

Abstract
Several compounds from Cinnamomum kotoense show anticancer activities. However, the detailed mechanisms of most compounds from C. kotoense remain unknown. In this study, we investigated the anticancer activity of obtusilactone A (OA) and (−)‐sesamin in lung cancer. Our results show that human Lon is upregulated in non‐small‐cell lung cancer (NSCLC) cell lines, and downregulation of Lon triggers caspase‐3 mediated apoptosis. Through enzyme‐based screening, we identified two small‐molecule compounds, obtusilactone A (OA) and (−)‐sesamin from C. kotoense, as potent Lon protease inhibitors. Obtusilactone A and (−)‐sesamin interact with Ser855 and Lys898 residues in the active site of the Lon protease according to molecular docking analysis. Thus, we suggest that cancer cytotoxicity of the compounds is partly due to the inhibitory effects on Lon protease. In addition, the compounds are able to cause DNA double‐strand breaks and activate checkpoints. Treatment with OA and (−)‐sesamin induced p53‐independent DNA damage responses in NSCLC cells, including G1/S checkpoint activation and apoptosis, as evidenced by phosphorylation of checkpoint proteins (H2AX, Nbs1, and Chk2), caspase‐3 cleavage, and sub‐G1 accumulation. In conclusion, OA and (−)‐sesamin act as both inhibitors of human mitochondrial Lon protease and DNA damage agents to activate the DNA damage checkpoints as well induce apoptosis in NSCLC cells. These dual functions open a bright avenue to develop more selective chemotherapy agents to overcome chemoresistance and sensitize cancer cells to other chemotherapeutics. (Cancer Sci 2010; 101: 2612–2620)
Lung cancer is a leading cause of cancer‐related death in both men and women in the USA( 1 ) and Taiwan. The survival rate after chemotherapy regimens and the drug development of molecular‐targeted agents for lung cancer therapy is still challenging.( 2 , 3 )
Lon is a highly conserved ATP‐dependent serine protease that has been identified from prokaryotes and to mitochondria of eukaryotes.( 4 ) In vivo studies show that Lon plays an important role in mitochondrial DNA maintenance and expression( 5 , 6 , 7 ) and regulation of mitochondria function.( 8 ) Mitochondrial function is a crucial determinant of cellular sensitivity to cancer therapeutic drugs because of its roles in mediating apoptosis.( 9 ) Enhanced mitochondrial biogenesis as well as tumorigenesis are linked to overexpression and increased proteolytic activity of Lon.( 8 , 10 ) Downregulation of Lon leads to loss of mitochondrial function, reduced cell proliferation capacity, and apoptosis.( 11 , 12 ) Deregulation of Lon leading to tumorigenesis has raised its potential as a target in the development of a novel drug in cancer therapy. To date, very few small‐molecule inhibitors of Lon protease have been found, although several peptide‐based proteasome inhibitors have been reported.( 13 , 14 )
Cell cycle checkpoints are sophisticated surveillance mechanisms that are used to monitor the integrity of DNA.( 15 , 16 ) After DNA lesions are sensed by sensor proteins, the kinase activities of ataxia talangiectasia mutated (ATM) and/or ATM and Rad3‐related (ATR) are stimulated. ATM is predominantly activated by DSBs, whereas ATR responds to stalled replication forks that can be induced by UV light and re‐replication.( 15 , 17 , 18 , 19 ) When DNA is damaged, ATM is activated and phosphorylates downstream H2AX on S139, Nbs1 on S343, and Chk2 on T68, leading to cell cycle arrest and DNA repair.( 9 , 16 , 17 , 20 ) Importantly, checkpoints can also activate apoptosis to eliminate damaged cells and protect the organism if DNA damage repair is impaired.( 21 , 22 , 23 ) Such mechanisms are extremely important for the maintenance of genome stability and prevention of tumorigenesis.
Cinnamomum kotoense Kanehira & Sasaki is a small evergreen tree endemic to Lanyu Island, Taiwan.( 24 ) Various constituents of C. kotoense showed antiproliferation activity on peripheral blood mononuclear cells and antitumor activity against HeLa cells.( 25 , 26 , 27 ) However, the cytotoxic properties and the detailed mechanisms of OA and (−)‐sesamin from C. kotoense are unknown. In this study, we have identified OA and (−)‐sesamin as novel inhibitors of human Lon. We have also shown that OA and (−)‐sesamin can cause DNA DSBs and induce DNA damage response of cell cycle G1/S‐phase arrest, and further cause cell death.
Materials and Methods
Cell culture, retroviral infections, and shRNA.
MRC‐5, HEL299, H1299, A549, H1437, or 293T cells were grown in DMEM containing 5% FBS and 5% super calf serum.
Silencing of endogenous Lon in H1299 cells was carried out by retroviral infection using pMKO vector( 28 ) that expressed Lon shRNA target sequences. The shRNA target sequences used were Lon GAAAGUUCGUCUCGCCCAGCC and AGGAGCAGCUAAAGAUCAUCA.( 14 )
Western blot analysis and antibodies.
Western blot analysis was carried out as described previously.( 18 ) Antibodies used in this study are described in Supporting Information.
Expression and purification of human Lon protease.
A plasmid encoding human Lon carrying an amino‐terminal hexa‐histidine tag was a gift from Dr Carolyn K. Suzuki (University of Medicine and Dentistry of New Jersey, NJ, USA).( 7 ) Human Lon was overexpressed in the Escherichia coli strain, Rosetta (Novagen, Darmstadt, Germany), and purified as previously described( 7 , 29 ) with some modifications.
Human Lon protease inhibition assay.
The inhibitory effect on proteolytic activity of human Lon was determined as described previously( 29 , 30 ) with minor modifications. The detailed procedure is described in Supporting Information.
Extraction and isolation.
