocarcinogenesis. In the present study, we used the MCA and SSH differential screening approaches to identify candidate genes in the A/J mouse model of adenoma. The A/J mouse model is unique and particularly useful, as the tumors induced by NNK mimic their human counterpart, bronchioloalveolar carcinoma (Fig. 1). Of course, there are several differences between A/J mouse adenoma and human lung adenocarcinoma at the early stage, including bronchioloalveolar carcinoma. Histologically, A/J mouse adenoma contains no fibrosis, whereas human lung adenocarcinoma at an early stage sometimes shows focal collapse or fibrosis. Clinicopathologically, A/J mouse adenoma never shows distant metastasis, whereas early stage human adenocarcinoma sometimes does so. However, both are at a very early stage of carcinogenesis, and the tumor cells are derived from type 2 pneumocytes.
First, we screened for hypermethylated genes in mouse adenomas and searched for their human counterparts. Although we selected 107 clones by differential screening, most of the clones did not match with real mouse genes. Therefore, more genes may be epigenetically associated with the A/J mouse adenocarcinogenenesis model. We speculated that the MCA‐SSH method would be very useful for screening hypermethylated sites, even though it is very complicated and has numerous steps. For example, several of the enzyme digestions might be incomplete, and many candidate sequences might therefore be lost. Therefore, it may be useful to apply another powerful method, such as MCA‐representational‐difference analysis, for differential analysis.
Finally, we identified SAMD14 as one of the most consistently downregulated genes in various human adenocarcinoma cell lines and small‐sized human lung adenocarcinomas. Genetically, SAMD14 is localized on chromosome 17q21,33. To date, no information about the hyper‐ or hypomethylation of SAMD14 in cancer has been available, and the function of this gene is not known. The promoter site of SAMD14 has a CG content of over 68%, and the CpG island of the SAMD14 promoter was highly methylated in most (4/5) of the cancer cell lines tested, and in PL16T but not in PL16B. Furthermore, the expression of SAMD14 in cell lines that showed hypermethylation in the promoter region was downregulated, whereas its expression was preserved in PL16B, which is an immortalized normal bronchial epithelial line without hypermethylation of SAMD14 (Fig. 6). However, our study showed that treatment with 5‐Aza rescued the downregulated expression of SAMD14 in A549 (Fig. 7). These results provide evidence that hypermethylation is at least one of the mechanisms responsible for silencing of the SAMD14 gene.
Furthermore, we analyzed the methylation status of the promoter region of SAMD14 in human adenocarcinomas and assessed its correlation with clinicopathological features. Our data show that hypermethylation of the promoter region within the SAMD14 CpG island is strictly limited to cancer tissue and is not detected in normal lung tissue (Table 3). The promoter region is significantly hypermethylated in early invasive adenocarcinomas (8/24, 33.3%), but not in in situ adenocarcinomas (0/7, 0%) (Table 4). As early stage lung adenocarcinoma tissues often contain normal lung epithelial components that express SAMD14, the true extent of promoter methylation as well as downregulation of expression in tumor tissue is likely to be higher than observed in the present study. However, it is interesting that we were unable to detect hypermethylation of the SAMD14 gene in in situ adenocarcinomas (types A and B). Downregulation of SAMD14 may be acquired in the course of malignant progression of pulmonary adenocarcinoma, although we could not find any histological differences between early invasive adenocarcinomas with and without hypermethylation of SAMD14. More detailed studies are needed to clarify the effects of hypermethylation of the SAMD14 promoter region on tumor histology.
It may be important that the SAMD14 protein contains the sterile α motif domain, which is an evolutionarily conserved protein‐binding domain involved in the regulation of numerous developmental processes among diverse eukaryotes.( 28 ) The sterile α motif domain, which extends over approximately 70 residues, is found in diverse eukaryotic organisms,( 29 ) and proteins containing it can be detected in various regions of the cell membrane, cytoplasm, and nucleus. The sterile α motif domain is involved in many different biological functions and binds a variety of proteins and RNA. For example, stromal interaction molecule 1 contains an extracellular sterile α motif domain and may bind extracellular proteins, whereas the functional apoptosis regulator BAR contains a sterile α motif domain that binds both Bcl‐Xl and Bcl‐2.
Although the function of SAMD14 is still unknown, the present study has demonstrated characteristic hypermethylation of its promoter region in early invasive pulmonary adenocarcinoma. SAMD14 is thought to be associated with malignant progression of pulmonary adenocarcinoma, and identification of the molecular species (protein or RNA) that binds to SAMD14 protein will be of great interest.
Supporting information
Sequences of five differentially methylated genes, identified by methylated CpG island amplification–suppressive subtractive hybridization differential screening. These sequences were matched with LOC674120, Kif21a, Samd14, EG436235, and Vwa1.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
The present work was supported in part by a Grant‐in‐Aid for Cancer Research (16‐1) from the Ministry of Health, Labor, and Welfare of Japan and a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (grant number 17590292).