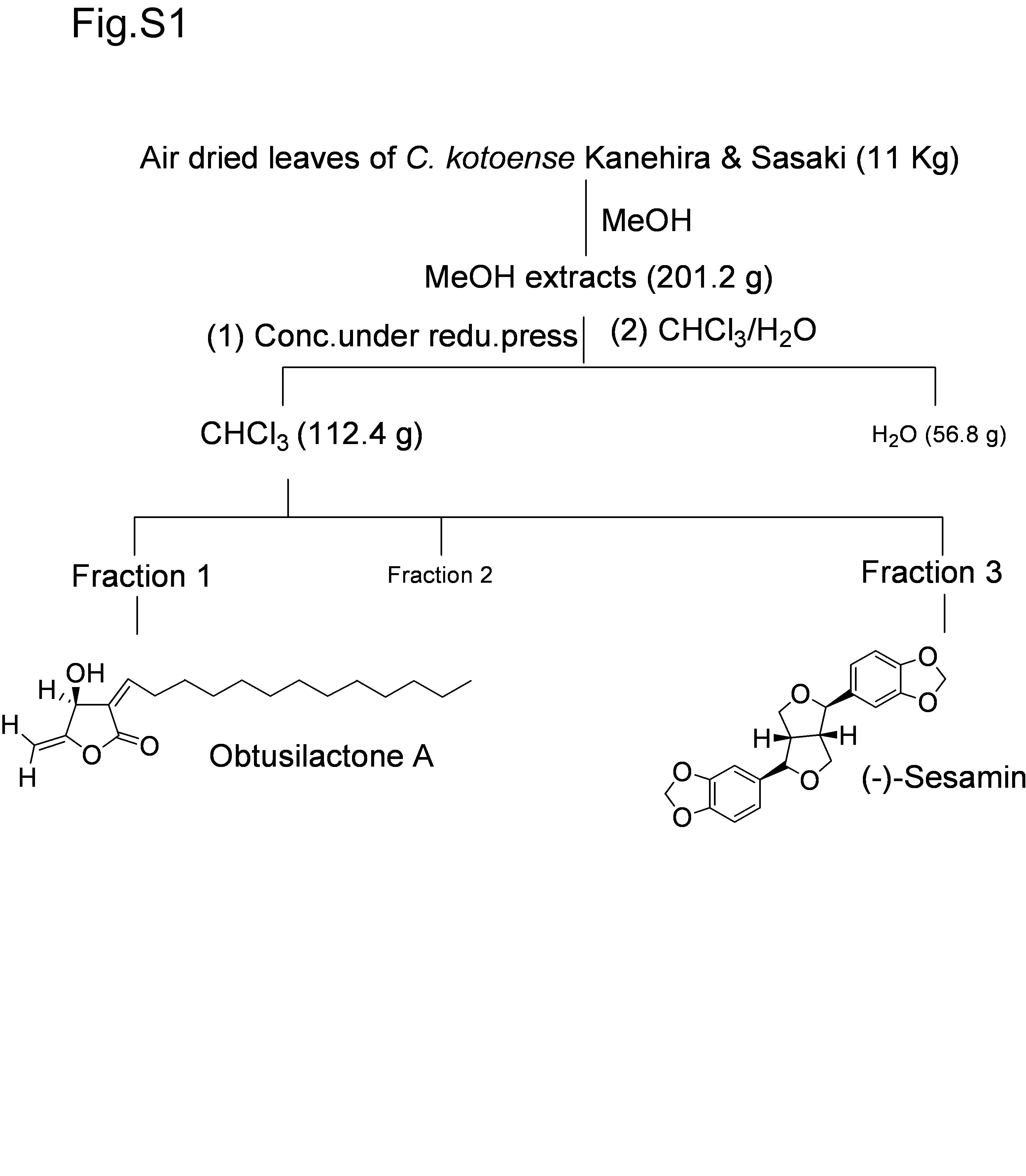
The detailed procedure for isolation of OA and (−)‐sesamin is described in Figure S1.
Homology modeling of human Lon protease and molecular docking.
The structure of human Lon proteolytic domain was generated with the MODELER program encoded in InsightII (Accelrys, San Diego, CA, USA) using the E. coli proteolytic domain (Protein Data Bank code 1RR9) as the template structure. The active site was docked with the inhibitors by using dock suite of Discovery Studio 2.0 (Accelrys). PyMOL software (DeLano Scientific, http://www.pymol.org) was used to show the final blocking structure of human Lon homology with the inhibitors.
Comet assay.
The protocol for comet–nuclear extract assay was described previously.( 31 , 32 ) The migration of DNA from the nucleus of each cell was measured under a fluorescence microscope (TE2000‐U; Nikon, Tokyo, Japan) with a computer program (http://tritekcorp.com/main_home.php) using the parameter of tail moment, which is defined as the product of the tail length and the fraction of total DNA in the tail.
Fluorescence‐activated cell sorting analysis.
Cells were washed twice with PBS, collected by trypsinization and fixed with 70% ethanol overnight at 4°C. After fixation, cells were stained with a buffer containing 38 mM sodium citrate, 10 μg/mL RNaseA, and 15 μg/mL propidium iodide (Sigma‐Aldrich, St Louis, MO, USA). The labeled cells were analyzed with a FACScan flow cytometer (Becton‐Dickinson, Mansfield, MA, USA) using Cellquest software and Modfit software (Becton‐Dickinson).
Cell viability assay.
The viability of cells was determined by a Trypan blue dye exclusion assay. Cells were treated with DMSO or different doses of OA in medium for 12 h. After incubation, cells exposed to 0.2% Trypan blue were counted in a hemocytometer.
Results
Lon is upregulated in lung cancer‐derived cells and down‐regulation of Lon causes cell death.
To detect human Lon protease, a polyclonal antiserum was raised in rabbits against the Lon. Three NSCLC cell lines plus two normal cells were examined for Lon protein abundance. We were unable to detect high amounts of Lon protein in whole cell lysates from normal human fetal lung fibroblasts (MRC‐5) and embryonic lung fibroblasts (HEL299, Fig. 1A), suggesting that Lon protein is quite low in normal cell lines. In contrast, Lon was detected as overexpressing in NSCLC cell lines (Fig. 1A). For example, H1299, A549, and H1437 showed a two‐ to fivefold increase of Lon expression compared with the normal cells (Fig. 1A).
Figure 1.
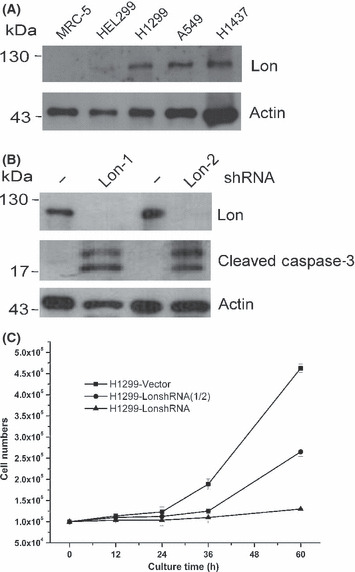
Level of Lon determines cell fate. (A) Abundance of Lon protease in normal and lung cancer cell lines. Whole cell lysates from normal lung fibroblasts, MRC‐5 and HEL299, and non‐small‐cell lung cancer cell lines, H1299, A549, and H1437, were immunoblotted with an antibody to Lon and antibody to actin as a loading control. (B) Downregulation of Lon induces caspase‐3‐dependent apoptosis. The expression of Lon protease was suppressed in cells by expressing two different shRNAs (Lon‐1 and Lon‐2) from the retroviral vector. Cell lysates were prepared from H1299 cells expressing shLon‐1, shLon‐2 or vector and immunoblotted with antibodies to Lon, anti‐cleaved caspase‐3 and actin as a loading control. (C) Downregulation of Lon suppresses cell proliferation and induces cytotoxicity in a dose‐dependent manner. The expression of Lon was inhibited in H1299 cells by expressing shRNA from the retroviral vector. LonshRNA and LonshRNA(1/2) represent cells that were infected by full and half amounts of the retrovirus expressing shRNA to get complete and half inhibition of Lon expression, respectively. Cell growth was measured under the different extent of Lon inhibition at the indicated time. Cell numbers were calculated using hemocytometers. Each value represents the mean ± SD.
To confirm that Lon depletion induces apoptosis, we inhibited expression of Lon by shRNA. Silencing Lon by shRNA results in an undetectable level of Lon in H1299 cells compared with the control cells (Fig. 1B). Indeed, in cell lysates taken from Lon‐depleted cells, active caspase‐3 could easily be detected (Fig. 1B). Therefore, we suggest that in H1299 cells, inhibition of Lon protease causes apoptotic cell death. To confirm this, we examined the effect of Lon inhibition on the proliferation of H1299 cells. In addition, to evaluate how different Lon expression levels contribute to the selectivity of cytotoxicity caused by Lon inhibition, we designed the experiment for different extents of Lon inhibition by shRNA. The data show that Lon suppression inhibits the proliferation of H1299 cells in a dose‐dependent manner. The percentage of viable cells was 8.3% or 45.5% of the control level following treatment with a complete or 50% inhibition after 60 h culture, respectively (Fig. 1C).
Preparation and characterization of recombinant human Lon protease.
To screen the inhibitors of Lon protease in vitro, the protein was overexpressed and purified from E. coli Rosetta. The homogeneity of the purified proteins was analyzed by SDS‐PAGE, by which Lon was a major band of approximately 110 kDa after purification (Fig. 2A). The recombinant Lon was able to be detected by anti‐human Lon antibody (Fig. S2, lane 2). In addition, the proteins were identified as human Lon by mass spectrometry analysis (Fig. S3). To determine the enzyme kinetics of Lon protease, the fluorogenic peptide Glt‐AAF‐MNA( 7 , 29 , 30 ) was used to monitor the real‐time activity of the enzyme. The K m and k cat of the enzyme using this substrate were measured to be 9.15 ± 1.95 μM and 68.3/min, respectively (Fig. 2B).
Figure 2.
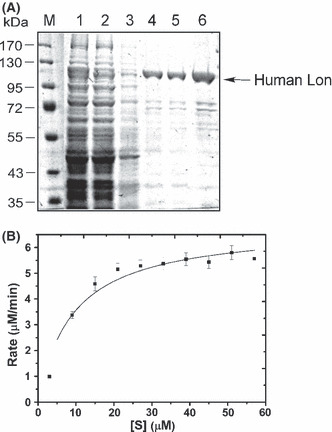
(A) Analysis of recombinant human Lon by SDS‐PAGE. Crude lysate from Escherichia coli Rosetta cells containing human Lon expression plasmid with (lane 1) or without (lane 2) isopropyl‐β‐D‐thiogalactoside induction was incubated with a Ni‐NTA affinity agarose gel. After binding, bound fractions were eluted by imidazole (lane 4, 200 mM; lane 5, 250 mM) from the column. Affinity purified human Lon was concentrated (lane 6). The arrow shows the recombinant human Lon. M, molecular mass markers. (B) Substrate saturation curve for the recombinant human Lon, incubated with indicated concentrations of fluorogenic substrate (Glt‐AAF‐MNA,[S]) in a real‐time manner (0–30 min). The velocity (Y‐axis) of the reaction was determined at various Glt‐AAF‐MNA concentrations (X‐axis). The values of K m and k cat were 9.15 ± 1.95 μM and 68.3/min, respectively, by fitting the data with the Michaelis–Menten equation using the KaleidaGraph program.
Discovery of human Lon protease inhibitors.
We screened a series of pure small compounds with anticancer activities isolated from C. kotoense. Two target compounds, OA and (−)‐sesamin (Fig. 3A), were recognized as significant inhibitors of Lon protease through an enzyme‐based screening. Subsequently, Lon and the fluorogenic substrate were used to evaluate the IC50 of the two compounds against the proteolytic activity. Incubation of Lon with OA and (−)‐sesamin for 30 min resulted in a dose‐dependent inhibition of the protease activity with an IC50 of 34.1 and 19.9 μM, respectively (Fig. 3B,C). These results suggest that OA and (−)‐sesamin act as significant inhibitors of Lon protease in vitro. To further explore OA and (−)‐sesamin inhibit Lon protease in vivo, we assessed the level of endogenous substrate of human Lon, mitochondrial aconitase,( 33 ) under various conditions. Western blot analysis showed that OA causes a significant accumulation of aconitase in a time‐ and dose‐dependent manner (Fig. 3D,E), suggesting that OA is a significant inhibitor of Lon protease in vivo. Taken together, we found that OA is a bona fide inhibitor of Lon protease in vitro and in vivo.
Figure 3.
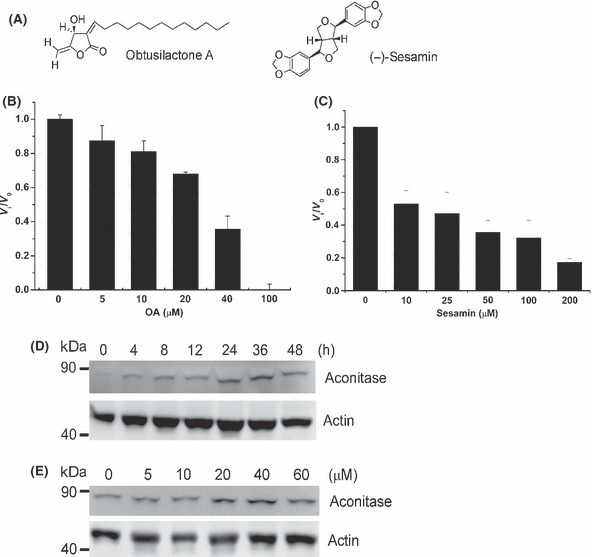
Obtusilactone A (OA) and (−)‐sesamin inhibit human Lon protease in vitro and in vivo. (A) Chemical structure of OA and (−)‐sesamin. (B) Dose‐dependent inhibition of Lon protease by OA. The inhibitory effect of OA on Lon protease was measured using FITC‐α‐casein as a substrate. Where v i is the initial velocity in the presence of inhibitor at concentration [I] and v 0 is the initial velocity in the absence of inhibitor. (C) Dose‐dependent inhibition of Lon protease by (−)‐sesamin. The same as (B) except OA is replaced by (−)‐sesamin. (D) Time‐dependent inhibition of Lon protease by OA in vivo. H1299 cells were treated with OA for the indicated time at the concentration of 40 μM. Whole cell lysates from the treated H1299 cells were immunoblotted with an antibody to aconitase and anti‐actin as a loading control. (E) Dose‐dependent inhibition of Lon protease by OA in vivo. H1299 cells were treated with OA for 12 h at the indicated concentration. Whole cell lysates from the treated H1299 cells were immunoblotted with an antibody to aconitase and anti‐actin as a loading control.