References
- 1. Parkin DM. Global cancer statistics in the year 2000. Lancet Oncol 2001; 2: 533–43. [DOI] [PubMed] [Google Scholar]
- 2. Takeshima Y, Nishisaka T, Kawano R et al . p16/CDKN2 gene and p53 gene alterations in Japanese non‐smoking female lung adenocarcinoma. Jpn J Cancer Res 1996; 87: 134–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Travis WD, Travis LB, Devesa SS. Lung cancer. Cancer 1995; 75: 191–202. [DOI] [PubMed] [Google Scholar]
- 4. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell 1996; 87: 159–70. [DOI] [PubMed] [Google Scholar]
- 5. Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet 1999; 21: 163–7. [DOI] [PubMed] [Google Scholar]
- 6. Laird PW. Oncogenic mechanisms mediated by DNA methylation. Mol Med Today 1997; 3: 223–9. [DOI] [PubMed] [Google Scholar]
- 7. Kerr KM, Galler JS, Hagen JA, Laird PW, Laird‐Offringa IA. The role of DNA methylation in the development and progression of lung adenocarcinoma. Dis Markers 2007; 23: 5–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3: 415–28. [DOI] [PubMed] [Google Scholar]
- 9. Azhikina TL, Sverdlov ED. Study of tissue‐specific CpG methylation of DNA in extended genomic loci. Biochemistry 2005; 70: 596–603. [DOI] [PubMed] [Google Scholar]
- 10. Palmisano WA, Divine KK, Saccomanno G et al . Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res 2000; 60: 5954–8. [PubMed] [Google Scholar]
- 11. Tsou JA, Galler JS, Wali A et al . DNA methylation profile of 28 potential marker loci in malignant mesothelioma. Lung Cancer 2007; 58: 220–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Widschwendter M, Jiang G, Woods C et al . DNA hypomethylation and ovarian cancer biology. Cancer Res 2004; 64: 4472–80. [DOI] [PubMed] [Google Scholar]
- 13. Noguchi M, Morikawa A, Kawasaki M et al . Small adenocarcinoma of the lung. Histologic characteristics and prognosis. Cancer 1995; 75: 2844–52. [DOI] [PubMed] [Google Scholar]
- 14. Okubo C, Morishita Y, Minami Y et al . Phenotypic characteristics of mouse lung adenoma induced by 4‐(methylnitrosamino)‐1‐(3‐pyridyl)‐1‐butanone. Mol Carcinog 2005; 42: 121–6. [DOI] [PubMed] [Google Scholar]
- 15. Brambilla E, Travis WD, Colby TV, Corrin B, Shimosato Y. The new World Health Organization classification of lung tumours. Eur Respir J 2001; 18: 1059–68. [DOI] [PubMed] [Google Scholar]
- 16. Giard DJ, Aaronson SA, Todaro GJ et al . In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst 1973; 51: 1417–23. [DOI] [PubMed] [Google Scholar]
- 17. Fogh J, Wright WC, Loveless JD. Absence of HeLa cell contamination in 169 cell lines derived from human tumors. J Natl Cancer Inst 1977; 58: 209–14. [DOI] [PubMed] [Google Scholar]
- 18. Teraoka S, Kyoizumi S, Seyama T, Yamakido M, Akiyama M. A novel SCID mouse model for studying spontaneous metastasis of human lung cancer to human tissue. Jpn J Cancer Res 1995; 86: 419–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gazdar AF, Oie HK, Shackleton CH et al . Establishment and characterization of a human adrenocortical carcinoma cell line that expresses multiple pathways of steroid biosynthesis. Cancer Res 1990; 50: 5488–96. [PubMed] [Google Scholar]
- 20. Shimada A, Kano J, Ishiyama T et al . Establishment of an immortalized cell line from a precancerous lesion of lung adenocarcinoma, and genes highly expressed in the early stages of lung adenocarcinoma development. Cancer Sci 2005; 96: 668–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Toyota M, Ho C, Ahuja N et al . Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res 1999; 59: 2307–12. [PubMed] [Google Scholar]
- 22. Aoyagi K, Tatsuta T, Nishigaki M et al . A faithful method for PCR‐mediated global mRNA amplification and its integration into microarray analysis on laser‐captured cells. Biochem Biophys Res Commun 2003; 300: 915–20. [DOI] [PubMed] [Google Scholar]
- 23. Shu Y, Iijima T, Sun W et al . The ACIN1 gene is hypermethylated in early stage lung adenocarcinoma. J Thorac Oncol 2006; 1: 160–7. [PubMed] [Google Scholar]
- 24. Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res 1994; 22: 2990–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Frommer M, McDonald LE, Millar DS et al . A genomic sequencing protocol that yields a positive display of 5‐methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA 1992; 89: 1827–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Takai D, Jones PA. The CpG island searcher: a new WWW resource. In Silico Biol 2003; 3: 235–40. [PubMed] [Google Scholar]
- 27. Merlo A, Herman JG, Mao L et al . 5′‐CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med 1995; 1: 686–92. [DOI] [PubMed] [Google Scholar]
- 28. Schultz J, Ponting CP, Hofmann K, Bork P. SAM as a protein interaction domain involved in developmental regulation. Protein Sci 1997; 6: 249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stapleton D, Balan I, Pawson T, Sicheri F. The crystal structure of an Eph receptor SAM domain reveals a mechanism for modular dimerization. Nat Struct Biol 1999; 6: 44–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequences of five differentially methylated genes, identified by methylated CpG island amplification–suppressive subtractive hybridization differential screening. These sequences were matched with LOC674120, Kif21a, Samd14, EG436235, and Vwa1.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item