To better understand the inhibitory mechanism of the compounds, we simulated the structure of human Lon P domain using the X‐ray crystal structure of E. coli Lon P domain( 34 ) as a template (Fig. 4A) and docked OA or (−)‐sesamin into the active site of human Lon (Fig. 4B). The active site of P domain carries the serine and lysine residues that form the catalytic dyad involved in the catalytic mechanism( 7 , 34 , 35 ) and are shown as Ser855 and Lys898 (Fig. 4). The carbonyl of γ‐lactone moiety of OA interacts with Ser855 and Lys898 residues (Fig. 4B, a). We also observed that the oxygen atoms of the furan ring and piperonyl group of (−)‐sesamin interacts with Ser855 and Lys898 residues (Fig. 4B, b).
Figure 4.
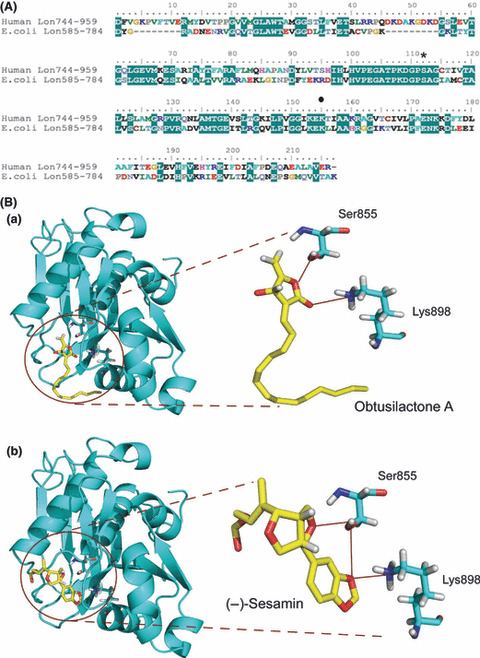
Modeling obtusilactone A (OA) or (−)‐sesamin in the active site of human Lon protease. (A) Multiple sequence alignment of Lon proteins. The sequences are taken from the GenBank database, including Homo sapiens Lon (Accession No.: NM_004793) and Escherichia coli Lon (Accession No.: P08177). Amino acid residues 744–959 in human Lon (Human Lon744–959) and 585–784 in E. coli Lon (E. coli Lon585–784) are used to align the sequences. The identical residues are highlighted in a green frame. Serine 855 (*) and lysine 898 (•), acting as the active site, are shown. (B) Homology modeling of human Lon proteolytic (P) domain and binding mode of inhibitors in the active site. The model of the P domain was used as the target for ducking simulation. Obtusilactone A (a) or (−)‐sesamin (b) was docked into the protease catalytic site.
Obtusilactone A and (−)‐sesamin induce DNA DSB damage responses.
(+)‐Sesamin was reported to have a genotoxic effect on human larynx carcinoma cells.( 36 ) Additionally, a study reported that sesamin induces growth arrest at the G1 phase in human MCF‐7 cells.( 37 ) Accordingly, we were very interested in whether OA and sesamin could damage DNA and further activate DNA damage response to induce apoptosis in a cancer cell line. Therefore, we carried out a neutral comet assay to investigate the compounds’ ability to cause DSBs.( 18 ) Indeed, sesamin‐treated H1299 cells showed the tail (comet) grown from the propidium iodide‐staining nuclei, whereas no tail was found in the untreated control (DMSO) (Fig. 5A, left panel), which is consistent with previous observations.( 36 ) The average tail moments for control, 50, and 100 μM sesamin treatments were 0.03 ± 0.04, 55.18 ± 16.16, and 83.53 ± 12.17, respectively (Fig. 5A, left panel). Interestingly, treatment using different concentrations of OA on the same cells also showed a positive result on comet assay, but induced responses more efficiently than sesamin (Fig. 5A, right panel). The average tail moments for control, 10, 20, and 50 μM OA were 0.03 ± 0.04, 18.56 ± 8.01, 46.27 ± 12.16, and 59.56 ± 14.17, respectively (Fig. 5A, right panel). This result allowed us to choose OA to follow up the consequence of the compound‐induced DNA damages.
Figure 5.
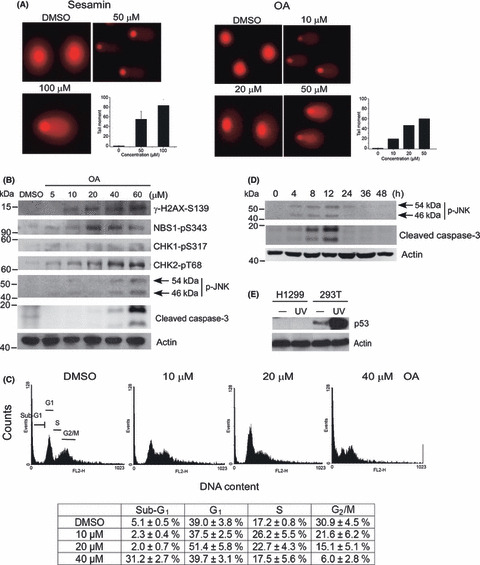
Obtusilactone A (OA) and (−)‐sesamin cause DNA double‐strand breaks (DSBs) and DNA damage response‐induced apoptosis. (A) Detection of DSBs in H1299 cells after treatment by comet assay. H1299 cells were treated with control (DMSO) or the indicated concentration of OA or (−)‐sesamin for 2 h. The circular spots in red are nuclei and the comet tails indicate the cells carrying the DNA damaged DSBs. Tail moment of cells with a comet tail is plotted. (B) Obtusilactone A activates the ATM‐CHK2 mediated checkpoint and JNK‐mediated apoptosis pathways. H1299 cells were treated with DMSO or the indicated concentration of OA for 4 h. Whole cell lysates from treated H1299 cells were immunoblotted with phosphospecific antibodies to Ser343 of Nbs1 (NBS1‐pS343), Ser317 of Chk1 (CHK1‐pS317), Thr68 of Chk2 (CHK2‐pT68), Ser139 of H2AX (γ‐H2AX‐S139), JNK (p‐JNK), or antibody to cleaved caspase‐3. Anti‐actin is a loading control. (C) Obtusilactone A induces G1/S cell cycle arrest and DNA damage‐induced apoptosis. H1299 cells were treated with DMSO or the indicated concentration of OA for 12 h. The determination of G1, S, G2/M, or sub‐G1 DNA content in H1299 cells by FACS analysis. The percentages of the cell cycle phase are shown at the bottom. Mean ± SD of three experiments. (D) Obtusilactone A activates JNK‐mediated apoptosis signaling. H1299 cells were treated with OA for the indicated time at the concentration of 40 μM. Whole cell lysates from the treated H1299 cells were immunoblotted with a phosphospecific antibody to JNK (p‐JNK) or antibody to cleaved caspase‐3 and anti‐actin as a loading control. (E) H1299 is a p53‐deficient cell line. Cell lysates were prepared from H1299 and 293T cells after mock treatment or exposure to UV (50 J/m2, 1 h after) and immunoblotted with anti‐p53 antibody. Actin was used as a loading control.
To confirm OA‐induced DSB formation, we next examined H2AX phosphorylation at Ser139 (γ‐H2AX) in the treated cells, which is an early cellular response to DSBs.( 38 ) As shown in Figure 5(B), OA‐induced γ‐H2AX formation is dose‐dependent. To further explore the activation of checkpoint pathways induced by OA, we next assessed activation of checkpoints under various concentration treatments. Western blot analysis revealed that OA induces significant phosphorylations of Nbs1 at Ser343 and Chk2 at Thr68 at 20 μM treatment but not a significant change in Chk1 phosphorylation at Ser317, suggesting that the DSBs induce ATM‐mediated checkpoint activation (Fig. 5B). The result not only indicates that OA causes DSB formation, but also shows that the ATM‐CHK2 pathway is activated in the treated cells.
Obtusilactone A induces cell cycle arrest and apoptosis.
To study the consequence responsible for OA‐induced DNA damage response, cell cycle progression of the treated cells was measured using FACS analysis. Interestingly, the cell cycle is arrested in S‐phase when treated with 10 μM OA for 12 h, and in G1 phase when treated with 20 μM or higher concentrations (Fig. 5C). This interesting finding about dual cell cycle arrests, however, needs further confirmation. Next, treatment with 40 μM OA began to significantly increase cell population in sub‐G1 (Fig. 5C), which means the cells were going through apoptosis and the DNA was broken into pieces. This result was confirmed by the activation of caspase‐3 cleavage (Fig. 5B) and the TUNEL assay (Fig. S4). To identify the mechanism of OA‐induced DNA damage and apoptosis and to evaluate the relevance of mitochondrial dysfunction to the DNA damage, activation of the intracellular ROS signaling pathway was examined. It has been shown that the increase in ROS level activates the MAPK signaling pathway, including the JNK and p38 pathways.( 39 , 40 ) As shown in Figure 5(B), we also observed the activation of the JNK pathway in OA‐treated cells at 40 μM, implying that OA‐induced apoptosis is mediated through the ROS signaling pathway. To further confirm this issue, a time‐course analysis was carried out using treatment with 40 μM OA. Again, we found that the time point of apoptosis induction, from 4 to 12 h after treatment, is the same as activation of the JNK pathway (Fig. 5D). We were unable to detect the signal after 12 h, which may be due to loss of cells that had undergone apoptosis. Notably, H1299 is a p53‐deficient cell line verified by Western blot analysis (Fig. 5E), implying that OA induces cell cycle arrest and cell death by way of a p53‐independent pathway.
Effect of OA on antiproliferation and cytotoxicity against lung cancer cell lines.
To evaluate the effect of OA on cell growth inhibition, we examined proliferation of the treated H1299 cells. The growth of H1299 cells was evaluated in the presence or absence of various concentrations of OA. As shown in Figure 6(A,B), OA inhibited the proliferation of H1299 cells in a dose‐dependent and time‐dependent manner; 40 μM OA had a cytostatic effect. To examine the selective cytotoxicity of OA to cancer cells, we measured the cell viability of A549 and H1299, as well as normal lung fibroblasts (Fig. 6C). The IC50 values of A549, H1299, and MRC‐5 were 26.50, 33.96, and 49.43 μM, respectively, after OA treatment for 12 h. The results suggested normal lung cell line MRC‐5 has the lowest cytotoxicity under the same circumstances.
Figure 6.
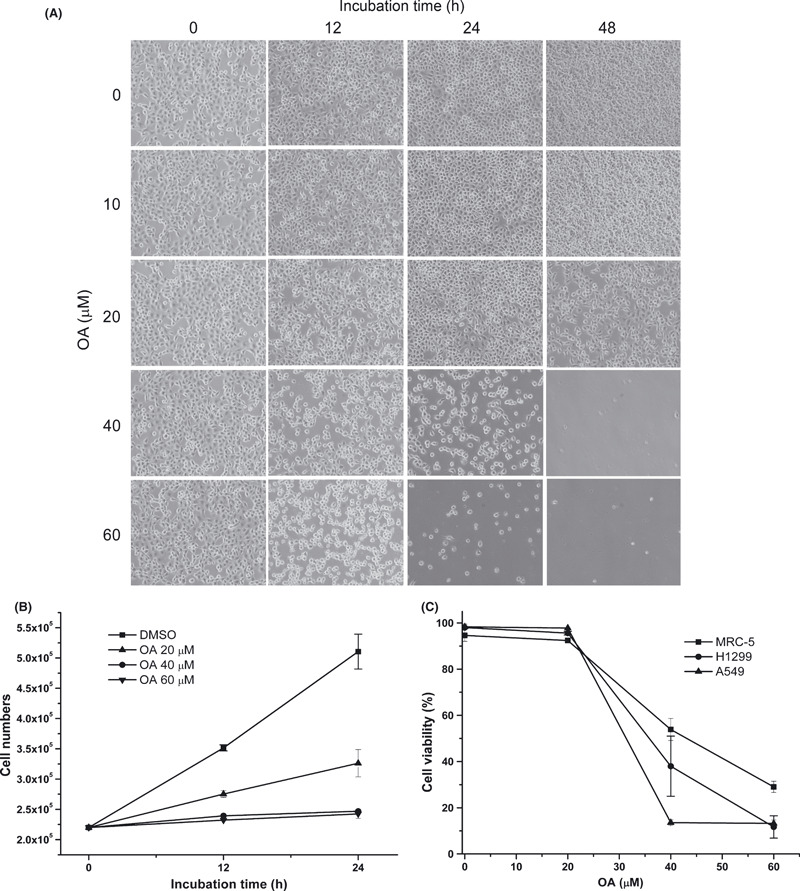
Obtusilactone A (OA) suppresses proliferation and induces selective cytotoxicity in lung cancer cell lines. (A) Cytotoxicity in H1299 cells treated with increasing concentrations of OA at the indicated incubation times. Cell images were captured using a Nikon Eclipse TE2000‐U inverted microscope. (B) Cell growth was measured under the different concentrations of OA at 12 and 24 h. Cell numbers were calculated using hemocytometers. Each value represents the mean ± SD. (C) Cytotoxicity in MRC‐5, A549, or H1299 cells treated with various concentrations of OA. The indicated cells were incubated with concentrations of OA (from 0, 20, 40, and 60 μM) for 12 h. Mean ± SD of three experiments.
Discussion
In this work, we investigated the level of mitochondrial Lon protease in NSCLC cell lines and found that Lon protein has a relatively low abundance in normal cells but has an increased expression in lung cancer cells. However, depletion of Lon using shRNA significantly causes apoptosis by activating caspase‐3, suggesting that Lon protease inhibition is a promising approach to tackling cancer cells. This study clearly showed that OA and/or (−)‐sesamin efficiently inhibit human Lon protease in vitro and in vivo. As shown in Figure 3(D,E), OA causes a significant accumulation of aconitase in a time‐ and dose‐dependent manner, suggesting that OA is a potent inhibitor of Lon protease in vivo. As Lon protease is a multiple‐function protein, we tried to clarify that the inhibition of protease activity‐dependent Lon function by OA and sesamin is associated with compound‐induced cytotoxicity. Thus, the phenotypes between shRNA‐treated cells and OA‐treated cells were compared. Our data show that both treatments share similar phenotypes in respect to degradation of mitochondrial aconitase (Fig. 3D,E), caspase‐3 activation (1, 5), growth suppression, and cell death (1, 6).
Sesamin is a major lignan constituent of sesame and effective in disease prevention such as cholesterol‐lowering( 41 ) and anticancer activities.( 37 , 42 , 43 ) Several published works have reported that sesamin inhibits DNA synthesis and cell cycle progression in G1 phase.( 37 , 42 ) However, the mechanism causing the inhibition is not well understood. Here we provide evidence showing that (−)‐sesamin‐induced G1 phase arrest is triggered by DNA DSBs. Interestingly, several coumarin and lignan derivatives including sesamin and asarinin inhibited DNA synthesis in human leukemia HL‐60 cells,( 42 ) both of which are potent inhibitors of Lon protease (( 44 ) and this work).
Double‐strand breaks are the most cytotoxic DNA lesions and activate the DNA damage response, which determine cell fate such as cell cycle arrest, repair, or apoptosis. Given that OA causes DNA DSB formation, we further investigated whether DSB formation is one possible mechanism associated with its cytotoxicity in H1299 cells. We found that OA activates DSBs in H1299 cells, evidenced by the comet assay and H2AX phosphorylation. It is noteworthy that no cell death was observed until 24 h after 20 μM OA treatment (Fig. 6A); thus, the formation of DSB was not a secondary event due to internucleosomal DNA cleavage in the late apoptotic phase. We also showed that OA activates checkpoint proteins, such as Nbs1 and Chk2 phosphorylation, but not Chk1 phosphorylation, suggesting that ATM is the major sensor kinase that initiates DNA DSB response. Obtusilactone A causes the cell cycle arrest in G1/S and apoptosis in the treated H1299 cells. Interestingly, OA causes S‐phase arrest at low concentrations (10 μM) and G1 arrest as well as apoptosis at high concentrations (20 and 40 μM, respectively) (Fig. 5C). Treatment with 10 μM OA does not cause cell death but is sufficient to activate the checkpoint to prevent G1/S‐phase progression in H1299 cells (Fig. 5B). The results are consistent with the results of OA‐induced proliferation inhibition (20 μM) and cell death (40 μM) (5, 6). This may be explained by the fact that the checkpoint activation and G1/S‐phase arrest starting from approximately 10–20 μM is responsible for the reduced growth rate. Such a unique feature, where one compound can induce two different cell cycle arrests, is worthy of further study. Taken together, OA‐induced DSBs activate the cell cycle arrest in G1/S and apoptosis, which is mediated by the ATM‐NBS1‐CHK2 pathway.
Severe DNA damage can induce apoptosis by both p53‐dependent and p53‐independent pathways, depending on the therapeutics and cell type.( 22 , 23 ) Our results showed that OA is cytotoxic against both p53‐deficient H1299 (5, 6) and p53 wild‐type A549 cells (Fig. 6C), suggesting that p53 function is not required for OA‐induced apoptosis in cancer cells. Therefore, OA‐induced apoptosis is ATM‐CHK2‐dependent but p53‐independent. Importantly, given that most cancers are defective in p53 or in p53‐related pathways,( 45 ) the p53‐independent apoptosis confers an advantage to OA as a novel chemotherapeutic agent.
The dual functions of OA to induce apoptosis give it great potential as a candidate for anticancer therapeutics. We have shown that OA both inhibits mitochondrial Lon protease (Fig. 3) and may generate ROS to causes a DNA DSB to activate DNA damage response, including checkpoint activation and apoptosis (Fig. 5). However, we still cannot exclude the possibility that OA‐induced DSB is due to the generation of excess ROS from dysfunctional mitochondria caused by Lon inhibition. Further experiments, such as the evaluation of ROS in Lon‐depleted and OA‐treated cells, are needed.
In conclusion, we showed that increased mitochondrial Lon expression may be a common and important step during tumorigenesis. Inhibition of the protein causes apoptosis, which has raised its potential as a target for anticancer therapy. Furthermore, the dual functions of OA to induce apoptosis give it unique advantages in overcoming drug resistance.
Abbreviations
- ATM
ataxia telangiectasia mutated
- ATR
ATM and Rad3‐related
- DSB
double‐strand break
- IPTG
isopropyl‐β‐D‐thiogalactoside
- NSCLC
non‐small‐cell lung cancer
- P
domain, proteolytic domain
- OA
obtusilactone A
- ROS
reactive oxygen species
Supporting information
Materials and Methods. Further detailed information regarding antibody production, mass spectrometry, compound isolation, and assays.
Fig. S1. Purification of obtusilactone A and (‐)‐sesamin from the leaves of Cinnamomum kotoense.
Fig. S2. Identification of the recombinant human Lon and specificity of the polyclonal antiserum against human Lon assayed by Western blot analysis. Crude whole cell lysate from 293T cells (lane 1) and affinity purified human Lon (lane 2) were separated by SDS‐PAGE and the blotted membrane was incubated with purified with anti‐human Lon antibody overnight at a 1:2000 dilution. The arrow shows the recognized human Lon. The Western blot analysis showed that the antibody against human Lon recognizes mainly a single band of ~110 kDa from the 293T cells extract pool (lane 1) that is exactly the same size as the predominant purified recombinant Lon (lane 2).
Fig. S3. Identification of the recombinant human mitochondrial Lon protease by mass spectrometry analysis. Amino acid sequence of human Lon protease is shown (Accession No. NM_004793). The amino acid sequences in red represent the peptides found from tandem mass spectrometry sequencing.
Fig. S4. Obtusilactone A induces apoptosis, revealed by TUNEL assay. H1299 cells were treated with DMSO (blank) or the indicated concentration of OA for 12 h. Apoptotic cells show a green fluorescence that can be detected using a standard fluorescein filter at 520 nm. All cells stained with propidium iodide show a red fluorescence at 620 nm.
Supporting info item
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Supporting info item
{kind=link}
Acknowledgments
The authors thank Dr Carolyn K. Suzuki (University of Medicine and Dentistry of New Jersey, NJ, USA) for the expression plasmid of human Lon protease and Liang‐Cheng Su for TUNEL assay. This work was supported by grants from Kaohsiung Medical University (KMU) Research Foundation (KMU‐Q097012 and Q098019), the Chi‐Mei Medical Center and KMU Research Foundation (98‐CM‐KMU‐09), the National Science Council (NSC 98‐2311‐B‐037‐001‐MY3), National Health Research Institutes (CA‐099‐PP‐41), and Department of Health (DOH99‐TD‐C‐111‐004), Taiwan to Alan Yueh‐Luen Lee.
References
- 1. Jemal A, Siegel R, Ward E et al. Cancer statistics, 2008. CA Cancer J Clin 2008; 58: 71–96. [DOI] [PubMed] [Google Scholar]
- 2. Adams J. The proteasome: structure, function, and role in the cell. Cancer Treat Rev 2003; 29(Suppl 1): 3–9. [DOI] [PubMed] [Google Scholar]
- 3. McConkey DJ, Zhu K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Updat 2008; 11: 164–79. [DOI] [PubMed] [Google Scholar]
- 4. Tsilibaris V, Maenhaut‐Michel G, Van Melderen L. Biological roles of the Lon ATP‐dependent protease. Res Microbiol 2006; 157: 701–13. [DOI] [PubMed] [Google Scholar]
- 5. Van Dyck L, Neupert W, Langer T. The ATP‐dependent PIM1 protease is required for the expression of intron‐containing genes in mitochondria. Genes Dev 1998; 12: 1515–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cheng X, Kanki T, Fukuoh A et al. PDIP38 associates with proteins constituting the mitochondrial DNA nucleoid. J Biochem (Tokyo) 2005; 138: 673–8. [DOI] [PubMed] [Google Scholar]
- 7. Liu T, Lu B, Lee I, Ondrovicova G, Kutejova E, Suzuki CK. DNA and RNA binding by the mitochondrial lon protease is regulated by nucleotide and protein substrate. J Biol Chem 2004; 279: 13902–10. [DOI] [PubMed] [Google Scholar]
- 8. Luciakova K, Sokolikova B, Chloupkova M, Nelson BD. Enhanced mitochondrial biogenesis is associated with increased expression of the mitochondrial ATP‐dependent Lon protease. FEBS Lett 1999; 444: 186–8. [DOI] [PubMed] [Google Scholar]
- 9. Sancar A, Lindsey‐Boltz LA, Unsal‐Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73: 39–85. [DOI] [PubMed] [Google Scholar]
- 10. Zhu Y, Wang M, Lin H, Huang C, Shi X, Luo J. Epidermal growth factor up‐regulates the transcription of mouse lon homology ATP‐dependent protease through extracellular signal‐regulated protein kinase‐ and phosphatidylinositol‐3‐kinase‐dependent pathways. Exp Cell Res 2002; 280: 97–106. [DOI] [PubMed] [Google Scholar]
- 11. Xue X, Zhu YF, Mao JP. Effect of RNA interference for Lon gene silencing on growth and apoptosis of human breast cancer MCF7 cells. Nan Fang Yi Ke Da Xue Xue Bao 2007; 27: 870–4. [PubMed] [Google Scholar]
- 12. Bota DA, Ngo JK, Davies KJ. Downregulation of the human Lon protease impairs mitochondrial structure and function and causes cell death. Free Radic Biol Med 2005; 38: 665–77. [DOI] [PubMed] [Google Scholar]
- 13. Frase H, Hudak J, Lee I. Identification of the proteasome inhibitor MG262 as a potent ATP‐dependent inhibitor of the Salmonella enterica serovar Typhimurium Lon protease. Biochemistry 2006; 45: 8264–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Granot Z, Kobiler O, Melamed‐Book N et al. Turnover of mitochondrial steroidogenic acute regulatory (StAR) protein by Lon protease: the unexpected effect of proteasome inhibitors. Mol Endocrinol 2007; 21: 2164–77. [DOI] [PubMed] [Google Scholar]
- 15. Melo J, Toczyski D. A unified view of the DNA‐damage checkpoint. Curr Opin Cell Biol 2002; 14: 237–45. [DOI] [PubMed] [Google Scholar]
- 16. Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature 2000; 408: 433–9. [DOI] [PubMed] [Google Scholar]
- 17. Shiloh Y. ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev 2001; 11: 71–7. [DOI] [PubMed] [Google Scholar]
- 18. Lee AY, Liu E, Wu X. The Mre11/Rad50/Nbs1 complex plays an important role in the prevention of DNA rereplication in mammalian cells. J Biol Chem 2007; 282: 32243–55. [DOI] [PubMed] [Google Scholar]
- 19. Liu E, Lee AY, Chiba T, Olson E, Sun P, Wu X. The ATR‐mediated S phase checkpoint prevents rereplication in mammalian cells when licensing control is disrupted. J Cell Biol 2007; 179: 643–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chou WC, Wang HC, Wong FH et al. Chk2‐dependent phosphorylation of XRCC1 in the DNA damage response promotes base excision repair. EMBO J 2008; 27: 3140–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clarke PR, Allan LA. Cell‐cycle control in the face of damage – a matter of life or death. Trends Cell Biol 2009; 19: 89–98. [DOI] [PubMed] [Google Scholar]
- 22. Norbury CJ, Zhivotovsky B. DNA damage‐induced apoptosis. Oncogene 2004; 23: 2797–808. [DOI] [PubMed] [Google Scholar]
- 23. Roos WP, Kaina B. DNA damage‐induced cell death by apoptosis. Trends Mol Med 2006; 12: 440–50. [DOI] [PubMed] [Google Scholar]
- 24. Liao JC, Huang TC, eds. Lauraceae in Flora of Taiwan, 2nd edn. Taipei: Editorial Committee of the Flora of Taiwan, 1996; 2: 443–83. [Google Scholar]
- 25. Chen CH, Lo WL, Liu YC, Chen CY. Chemical and cytotoxic constituents from the leaves of Cinnamomum kotoense . J Nat Prod 2006; 69: 927–33. [DOI] [PubMed] [Google Scholar]
- 26. Chen CY. Butanolides from the Stems of Cinnamomum kotoense . Nat Prod Commun 2006; 1: 453–5. [Google Scholar]
- 27. Chen FC, Peng CF, Tsai IL, Chen IS. Antitubercular constituents from the stem wood of Cinnamomum kotoense . J Nat Prod 2005; 68: 1318–23. [DOI] [PubMed] [Google Scholar]
- 28. Masutomi K, Yu EY, Khurts S et al. Telomerase maintains telomere structure in normal human cells. Cell 2003; 114: 241–53. [DOI] [PubMed] [Google Scholar]
- 29. Lee AY, Hsu CH, Wu SH. Functional domains of Brevibacillus thermoruber lon protease for oligomerization and DNA binding: role of N‐terminal and sensor and substrate discrimination domains. J Biol Chem 2004; 279: 34903–12. [DOI] [PubMed] [Google Scholar]
- 30. Lee AY, Tsay SS, Chen MY, Wu SH. Identification of a gene encoding Lon protease from Brevibacillus thermoruber WR‐249 and biochemical characterization of its thermostable recombinant enzyme. Eur J Biochem 2004; 271: 834–44. [DOI] [PubMed] [Google Scholar]
- 31. Chang YC, Jan KY, Cheng CA, Liao CB, Liu YC. Direct involvement of the tumor suppressor p53 in nucleotide excision repair. DNA Repair (Amst) 2008; 7: 751–61. [DOI] [PubMed] [Google Scholar]
- 32. Yen CY, Chiu CC, Chang FR et al. 4beta‐Hydroxywithanolide E from Physalis peruviana (golden berry) inhibits growth of human lung cancer cells through DNA damage, apoptosis and G2/M arrest. BMC Cancer 2010; 10: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bota DA, Davies KJ. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP‐stimulated mechanism. Nat Cell Biol 2002; 4: 674–80. [DOI] [PubMed] [Google Scholar]
- 34. Botos I, Melnikov EE, Cherry S et al. The catalytic domain of Escherichia coli Lon protease has a unique fold and a Ser‐Lys dyad in the active site. J Biol Chem 2004; 279: 8140–8. [DOI] [PubMed] [Google Scholar]
- 35. Besche H, Zwickl P. The Thermoplasma acidophilum Lon protease has a Ser‐Lys dyad active site. Eur J Biochem 2004; 271: 4361–5. [DOI] [PubMed] [Google Scholar]
- 36. Garcez FR, Garcez WS, Martins M et al. Cytotoxic and genotoxic butanolides and lignans from Aiouea trinervis . Planta Med 2005; 71: 923–7. [DOI] [PubMed] [Google Scholar]
- 37. Yokota T, Matsuzaki Y, Koyama M et al. Sesamin, a lignan of sesame, down‐regulates cyclin D1 protein expression in human tumor cells. Cancer Sci 2007; 98: 1447–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double‐strand breaks. J Biol Chem 2001; 276: 42462–7. [DOI] [PubMed] [Google Scholar]
- 39. Matsuzawa A, Ichijo H. Stress‐responsive protein kinases in redox‐regulated apoptosis signaling. Antioxid Redox Signal 2005; 7: 472–81. [DOI] [PubMed] [Google Scholar]
- 40. Sakon S, Xue X, Takekawa M et al. NF‐[kappa]B inhibits TNF‐induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J 2003; 22: 3898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ide T, Ashakumary L, Takahashi Y, Kushiro M, Fukuda N, Sugano M. Sesamin, a sesame lignan, decreases fatty acid synthesis in rat liver accompanying the down‐regulation of sterol regulatory element binding protein‐1. Biochim Biophys Acta 2001; 1534: 1–13. [DOI] [PubMed] [Google Scholar]
- 42. Ju Y, Still CC, Sacalis JN, Li J, Ho CT. Cytotoxic coumarins and lignans from extracts of the northern prickly ash (Zanthoxylum americanum). Phytother Res 2001; 15: 441–3. [DOI] [PubMed] [Google Scholar]
- 43. Hibasami H, Fujikawa T, Takeda H et al. Induction of apoptosis by Acanthopanax senticosus HARMS and its component, sesamin in human stomach cancer KATO III cells. Oncol Rep 2000; 7: 1213–6. [DOI] [PubMed] [Google Scholar]
- 44. Bayot A, Basse N, Lee I et al. Towards the control of intracellular protein turnover: mitochondrial Lon protease inhibitors versus proteasome inhibitors. Biochimie 2008; 90: 260–9. [DOI] [PubMed] [Google Scholar]
- 45. Sherr CJ. Principles of tumor suppression. Cell 2004; 116: 235–46. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and Methods. Further detailed information regarding antibody production, mass spectrometry, compound isolation, and assays.
Fig. S1. Purification of obtusilactone A and (‐)‐sesamin from the leaves of Cinnamomum kotoense.
Fig. S2. Identification of the recombinant human Lon and specificity of the polyclonal antiserum against human Lon assayed by Western blot analysis. Crude whole cell lysate from 293T cells (lane 1) and affinity purified human Lon (lane 2) were separated by SDS‐PAGE and the blotted membrane was incubated with purified with anti‐human Lon antibody overnight at a 1:2000 dilution. The arrow shows the recognized human Lon. The Western blot analysis showed that the antibody against human Lon recognizes mainly a single band of ~110 kDa from the 293T cells extract pool (lane 1) that is exactly the same size as the predominant purified recombinant Lon (lane 2).
Fig. S3. Identification of the recombinant human mitochondrial Lon protease by mass spectrometry analysis. Amino acid sequence of human Lon protease is shown (Accession No. NM_004793). The amino acid sequences in red represent the peptides found from tandem mass spectrometry sequencing.
Fig. S4. Obtusilactone A induces apoptosis, revealed by TUNEL assay. H1299 cells were treated with DMSO (blank) or the indicated concentration of OA for 12 h. Apoptotic cells show a green fluorescence that can be detected using a standard fluorescein filter at 520 nm. All cells stained with propidium iodide show a red fluorescence at 620 nm.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item